Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis–From Research to Clinical Practice

Abstract
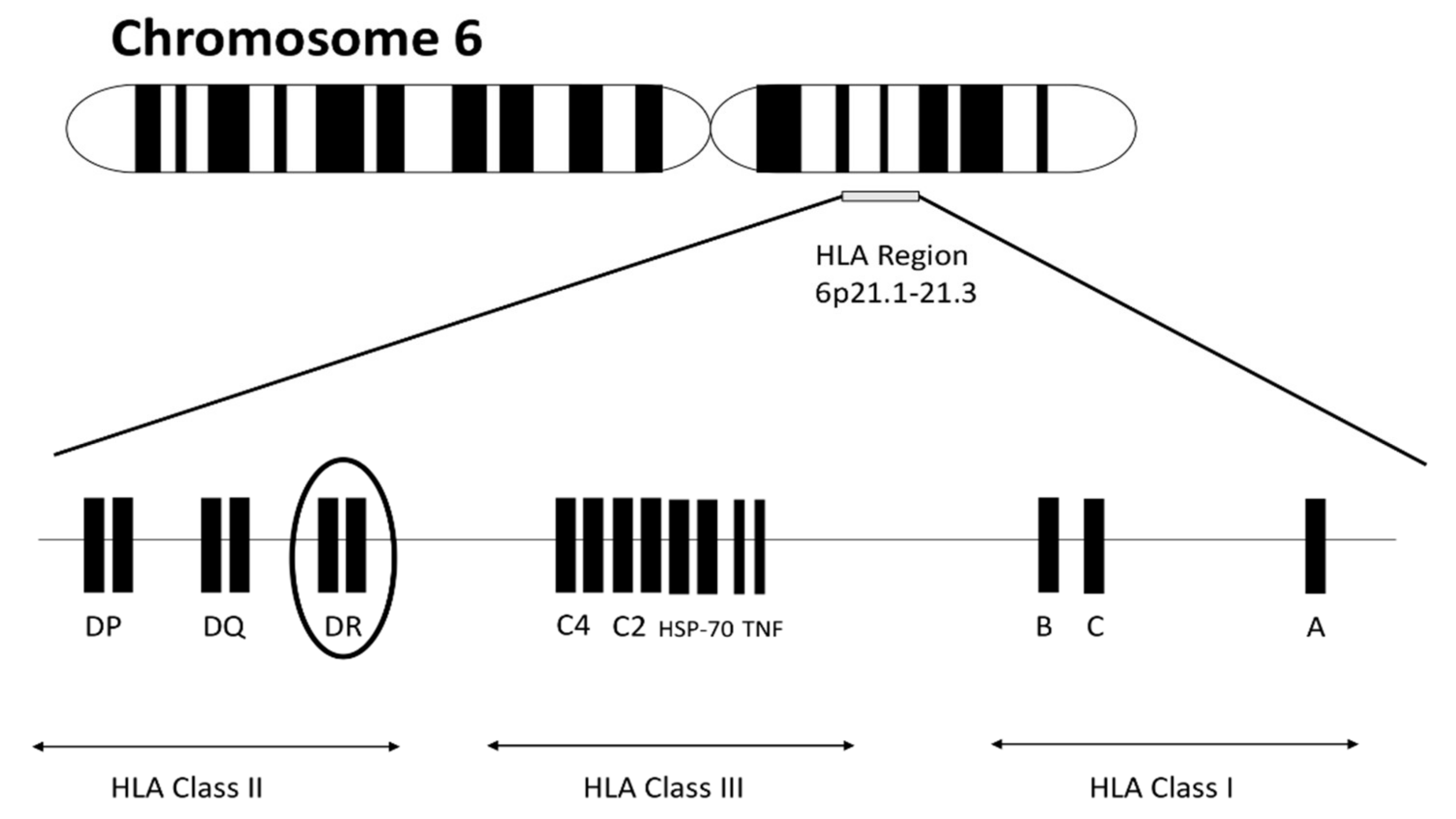
1. Introduction
- central, in which the self-reactive B and T cells are deleted during maturation in the thymus and bone marrow, respectively,
- peripheral, occurring through one of three mechanisms: clonal deletion (usually via apoptosis), induction of anergy (functional inactivation without cell death), or suppression of lymphocytes activation either by regulatory T cells or by clonal ignorance [3].
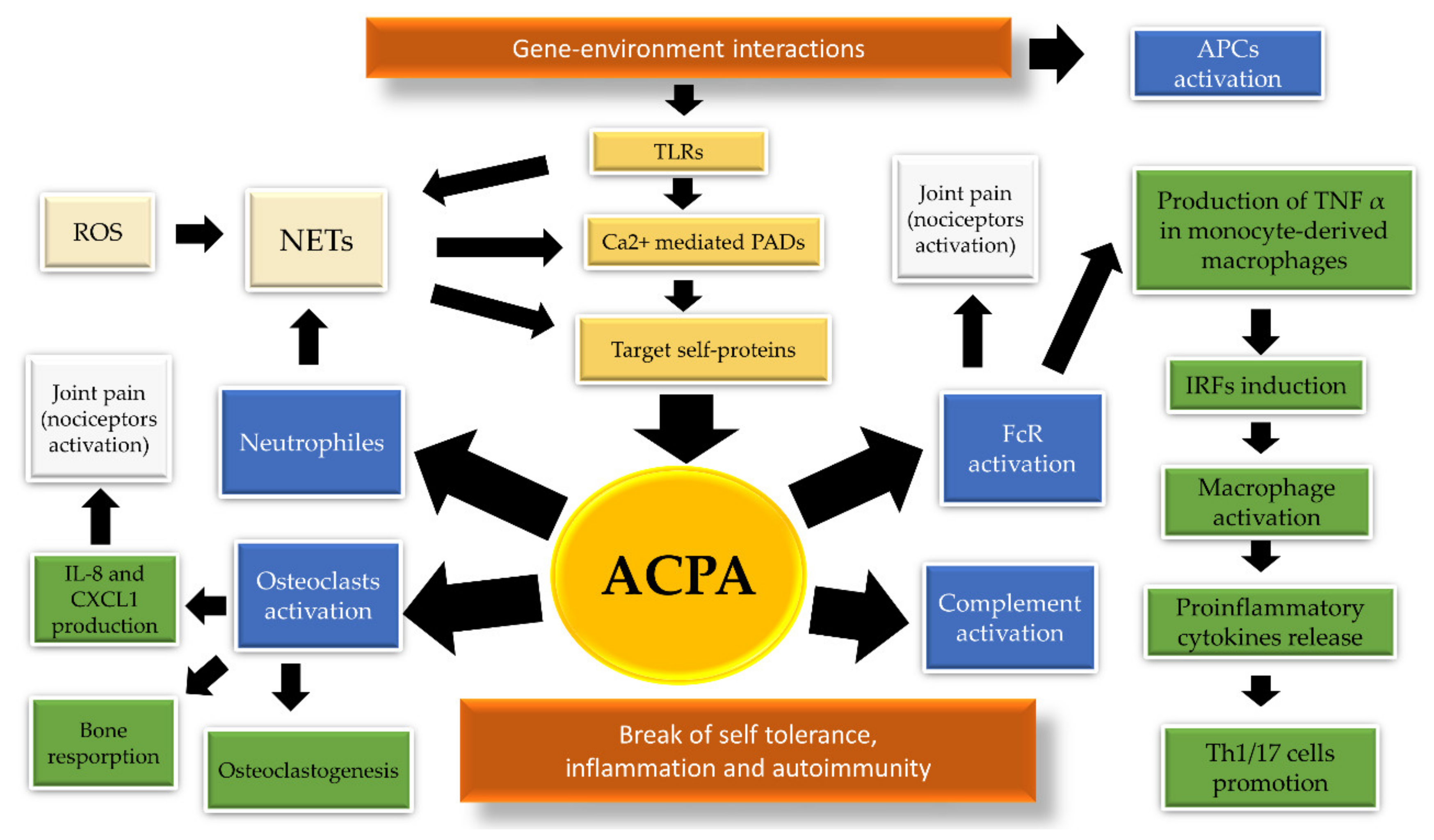
2. The Pathogenic Link between ACPA and HLA in Rheumatoid Arthritis
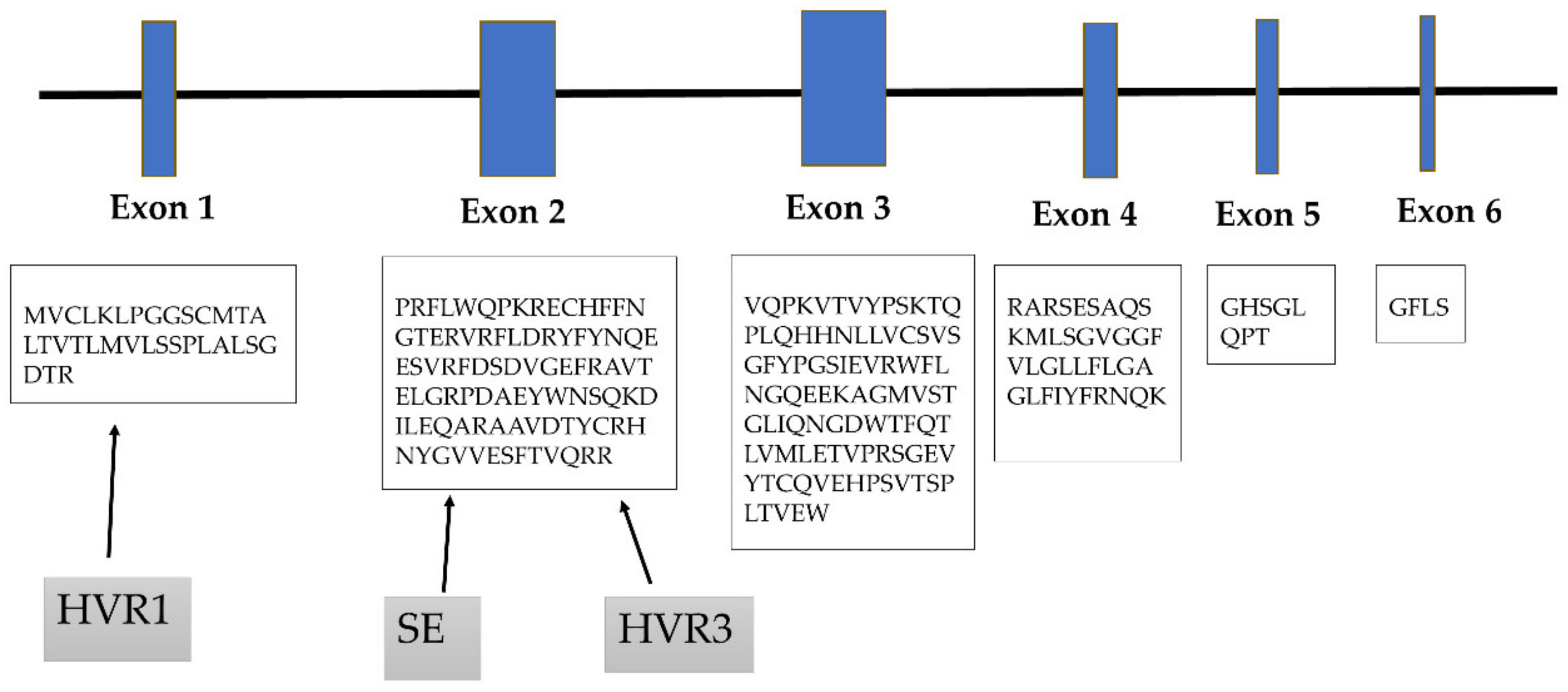
3. Role of Hypervariable Regions
4. Shared Epitope Hypothesis
- DRB1 *0401, *0409, *0413, *0416, *0421, *1419, *1421 (encoding 70QKRAA74 sequence),
- DRB1 *0101, *0102, *0105 *0404, *0405, *0408, *0410, *0419, *1402, *1406, *1409, *1413, *1417, *1420 (encoding 70QRRAA74)
- DRB1 *1001 (encoding 70RRRAA74) [48].
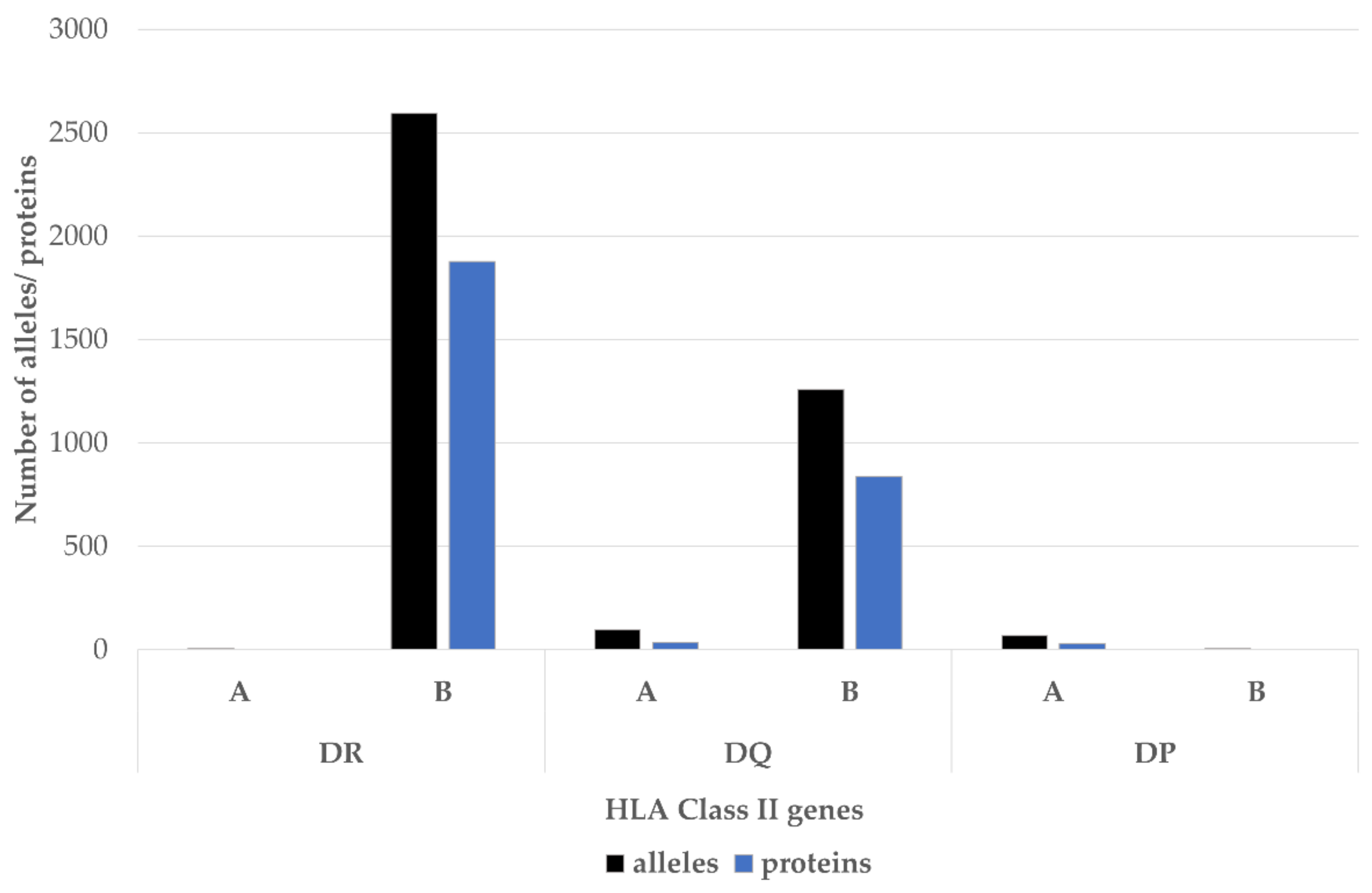
5. HLA-DRB1 Alleles Other than the SE
6. Protective Alleles
7. Ethnic Differences
8. Peptide Binding Affinity
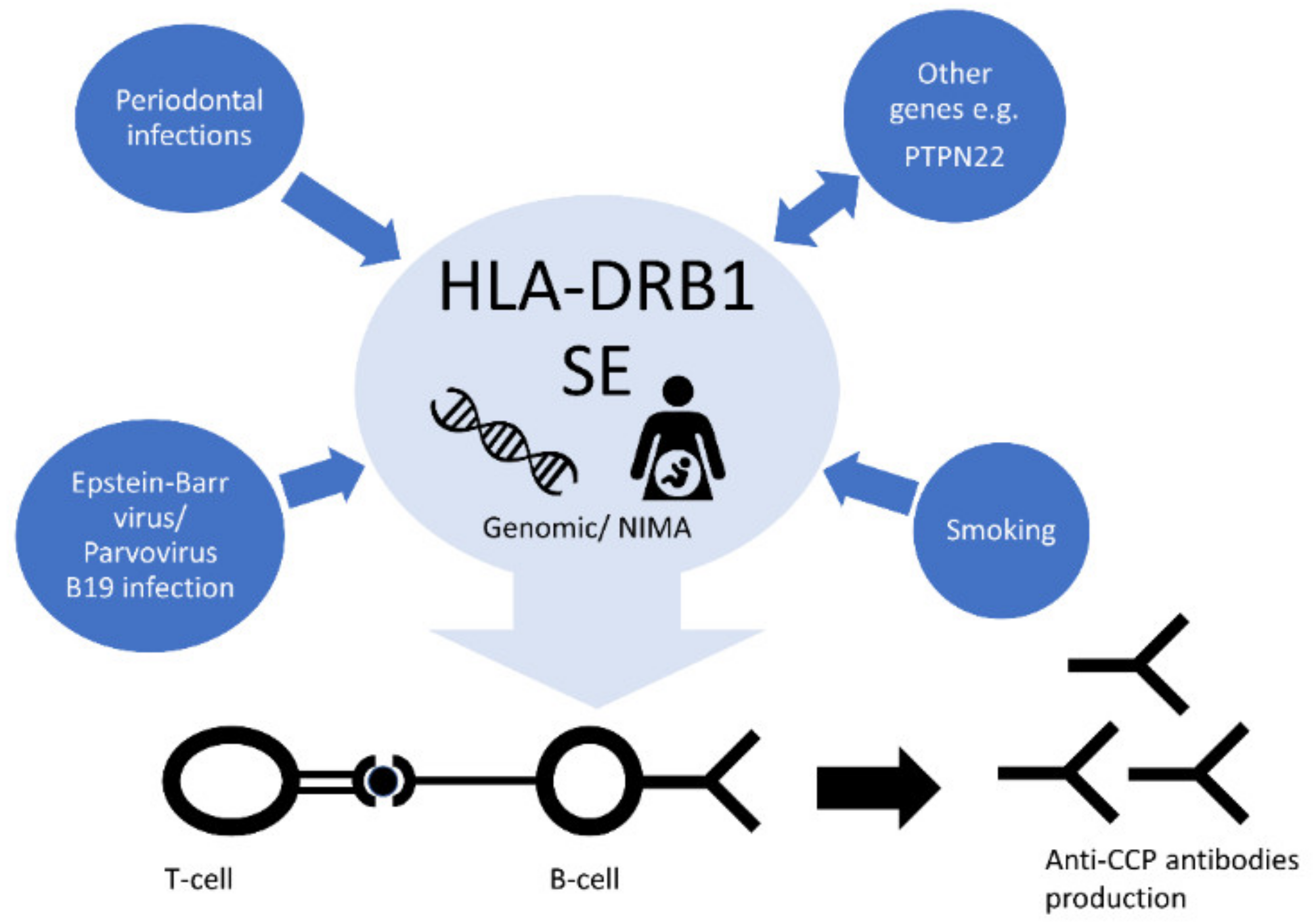
9. Genetic and Environmental Risk Factor’s Interactions
9.1. Genetic Interactions
9.2. Smoking
9.3. Alcohol Consumption
9.4. Viral Infections
9.5. Periodontal Infections
10. Microchimerism and Non-Inherited Maternal Antigens
11. Significance of HLA-DRB1 Methylation Status
12. Associations with Clinical Presentations
12.1. Mortality Risk
12.2. Risk of Radiographic Progression
12.3. Extra-Articular Manifestations
12.4. Pulmonary Fibrosis
12.5. Follicular Lymphoma
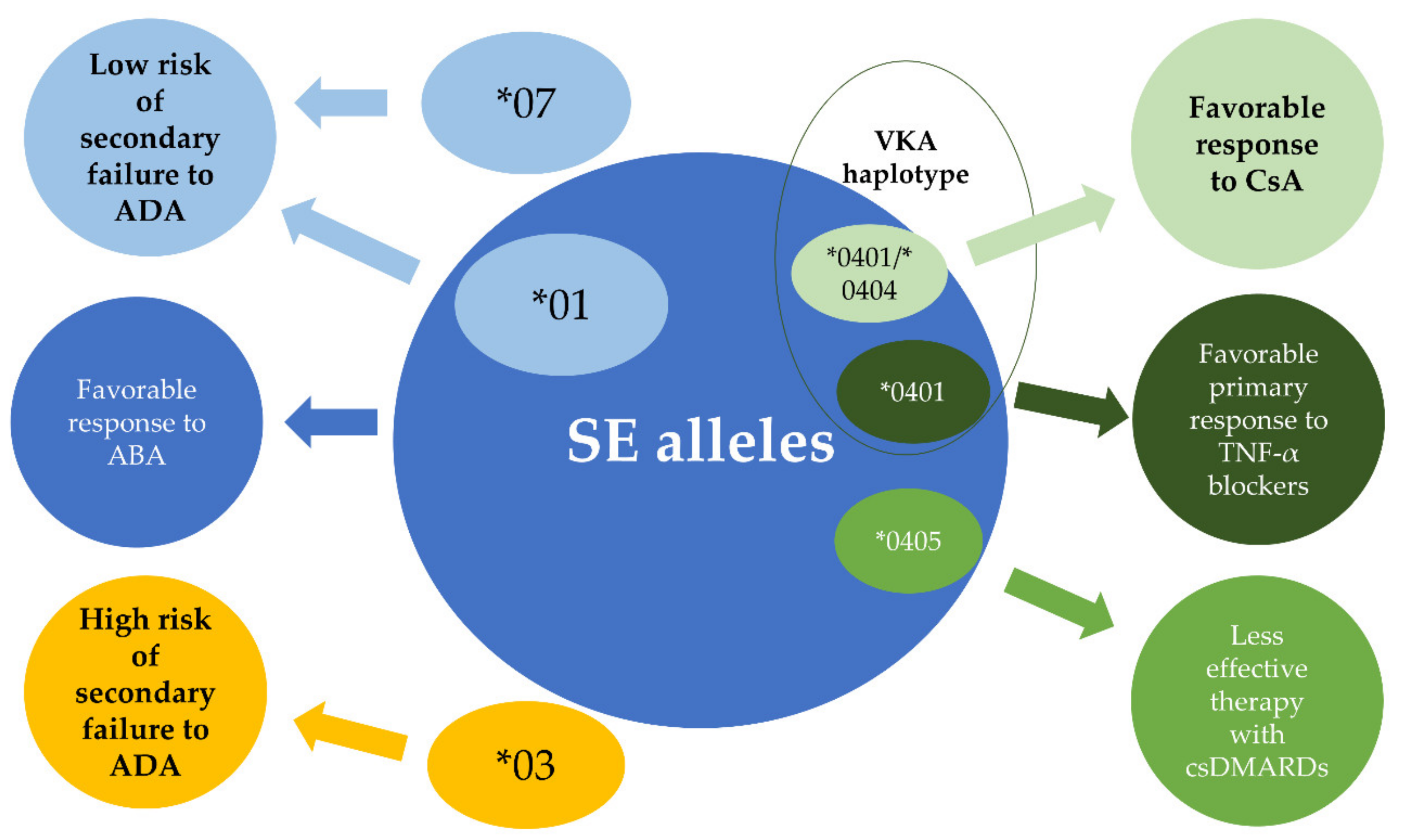
13. HLA-DRB 1 and Response to Treatment
13.1. Conventional Synthetic Disease-Modifying Anti-Rheumatic Drugs and Cyclosporine A
13.2. TNF- α Blockers
13.3. Abatacept
14. The Challenges Affecting the Implement HLA-DRB1 Genotyping in Clinical Practice
14.1. HLA Genotyping
14.2. Non-Mendelian Inheritance Pattern of RA–A Problem to Solve
14.3. Complex Pharmacogenetics of Anti-TNF Treatment Response
15. The Bumpy Road to Diagnostic Utility of HLA-DRB1
16. Concluding Remarks
Funding
Conflicts of Interest
References
- Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Incidence and prevalence of rheumatoid arthritis, based on the 1987 American College of Rheumatology criteria: A systematic review. Semin. Arthritis Rheum. 2006, 36, 182–188. [Google Scholar] [CrossRef]
- Jerne, N.K. The somatic generation of immune recognition. 1971. Eur. J. Immunol. 2004, 34, 1234–1242. [Google Scholar] [CrossRef] [PubMed]
- Szaflarska, A.; Rutkowska-Zapala, M.; Kowalczyk, D. Immune tolerance mechanisms—Brief review. Prz. Lek. 2015, 72, 765–769. [Google Scholar] [PubMed]
- Van Venrooij, W.J.; van Beers, J.J.; Pruijn, G.J. Anti-CCP Antibody, a Marker for the Early Detection of Rheumatoid Arthritis. Ann. N. Y. Acad. Sci. 2008, 1143, 268–285. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K. Genetics of rheumatoid arthritis: Confronting complexity. Arthritis Res. 1999, 1, 37–44. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Raychaudhuri, S. Recent advances in the genetics of rheumatoid arthritis. Curr. Opin. Rheumatol. 2010, 22, 109–118. [Google Scholar] [CrossRef]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Svendsen, A.J.; Kyvik, K.O.; Houen, G.; Junker, P.; Christensen, K.; Christiansen, L.; Nielsen, C.; Skytthe, A.; Hjelmborg, J.V. On the origin of rheumatoid arthritis: The impact of environment and genes—A population based twin study. PLoS ONE 2013, 8, e57304. [Google Scholar] [CrossRef]
- Van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.; Huizinga, T.W.; Thomson, W.; Worthington, J.; van der Helm-van Mil, A.H.; de Vries, R.R. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009, 60, 916–923. [Google Scholar] [CrossRef]
- Frisell, T.; Holmqvist, M.; Kallberg, H.; Klareskog, L.; Alfredsson, L.; Askling, J. Familial risks and heritability of rheumatoid arthritis: Role of rheumatoid factor/anti-citrullinated protein antibody status, number and type of affected relatives, sex, and age. Arthritis Rheum. 2013, 65, 2773–2782. [Google Scholar] [CrossRef] [PubMed]
- Deighton, C.M.; Walker, D.J.; Griffiths, I.D.; Roberts, D.F. The contribution of HLA to rheumatoid arthritis. Clin. Genet. 1989, 36, 178–182. [Google Scholar] [CrossRef]
- Okada, Y.; Suzuki, A.; Ikari, K.; Terao, C.; Kochi, Y.; Ohmura, K.; Higasa, K.; Akiyama, M.; Ashikawa, K.; Kanai, M.; et al. Contribution of a Non-classical HLA Gene, HLA-DOA, to the Risk of Rheumatoid Arthritis. Am. J. Hum. Genet. 2016, 99, 366–374. [Google Scholar] [CrossRef]
- Lard, L.R.; Boers, M.; Verhoeven, A.; Vos, K.; Visser, H.; Hazes, J.M.; Zwinderman, A.H.; Schreuder, G.M.; Breedveld, F.C.; De Vries, R.R.; et al. Early and aggressive treatment of rheumatoid arthritis patients affects the association of HLA class II antigens with progression of joint damage. Arthritis Rheum. 2002, 46, 899–905. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, J.R.; Nepom, B.S.; Haire, C.; Gersuk, V.H.; Gaur, L.; Moore, G.F.; Drymalski, W.; Palmer, W.; Eckhoff, P.J.; Klassen, L.W.; et al. HLA-DRB1 typing in rheumatoid arthritis: Predicting response to specific treatments. Ann. Rheum. Dis. 1998, 57, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Rantapaa-Dahlqvist, S.; de Jong, B.A.; Berglin, E.; Hallmans, G.; Wadell, G.; Stenlund, H.; Sundin, U.; van Venrooij, W.J. Antibodies against cyclic citrullinated peptide and IgA rheumatoid factor predict the development of rheumatoid arthritis. Arthritis Rheum. 2003, 48, 2741–2749. [Google Scholar] [CrossRef]
- Chibnik, L.B.; Mandl, L.A.; Costenbader, K.H.; Schur, P.H.; Karlson, E.W. Comparison of threshold cutpoints and continuous measures of anti-cyclic citrullinated peptide antibodies in predicting future rheumatoid arthritis. J. Rheumatol. 2009, 36, 706–711. [Google Scholar] [CrossRef][Green Version]
- Laurent, L.; Clavel, C.; Lemaire, O.; Anquetil, F.; Cornillet, M.; Zabraniecki, L.; Nogueira, L.; Fournie, B.; Serre, G.; Sebbag, M. Fcgamma receptor profile of monocytes and macrophages from rheumatoid arthritis patients and their response to immune complexes formed with autoantibodies to citrullinated proteins. Ann. Rheum. Dis. 2011, 70, 1052–1059. [Google Scholar] [CrossRef]
- Sokolove, J.; Zhao, X.; Chandra, P.E.; Robinson, W.H. Immune complexes containing citrullinated fibrinogen costimulate macrophages via Toll-like receptor 4 and Fcgamma receptor. Arthritis Rheum. 2011, 63, 53–62. [Google Scholar] [CrossRef]
- Laurent, L.; Anquetil, F.; Clavel, C.; Ndongo-Thiam, N.; Offer, G.; Miossec, P.; Pasquali, J.L.; Sebbag, M.; Serre, G. IgM rheumatoid factor amplifies the inflammatory response of macrophages induced by the rheumatoid arthritis-specific immune complexes containing anticitrullinated protein antibodies. Ann. Rheum. Dis. 2015, 74, 1425–1431. [Google Scholar] [CrossRef]
- Anquetil, F.; Clavel, C.; Offer, G.; Serre, G.; Sebbag, M. IgM and IgA rheumatoid factors purified from rheumatoid arthritis sera boost the Fc receptor- and complement-dependent effector functions of the disease-specific anti-citrullinated protein autoantibodies. J. Immunol. 2015, 194, 3664–3674. [Google Scholar] [CrossRef] [PubMed]
- Trouw, L.A.; Haisma, E.M.; Levarht, E.W.; van der Woude, D.; Ioan-Facsinay, A.; Daha, M.R.; Huizinga, T.W.; Toes, R.E. Anti-cyclic citrullinated peptide antibodies from rheumatoid arthritis patients activate complement via both the classical and alternative pathways. Arthritis Rheum. 2009, 60, 1923–1931. [Google Scholar] [CrossRef] [PubMed]
- Khandpur, R.; Carmona-Rivera, C.; Vivekanandan-Giri, A.; Gizinski, A.; Yalavarthi, S.; Knight, J.S.; Friday, S.; Li, S.; Patel, R.M.; Subramanian, V.; et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci. Transl. Med. 2013, 5, 178ra140. [Google Scholar] [CrossRef] [PubMed]
- Wigerblad, G.; Bas, D.B.; Fernades-Cerqueira, C.; Krishnamurthy, A.; Nandakumar, K.S.; Rogoz, K.; Kato, J.; Sandor, K.; Su, J.; Jimenez-Andrade, J.M.; et al. Autoantibodies to citrullinated proteins induce joint pain independent of inflammation via a chemokine-dependent mechanism. Ann. Rheum. Dis. 2016, 75, 730–738. [Google Scholar] [CrossRef]
- Catrina, A.I.; Svensson, C.I.; Malmstrom, V.; Schett, G.; Klareskog, L. Mechanisms leading from systemic autoimmunity to joint-specific disease in rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 79–86. [Google Scholar] [CrossRef]
- Van de Stadt, L.A.; de Koning, M.H.; van de Stadt, R.J.; Wolbink, G.; Dijkmans, B.A.; Hamann, D.; van Schaardenburg, D. Development of the anti-citrullinated protein antibody repertoire prior to the onset of rheumatoid arthritis. Arthritis Rheum. 2011, 63, 3226–3233. [Google Scholar] [CrossRef]
- Suwannalai, P.; van de Stadt, L.A.; Radner, H.; Steiner, G.; El-Gabalawy, H.S.; Zijde, C.M.; van Tol, M.J.; van Schaardenburg, D.; Huizinga, T.W.; Toes, R.E.; et al. Avidity maturation of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2012, 64, 1323–1328. [Google Scholar] [CrossRef]
- Scherer, H.U.; van der Woude, D.; Ioan-Facsinay, A.; el Bannoudi, H.; Trouw, L.A.; Wang, J.; Haupl, T.; Burmester, G.R.; Deelder, A.M.; Huizinga, T.W.; et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum. 2010, 62, 1620–1629. [Google Scholar] [CrossRef]
- Hafkenscheid, L.; Bondt, A.; Scherer, H.U.; Huizinga, T.W.; Wuhrer, M.; Toes, R.E.; Rombouts, Y. Structural Analysis of Variable Domain Glycosylation of Anti-Citrullinated Protein Antibodies in Rheumatoid Arthritis Reveals the Presence of Highly Sialylated Glycans. Mol. Cell. Proteom. MCP 2017, 16, 278–287. [Google Scholar] [CrossRef]
- Kissel, T.; van Schie, K.A.; Hafkenscheid, L.; Lundquist, A.; Kokkonen, H.; Wuhrer, M.; Huizinga, T.W.; Scherer, H.U.; Toes, R.; Rantapaa-Dahlqvist, S. On the presence of HLA-SE alleles and ACPA-IgG variable domain glycosylation in the phase preceding the development of rheumatoid arthritis. Ann. Rheum. Dis. 2019. [Google Scholar] [CrossRef]
- Hafkenscheid, L.; de Moel, E.; Smolik, I.; Tanner, S.; Meng, X.; Jansen, B.C.; Bondt, A.; Wuhrer, M.; Huizinga, T.W.J.; Toes, R.E.M.; et al. N-Linked Glycans in the Variable Domain of IgG Anti-Citrullinated Protein Antibodies Predict the Development of Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1626–1633. [Google Scholar] [CrossRef] [PubMed]
- Vergroesen, R.D.; Slot, L.M.; Hafkenscheid, L.; Koning, M.T.; Scherer, H.U.; Toes, R.E.M. Response to: Acquiring new N-glycosylation sites in variable regions of immunoglobulin genes by somatic hypermutation is a common feature of autoimmune diseases’ by Visser et al. Ann. Rheum. Dis. 2018, 77, e70. [Google Scholar] [CrossRef] [PubMed]
- Stastny, P. Mixed lymphocyte cultures in rheumatoid arthritis. J. Clin. Investig. 1976, 57, 1148–1157. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Halliwell, J.A.; Hayhurst, J.D.; Flicek, P.; Parham, P.; Marsh, S.G. The IPD and IMGT/HLA database: Allele variant databases. Nucleic Acids Res. 2015, 43, D423–D431. [Google Scholar] [CrossRef]
- Reinsmoen, N.L.; Bach, F.H. Structural model for T-cell recognition of HLA class II-associated alloepitopes. Hum. Immunol. 1990, 27, 51–72. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Reviron, D.; Perdriger, A.; Toussirot, E.; Wendling, D.; Balandraud, N.; Guis, S.; Semana, G.; Tiberghien, P.; Mercier, P.; Roudier, J. Influence of shared epitope-negative HLA-DRB1 alleles on genetic susceptibility to rheumatoid arthritis. Arthritis Rheum. 2001, 44, 535–540. [Google Scholar] [CrossRef]
- Ting, Y.T.; Petersen, J.; Ramarathinam, S.H.; Scally, S.W.; Loh, K.L.; Thomas, R.; Suri, A.; Baker, D.G.; Purcell, A.W.; Reid, H.H.; et al. The interplay between citrullination and HLA-DRB1 polymorphism in shaping peptide binding hierarchies in rheumatoid arthritis. J. Biol. Chem. 2018, 293, 3236–3251. [Google Scholar] [CrossRef]
- Kolfenbach, J.R.; Deane, K.D.; Derber, L.A.; O’Donnell, C.; Weisman, M.H.; Buckner, J.H.; Gersuk, V.H.; Wei, S.; Mikuls, T.R.; O’Dell, J.; et al. A prospective approach to investigating the natural history of preclinical rheumatoid arthritis (RA) using first-degree relatives of probands with RA. Arthritis Rheum. 2009, 61, 1735–1742. [Google Scholar] [CrossRef]
- Kallberg, H.; Padyukov, L.; Plenge, R.M.; Ronnelid, J.; Gregersen, P.K.; van der Helm-van Mil, A.H.; Toes, R.E.; Huizinga, T.W.; Klareskog, L.; Alfredsson, L.; et al. Gene-gene and gene-environment interactions involving HLA-DRB1, PTPN22, and smoking in two subsets of rheumatoid arthritis. Am. J. Hum. Genet. 2007, 80, 867–875. [Google Scholar] [CrossRef]
- Cruz, G.I.; Shao, X.; Quach, H.; Ho, K.A.; Sterba, K.; Noble, J.A.; Patsopoulos, N.A.; Busch, M.P.; Triulzi, D.J.; Wong, W.S.; et al. Increased risk of rheumatoid arthritis among mothers with children who carry DRB1 risk-associated alleles. Ann. Rheum. Dis. 2017, 76, 1405–1410. [Google Scholar] [CrossRef] [PubMed]
- Jawaheer, D.; Thomson, W.; MacGregor, A.J.; Carthy, D.; Davidson, J.; Dyer, P.A.; Silman, A.J.; Ollier, W.E. “Homozygosity” for the HLA-DR shared epitope contributes the highest risk for rheumatoid arthritis concordance in identical twins. Arthritis Rheum. 1994, 37, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Huizinga, T.W.; Amos, C.I.; van der Helm-van Mil, A.H.; Chen, W.; van Gaalen, F.A.; Jawaheer, D.; Schreuder, G.M.; Wener, M.; Breedveld, F.C.; Ahmad, N.; et al. Refining the complex rheumatoid arthritis phenotype based on specificity of the HLA-DRB1 shared epitope for antibodies to citrullinated proteins. Arthritis Rheum. 2005, 52, 3433–3438. [Google Scholar] [CrossRef]
- Hensvold, A.H.; Magnusson, P.K.; Joshua, V.; Hansson, M.; Israelsson, L.; Ferreira, R.; Jakobsson, P.J.; Holmdahl, R.; Hammarstrom, L.; Malmstrom, V.; et al. Environmental and genetic factors in the development of anticitrullinated protein antibodies (ACPAs) and ACPA-positive rheumatoid arthritis: An epidemiological investigation in twins. Ann. Rheum. Dis. 2015, 74, 375–380. [Google Scholar] [CrossRef]
- Wagner, U.; Kaltenhauser, S.; Pierer, M.; Seidel, W.; Troltzsch, M.; Hantzschel, H.; Kalden, J.R.; Wassmuth, R. Prospective analysis of the impact of HLA-DR and -DQ on joint destruction in recent-onset rheumatoid arthritis. Rheumatology 2003, 42, 553–562. [Google Scholar] [CrossRef][Green Version]
- Farragher, T.M.; Goodson, N.J.; Naseem, H.; Silman, A.J.; Thomson, W.; Symmons, D.; Barton, A. Association of the HLA-DRB1 gene with premature death, particularly from cardiovascular disease, in patients with rheumatoid arthritis and inflammatory polyarthritis. Arthritis Rheum. 2008, 58, 359–369. [Google Scholar] [CrossRef]
- Del Rincon, I.; Battafarano, D.F.; Arroyo, R.A.; Murphy, F.T.; Escalante, A. Heterogeneity between men and women in the influence of the HLA-DRB1 shared epitope on the clinical expression of rheumatoid arthritis. Arthritis Rheum. 2002, 46, 1480–1488. [Google Scholar] [CrossRef]
- Newton, J.L.; Harney, S.M.; Wordsworth, B.P.; Brown, M.A. A review of the MHC genetics of rheumatoid arthritis. Genes Immun. 2004, 5, 151–157. [Google Scholar] [CrossRef]
- Thomson, W.; Harrison, B.; Ollier, B.; Wiles, N.; Payton, T.; Barrett, J.; Symmons, D.; Silman, A. Quantifying the exact role of HLA-DRB1 alleles in susceptibility to inflammatory polyarthritis: Results from a large, population-based study. Arthritis Rheum. 1999, 42, 757–762. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Hajeer, A.H.; Dababneh, A.; Makki, R.; Garcia-Porrua, C.; Thomson, W.; Ollier, W. Seronegative rheumatoid arthritis in elderly and polymyalgia rheumatica have similar patterns of HLA association. J. Rheumatol. 2001, 28, 122–125. [Google Scholar]
- Hinks, A.; Marion, M.C.; Cobb, J.; Comeau, M.E.; Sudman, M.; Ainsworth, H.C.; Bowes, J.; Juvenile Idiopathic Arthritis Consortium for Immunochip; Becker, M.L.; Bohnsack, J.F.; et al. The Genetic Profile of Rheumatoid Factor-Positive Polyarticular Juvenile Idiopathic Arthritis Resembles That of Adult Rheumatoid Arthritis. Arthritis Rheum. 2018. [Google Scholar] [CrossRef]
- Okada, Y.; Kim, K.; Han, B.; Pillai, N.E.; Ong, R.T.; Saw, W.Y.; Luo, M.; Jiang, L.; Yin, J.; Bang, S.Y.; et al. Risk for ACPA-positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Hum. Mol. Genet. 2014, 23, 6916–6926. [Google Scholar] [CrossRef]
- Nagafuchi, Y.; Shoda, H.; Sumitomo, S.; Nakachi, S.; Kato, R.; Tsuchida, Y.; Tsuchiya, H.; Sakurai, K.; Hanata, N.; Tateishi, S.; et al. Immunophenotyping of rheumatoid arthritis reveals a linkage between HLA-DRB1 genotype, CXCR4 expression on memory CD4(+) T cells, and disease activity. Sci. Rep. 2016, 6, 29338. [Google Scholar] [CrossRef]
- Alvandpur, N.; Tabatabaei, R.; Tahamoli-Roudsari, A.; Basiri, Z.; Behzad, M.; Rezaeepoor, M.; Roshanaei, G.; Hajilooi, M.; Solgi, G. Circulating IFN-gamma producing CD4+ T cells and IL-17A producing CD4+ T cells, HLA-shared epitope and ACPA may characterize the clinical response to therapy in rheumatoid arthritis patients. Hum. Immunol. 2020. [Google Scholar] [CrossRef]
- Guthrie, K.A.; Tishkevich, N.R.; Nelson, J.L. Non-inherited maternal human leukocyte antigen alleles in susceptibility to familial rheumatoid arthritis. Ann. Rheum. Dis. 2009, 68, 107–109. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Bitoun, S.; Roques, P.; Maillere, B.; Le Grand, R.; Mariette, X. Valine 11 and phenylalanine 13 have a greater impact on the T-cell response to citrullinated peptides than the 70-74 shared epitope of the DRB1 molecule in macaques. Ann. Rheum. Dis. 2019, 78, 917–921. [Google Scholar] [CrossRef] [PubMed]
- Van Steenbergen, H.W.; Raychaudhuri, S.; Rodriguez-Rodriguez, L.; Rantapaa-Dahlqvist, S.; Berglin, E.; Toes, R.E.; Huizinga, T.W.; Fernandez-Gutierrez, B.; Gregersen, P.K.; van der Helm-van Mil, A.H. Association of valine and leucine at HLA-DRB1 position 11 with radiographic progression in rheumatoid arthritis, independent of the shared epitope alleles but not independent of anti-citrullinated protein antibodies. Arthritis Rheumatol. 2015, 67, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Milicic, A.; Lee, D.; Brown, M.A.; Darke, C.; Wordsworth, B.P. HLA-DR/DQ haplotype in rheumatoid arthritis: Novel allelic associations in UK Caucasians. J. Rheumatol. 2002, 29, 1821–1826. [Google Scholar]
- Furuya, T.; Hakoda, M.; Ichikawa, N.; Higami, K.; Nanke, Y.; Yago, T.; Kobashigawa, T.; Tokunaga, K.; Tsuchiya, N.; Kamatani, N.; et al. Differential association of HLA-DRB1 alleles in Japanese patients with early rheumatoid arthritis in relationship to autoantibodies to cyclic citrullinated peptide. Clin. Exp. Rheumatol. 2007, 25, 219–224. [Google Scholar]
- Mackie, S.L.; Taylor, J.C.; Martin, S.G.; Consortium, Y.; Consortium, U.; Wordsworth, P.; Steer, S.; Wilson, A.G.; Worthington, J.; Emery, P.; et al. A spectrum of susceptibility to rheumatoid arthritis within HLA-DRB1: Stratification by autoantibody status in a large UK population. Genes Immun. 2012, 13, 120–128. [Google Scholar] [CrossRef]
- Van der Helm-van Mil, A.H.; Huizinga, T.W.; Schreuder, G.M.; Breedveld, F.C.; de Vries, R.R.; Toes, R.E. An independent role of protective HLA class II alleles in rheumatoid arthritis severity and susceptibility. Arthritis Rheum. 2005, 52, 2637–2644. [Google Scholar] [CrossRef] [PubMed]
- Balsa, A.; Cabezon, A.; Orozco, G.; Cobo, T.; Miranda-Carus, E.; Lopez-Nevot, M.A.; Vicario, J.L.; Martin-Mola, E.; Martin, J.; Pascual-Salcedo, D. Influence of HLA DRB1 alleles in the susceptibility of rheumatoid arthritis and the regulation of antibodies against citrullinated proteins and rheumatoid factor. Arthritis Res. Ther. 2010, 12, R62. [Google Scholar] [CrossRef] [PubMed]
- Van der Woude, D.; Lie, B.A.; Lundstrom, E.; Balsa, A.; Feitsma, A.L.; Houwing-Duistermaat, J.J.; Verduijn, W.; Nordang, G.B.; Alfredsson, L.; Klareskog, L.; et al. Protection against anti-citrullinated protein antibody-positive rheumatoid arthritis is predominantly associated with HLA-DRB1*1301: A meta-analysis of HLA-DRB1 associations with anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis in four European populations. Arthritis Rheum. 2010, 62, 1236–1245. [Google Scholar] [CrossRef] [PubMed]
- Oka, S.; Furukawa, H.; Kawasaki, A.; Shimada, K.; Sugii, S.; Hashimoto, A.; Komiya, A.; Fukui, N.; Ito, S.; Nakamura, T.; et al. Protective effect of the HLA-DRB1*13:02 allele in Japanese rheumatoid arthritis patients. PLoS ONE 2014, 9, e99453. [Google Scholar] [CrossRef]
- Yang, M.; Kuang, X.; Li, J.; Pan, Y.; Tan, M.; Lu, B.; Cheng, Q.; Wu, L.; Pang, G. Meta-analysis of the association of HLA-DRB1 with rheumatoid arthritis in Chinese populations. BMC Musculoskelet. Disord. 2013, 14, 307. [Google Scholar] [CrossRef]
- Balandraud, N.; Picard, C.; Reviron, D.; Landais, C.; Toussirot, E.; Lambert, N.; Telle, E.; Charpin, C.; Wendling, D.; Pardoux, E.; et al. HLA-DRB1 genotypes and the risk of developing anti citrullinated protein antibody (ACPA) positive rheumatoid arthritis. PLoS ONE 2013, 8, e64108. [Google Scholar] [CrossRef]
- Han, B.; Diogo, D.; Eyre, S.; Kallberg, H.; Zhernakova, A.; Bowes, J.; Padyukov, L.; Okada, Y.; Gonzalez-Gay, M.A.; Rantapaa-Dahlqvist, S.; et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am. J. Hum. Genet. 2014, 94, 522–532. [Google Scholar] [CrossRef]
- Verpoort, K.N.; van Gaalen, F.A.; van der Helm-van Mil, A.H.; Schreuder, G.M.; Breedveld, F.C.; Huizinga, T.W.; de Vries, R.R.; Toes, R.E. Association of HLA-DR3 with anti-cyclic citrullinated peptide antibody-negative rheumatoid arthritis. Arthritis Rheum. 2005, 52, 3058–3062. [Google Scholar] [CrossRef]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Terao, C.; Yano, K.; Ikari, K.; Furu, M.; Yamakawa, N.; Yoshida, S.; Hashimoto, M.; Ito, H.; Fujii, T.; Ohmura, K.; et al. Brief Report: Main Contribution of DRB1*04:05 Among the Shared Epitope Alleles and Involvement of DRB1 Amino Acid Position 57 in Association With Joint Destruction in Anti-Citrullinated Protein Antibody-Positive Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 1744–1750. [Google Scholar] [CrossRef] [PubMed]
- Ferucci, E.D.; Templin, D.W.; Lanier, A.P. Rheumatoid arthritis in American Indians and Alaska Natives: A review of the literature. Semin. Arthritis Rheum. 2005, 34, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Vega, A.M.; Anaya, J.M. Meta-analysis of HLA-DRB1 polymorphism in Latin American patients with rheumatoid arthritis. Autoimmun. Rev. 2007, 6, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Cruz-Tapias, P.; Perez-Fernandez, O.M.; Rojas-Villarraga, A.; Rodriguez-Rodriguez, A.; Arango, M.T.; Anaya, J.M. Shared HLA Class II in Six Autoimmune Diseases in Latin America: A Meta-Analysis. Autoimmune Dis. 2012, 2012, 569728. [Google Scholar] [CrossRef]
- De Vries, N.; Ronningen, K.S.; Tilanus, M.G.; Bouwens-Rombouts, A.; Segal, R.; Egeland, T.; Thorsby, E.; van de Putte, L.B.; Brautbar, C. HLA-DR1 and rheumatoid arthritis in Israeli Jews: Sequencing reveals that DRB1*0102 is the predominant HLA-DR1 subtype. Tissue Antigens 1993, 41, 26–30. [Google Scholar] [CrossRef]
- Hughes, L.B.; Morrison, D.; Kelley, J.M.; Padilla, M.A.; Vaughan, L.K.; Westfall, A.O.; Dwivedi, H.; Mikuls, T.R.; Holers, V.M.; Parrish, L.A.; et al. The HLA-DRB1 shared epitope is associated with susceptibility to rheumatoid arthritis in African Americans through European genetic admixture. Arthritis Rheum. 2008, 58, 349–358. [Google Scholar] [CrossRef]
- Singwe-Ngandeu, M.; Finckh, A.; Bas, S.; Tiercy, J.M.; Gabay, C. Diagnostic value of anti-cyclic citrullinated peptides and association with HLA-DRB1 shared epitope alleles in African rheumatoid arthritis patients. Arthritis Res. Ther. 2010, 12, R36. [Google Scholar] [CrossRef]
- Kochi, Y.; Yamada, R.; Kobayashi, K.; Takahashi, A.; Suzuki, A.; Sekine, A.; Mabuchi, A.; Akiyama, F.; Tsunoda, T.; Nakamura, Y.; et al. Analysis of single-nucleotide polymorphisms in Japanese rheumatoid arthritis patients shows additional susceptibility markers besides the classic shared epitope susceptibility sequences. Arthritis Rheum. 2004, 50, 63–71. [Google Scholar] [CrossRef]
- Lee, H.S.; Lee, K.W.; Song, G.G.; Kim, H.A.; Kim, S.Y.; Bae, S.C. Increased susceptibility to rheumatoid arthritis in Koreans heterozygous for HLA-DRB1*0405 and *0901. Arthritis Rheum. 2004, 50, 3468–3475. [Google Scholar] [CrossRef]
- Jun, K.R.; Choi, S.E.; Cha, C.H.; Oh, H.B.; Heo, Y.S.; Ahn, H.Y.; Lee, K.J. Meta-analysis of the association between HLA-DRB1 allele and rheumatoid arthritis susceptibility in Asian populations. J. Korean Med. Sci. 2007, 22, 973–980. [Google Scholar] [CrossRef]
- Sepulveda-Delgado, J.; Rizo-Pinto, A.; Granados-Arriola, J.; Mena-Vela, B.A.; Cetina-Diaz, J.H.; Garcia-Silva, R.; Hernandez-Dono, S.; Cruz-Salvatierra, M.A.; Perez-Tirado, J.M.; Vazquez-Guzman, C.; et al. Role of HLA-DRB1*04 in the susceptibility and HLA-DRB1*08 in the protection for development of rheumatoid arthritis in a population of Southern Mexico: Brief report. Clin. Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Rossjohn, J.; Gras, S.; Miles, J.J.; Turner, S.J.; Godfrey, D.I.; McCluskey, J. T cell antigen receptor recognition of antigen-presenting molecules. Annu. Rev. Immunol. 2015, 33, 169–200. [Google Scholar] [CrossRef] [PubMed]
- Scally, S.W.; Petersen, J.; Law, S.C.; Dudek, N.L.; Nel, H.J.; Loh, K.L.; Wijeyewickrema, L.C.; Eckle, S.B.; van Heemst, J.; Pike, R.N.; et al. A molecular basis for the association of the HLA-DRB1 locus, citrullination, and rheumatoid arthritis. J. Exp. Med. 2013, 210, 2569–2582. [Google Scholar] [CrossRef] [PubMed]
- Jones, E.Y.; Fugger, L.; Strominger, J.L.; Siebold, C. MHC class II proteins and disease: A structural perspective. Nat. Rev. Immunol. 2006, 6, 271–282. [Google Scholar] [CrossRef]
- Foley, P.J.; McGrath, D.S.; Puscinska, E.; Petrek, M.; Kolek, V.; Drabek, J.; Lympany, P.A.; Pantelidis, P.; Welsh, K.I.; Zielinski, J.; et al. Human leukocyte antigen-DRB1 position 11 residues are a common protective marker for sarcoidosis. Am. J. Respir. Cell Mol. Biol. 2001, 25, 272–277. [Google Scholar] [CrossRef]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef]
- Sidney, J.; Becart, S.; Zhou, M.; Duffy, K.; Lindvall, M.; Moore, E.C.; Moore, E.L.; Rao, T.; Rao, N.; Nielsen, M.; et al. Citrullination only infrequently impacts peptide binding to HLA class II MHC. PLoS ONE 2017, 12, e0177140. [Google Scholar] [CrossRef]
- Kampstra, A.S.; van Heemst, J.; Moustakas, A.K.; Papadopoulos, G.K.; Huizinga, T.W.; Toes, R.E. The increased ability to present citrullinated peptides is not unique to HLA-SE molecules: Arginine-to-citrulline conversion also enhances peptide affinity for HLA-DQ molecules. Arthritis Res. Ther. 2016, 18, 254. [Google Scholar] [CrossRef]
- Holoshitz, J. The rheumatoid arthritis HLA-DRB1 shared epitope. Curr. Opin. Rheumatol. 2010, 22, 293–298. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Ikari, K.; Hashimoto, M.; Ohmura, K.; Tanaka, M.; Ito, H.; Taniguchi, A.; Yamanaka, H.; Mimori, T.; Terao, C. Shared epitope defines distinct associations of cigarette smoking with levels of anticitrullinated protein antibody and rheumatoid factor. Ann. Rheum. Dis. 2019. [Google Scholar] [CrossRef]
- Mahdi, H.; Fisher, B.A.; Kallberg, H.; Plant, D.; Malmstrom, V.; Ronnelid, J.; Charles, P.; Ding, B.; Alfredsson, L.; Padyukov, L.; et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat. Genet. 2009, 41, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Sundstrom, B.; Johansson, I.; Rantapaa-Dahlqvist, S. Interaction between dietary sodium and smoking increases the risk for rheumatoid arthritis: Results from a nested case-control study. Rheumatology 2015, 54, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Hedstrom, A.K.; Hossjer, O.; Klareskog, L.; Alfredsson, L. Interplay between alcohol, smoking and HLA genes in RA aetiology. RMD Open 2019, 5, e000893. [Google Scholar] [CrossRef]
- Balandraud, N.; Roudier, J. Epstein-Barr virus and rheumatoid arthritis. Jt. Bone Spine Rev. Rhum. 2018, 85, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Roudier, J.; Petersen, J.; Rhodes, G.H.; Luka, J.; Carson, D.A. Susceptibility to rheumatoid arthritis maps to a T-cell epitope shared by the HLA-Dw4 DR beta-1 chain and the Epstein-Barr virus glycoprotein gp110. Proc. Natl. Acad. Sci. USA 1989, 86, 5104–5108. [Google Scholar] [CrossRef] [PubMed]
- Pratesi, F.; Tommasi, C.; Anzilotti, C.; Chimenti, D.; Migliorini, P. Deiminated Epstein-Barr virus nuclear antigen 1 is a target of anti-citrullinated protein antibodies in rheumatoid arthritis. Arthritis Rheum. 2006, 54, 733–741. [Google Scholar] [CrossRef]
- Kerr, J.R.; Cartron, J.P.; Curran, M.D.; Moore, J.E.; Elliott, J.R.; Mollan, R.A. A study of the role of parvovirus B19 in rheumatoid arthritis. Br. J. Rheumatol. 1995, 34, 809–813. [Google Scholar] [CrossRef]
- Chen, Y.S.; Chou, P.H.; Li, S.N.; Tsai, W.C.; Lin, K.H.; Tsai, K.B.; Yen, J.H.; Liu, H.W. Parvovirus B19 infection in patients with rheumatoid arthritis in Taiwan. J. Rheumatol. 2006, 33, 887–891. [Google Scholar]
- Kharlamova, N.; Jiang, X.; Sherina, N.; Potempa, B.; Israelsson, L.; Quirke, A.M.; Eriksson, K.; Yucel-Lindberg, T.; Venables, P.J.; Potempa, J.; et al. Antibodies to Porphyromonas gingivalis Indicate Interaction Between Oral Infection, Smoking, and Risk Genes in Rheumatoid Arthritis Etiology. Arthritis Rheumatol. 2016, 68, 604–613. [Google Scholar] [CrossRef]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef]
- Xu, H.; Yin, J. HLA risk alleles and gut microbiome in ankylosing spondylitis and rheumatoid arthritis. Best Pract. Res. Clin. Rheumatol. 2020, 101499. [Google Scholar] [CrossRef] [PubMed]
- Rak, J.M.; Maestroni, L.; Balandraud, N.; Guis, S.; Boudinet, H.; Guzian, M.C.; Yan, Z.; Azzouz, D.; Auger, I.; Roudier, C.; et al. Transfer of the shared epitope through microchimerism in women with rheumatoid arthritis. Arthritis Rheum. 2009, 60, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Molitor, M.L.; Haynes, L.D.; Jankowska-Gan, E.; Mulder, A.; Burlingham, W.J. HLA class I noninherited maternal antigens in cord blood and breast milk. Hum. Immunol. 2004, 65, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, D.W.; Zickwolf, G.K.; Weil, G.J.; Sylvester, S.; DeMaria, M.A. Male fetal progenitor cells persist in maternal blood for as long as 27 years postpartum. Proc. Natl. Acad. Sci. USA 1996, 93, 705–708. [Google Scholar] [CrossRef]
- Roh, E.Y.; Yoon, J.H.; Shin, S.; Song, E.Y.; Chung, H.Y.; Park, M.H. Frequency of fetal-maternal microchimerism: An analysis of the HLA-DRB1 gene in cord blood and maternal sample pairs. J. Matern. Fetal Neonatal Med. Off. J. Eur. Assoc. Perinat. Med. Fed. Asia Ocean. Perinat. Soc. Int. Soc. Perinat. Obs. 2017, 30, 2613–2619. [Google Scholar] [CrossRef]
- Lambert, N.C.; Erickson, T.D.; Yan, Z.; Pang, J.M.; Guthrie, K.A.; Furst, D.E.; Nelson, J.L. Quantification of maternal microchimerism by HLA-specific real-time polymerase chain reaction: Studies of healthy women and women with scleroderma. Arthritis Rheum. 2004, 50, 906–914. [Google Scholar] [CrossRef]
- Nelson, J.L.; Gillespie, K.M.; Lambert, N.C.; Stevens, A.M.; Loubiere, L.S.; Rutledge, J.C.; Leisenring, W.M.; Erickson, T.D.; Yan, Z.; Mullarkey, M.E.; et al. Maternal microchimerism in peripheral blood in type 1 diabetes and pancreatic islet beta cell microchimerism. Proc. Natl. Acad. Sci. USA 2007, 104, 1637–1642. [Google Scholar] [CrossRef]
- Harney, S.; Newton, J.; Milicic, A.; Brown, M.A.; Wordsworth, B.P. Non-inherited maternal HLA alleles are associated with rheumatoid arthritis. Rheumatology 2003, 42, 171–174. [Google Scholar] [CrossRef]
- Johnson, K.L.; McAlindon, T.E.; Mulcahy, E.; Bianchi, D.W. Microchimerism in a female patient with systemic lupus erythematosus. Arthritis Rheum. 2001, 44, 2107–2111. [Google Scholar] [CrossRef]
- Van Heemst, J.; Jansen, D.T.; Polydorides, S.; Moustakas, A.K.; Bax, M.; Feitsma, A.L.; Bontrop-Elferink, D.G.; Baarse, M.; van der Woude, D.; Wolbink, G.J.; et al. Crossreactivity to vinculin and microbes provides a molecular basis for HLA-based protection against rheumatoid arthritis. Nat. Commun. 2015, 6, 6681. [Google Scholar] [CrossRef]
- Mold, J.E.; Michaelsson, J.; Burt, T.D.; Muench, M.O.; Beckerman, K.P.; Busch, M.P.; Lee, T.H.; Nixon, D.F.; McCune, J.M. Maternal alloantigens promote the development of tolerogenic fetal regulatory T cells in utero. Science 2008, 322, 1562–1565. [Google Scholar] [CrossRef] [PubMed]
- Burlingham, W.J.; Grailer, A.P.; Heisey, D.M.; Claas, F.H.; Norman, D.; Mohanakumar, T.; Brennan, D.C.; de Fijter, H.; van Gelder, T.; Pirsch, J.D.; et al. The effect of tolerance to noninherited maternal HLA antigens on the survival of renal transplants from sibling donors. N. Engl. J. Med. 1998, 339, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Claas, F.H.; Gijbels, Y.; van der Velden-de Munck, J.; van Rood, J.J. Induction of B cell unresponsiveness to noninherited maternal HLA antigens during fetal life. Science 1988, 241, 1815–1817. [Google Scholar] [CrossRef] [PubMed]
- Van Rood, J.J.; Loberiza, F.R., Jr.; Zhang, M.J.; Oudshoorn, M.; Claas, F.; Cairo, M.S.; Champlin, R.E.; Gale, R.P.; Ringden, O.; Hows, J.M.; et al. Effect of tolerance to noninherited maternal antigens on the occurrence of graft-versus-host disease after bone marrow transplantation from a parent or an HLA-haploidentical sibling. Blood 2002, 99, 1572–1577. [Google Scholar] [CrossRef]
- Ten Wolde, S.; Breedveld, F.C.; de Vries, R.R.; D’Amaro, J.; Rubenstein, P.; Schreuder, G.M.; Claas, F.H.; van Rood, J.J. Influence of non-inherited maternal HLA antigens on occurrence of rheumatoid arthritis. Lancet 1993, 341, 200–202. [Google Scholar]
- Pani, M.A.; Van Autreve, J.; Van der Auwera, B.J.; Gorus, F.K.; Badenhoop, K. Non-transmitted maternal HLA DQ2 or DQ8 alleles and risk of Type I diabetes in offspring: The importance of foetal or post partum exposure to diabetogenic molecules. Diabetologia 2002, 45, 1340–1343. [Google Scholar] [CrossRef][Green Version]
- Van der Horst-Bruinsma, I.E.; Hazes, J.M.; Schreuder, G.M.; Radstake, T.R.; Barrera, P.; van de Putte, L.B.; Mustamu, D.; van Schaardenburg, D.; Breedveld, F.C.; de Vries, R.R. Influence of non-inherited maternal HLA-DR antigens on susceptibility to rheumatoid arthritis. Ann. Rheum. Dis. 1998, 57, 672–675. [Google Scholar] [CrossRef]
- Zong, W.; Ge, Y.; Han, Y.; Yang, X.; Li, Q.; Chen, M. Hypomethylation of HLA-DRB1 and its clinical significance in psoriasis. Oncotarget 2017, 8, 12323–12332. [Google Scholar] [CrossRef]
- Kular, L.; Liu, Y.; Ruhrmann, S.; Zheleznyakova, G.; Marabita, F.; Gomez-Cabrero, D.; James, T.; Ewing, E.; Linden, M.; Gornikiewicz, B.; et al. DNA methylation as a mediator of HLA-DRB1*15:01 and a protective variant in multiple sclerosis. Nat. Commun. 2018, 9, 2397. [Google Scholar] [CrossRef]
- Miller, S.; Tsou, P.S.; Coit, P.; Gensterblum-Miller, E.; Renauer, P.; Rohraff, D.M.; Kilian, N.C.; Schonfeld, M.; Sawalha, A.H. Hypomethylation of STAT1 and HLA-DRB1 is associated with type-I interferon-dependent HLA-DRB1 expression in lupus CD8+ T cells. Ann. Rheum. Dis. 2019, 78, 519–528. [Google Scholar] [CrossRef]
- Weyand, C.M.; Goronzy, J.J. Disease mechanisms in rheumatoid arthritis: Gene dosage effect of HLA-DR haplotypes. J. Lab. Clin. Med. 1994, 124, 335–338. [Google Scholar] [PubMed]
- Marotte, H.; Pallot-Prades, B.; Grange, L.; Tebib, J.; Gaudin, P.; Alexandre, C.; Blond, J.L.; Cazalis, M.A.; Mougin, B.; Miossec, P. The shared epitope is a marker of severity associated with selection for, but not with response to, infliximab in a large rheumatoid arthritis population. Ann. Rheum. Dis. 2006, 65, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Plant, D.; Han, B.; Fu, B.; Yarwood, A.; Thomson, W.; Symmons, D.P.; Worthington, J.; Young, A.; Hyrich, K.L.; et al. Association of HLA-DRB1 haplotypes with rheumatoid arthritis severity, mortality, and treatment response. JAMA 2015, 313, 1645–1656. [Google Scholar] [CrossRef]
- Zhao, M.; Mauer, L.; Sayles, H.; Cannon, G.W.; Reimold, A.; Kerr, G.S.; Baker, J.F.; Thiele, G.M.; England, B.R.; Mikuls, T.R. HLA-DRB1 Haplotypes, Shared Epitope, and Disease Outcomes in US Veterans with Rheumatoid Arthritis. J. Rheumatol. 2019, 46, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Plant, M.J.; Williams, A.L.; O’Sullivan, M.M.; Lewis, P.A.; Coles, E.C.; Jessop, J.D. Relationship between time-integrated C-reactive protein levels and radiologic progression in patients with rheumatoid arthritis. Arthritis Rheum. 2000, 43, 1473–1477. [Google Scholar] [CrossRef]
- Knevel, R.; Grondal, G.; Huizinga, T.W.; Visser, A.W.; Jonsson, H.; Vikingsson, A.; Geirsson, A.J.; Steinsson, K.; van der Helm-van Mil, A.H. Genetic predisposition of the severity of joint destruction in rheumatoid arthritis: A population-based study. Ann. Rheum. Dis. 2012, 71, 707–709. [Google Scholar] [CrossRef]
- Ling, S.F.; Viatte, S.; Lunt, M.; Van Sijl, A.M.; Silva-Fernandez, L.; Symmons, D.P.; Young, A.; Macgregor, A.J.; Barton, A. HLA-DRB1 Amino Acid Positions 11/13, 71, and 74 Are Associated With Inflammation Level, Disease Activity, and the Health Assessment Questionnaire Score in Patients With Inflammatory Polyarthritis. Arthritis Rheumatol. 2016, 68, 2618–2628. [Google Scholar] [CrossRef]
- Traylor, M.; Knevel, R.; Cui, J.; Taylor, J.; Harm-Jan, W.; Conaghan, P.G.; Cope, A.P.; Curtis, C.; Emery, P.; Newhouse, S.; et al. Genetic associations with radiological damage in rheumatoid arthritis: Meta-analysis of seven genome-wide association studies of 2,775 cases. PLoS ONE 2019, 14, e0223246. [Google Scholar] [CrossRef]
- Roudier, J. HLA-DRB1 genes and extraarticular rheumatoid arthritis. Arthritis Res. Ther. 2006, 8, 103. [Google Scholar] [CrossRef]
- Turesson, C.; Schaid, D.J.; Weyand, C.M.; Jacobsson, L.T.; Goronzy, J.J.; Petersson, I.F.; Sturfelt, G.; Nyhall-Wahlin, B.M.; Truedsson, L.; Dechant, S.A.; et al. The impact of HLA-DRB1 genes on extra-articular disease manifestations in rheumatoid arthritis. Arthritis Res. Ther. 2005, 7, R1386–R1393. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.D.; David-Vaudey, E.; Pai, M.; Lum, R.F.; Criswell, L.A. Particular HLA-DRB1 shared epitope genotypes are strongly associated with rheumatoid vasculitis. Arthritis Rheum. 2004, 50, 3476–3484. [Google Scholar] [CrossRef]
- Gonzalez-Juanatey, C.; Testa, A.; Garcia-Castelo, A.; Garcia-Porrua, C.; Llorca, J.; Vidan, J.; Hajeer, A.H.; Ollier, W.E.; Mattey, D.L.; Gonzalez-Gay, M.A. HLA-DRB1 status affects endothelial function in treated patients with rheumatoid arthritis. Am. J. Med. 2003, 114, 647–652. [Google Scholar] [CrossRef]
- Young, A.; Koduri, G.; Batley, M.; Kulinskaya, E.; Gough, A.; Norton, S.; Dixey, J.; Early Rheumatoid Arthritis Study, g. Mortality in rheumatoid arthritis. Increased in the early course of disease, in ischaemic heart disease and in pulmonary fibrosis. Rheumatology 2007, 46, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Ennis, H.; Gupta, A.; Dawson, J.; Lunt, M.; Thomson, W.; Herrick, A. HLA-DRB1 associations with rheumatoid arthritis-related pulmonary fibrosis. Scand. J. Rheumatol. 2014, 43, 75–76. [Google Scholar] [CrossRef] [PubMed]
- Migita, K.; Nakamura, T.; Koga, T.; Eguchi, K. HLA-DRB1 alleles and rheumatoid arthritis-related pulmonary fibrosis. J. Rheumatol. 2010, 37, 205–207. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chen, Y.J.; Chang, Y.T.; Wang, C.B.; Wu, C.Y. The risk of cancer in patients with rheumatoid arthritis: A nationwide cohort study in Taiwan. Arthritis Rheum. 2011, 63, 352–358. [Google Scholar] [CrossRef]
- Baecklund, F.; Foo, J.N.; Askling, J.; Eloranta, S.; Glimelius, I.; Liu, J.; Hjalgrim, H.; Rosenquist, R.; Padyukov, L.; Smedby, K.E. Possible Interaction Between Cigarette Smoking and HLA-DRB1 Variation in the Risk of Follicular Lymphoma. Am. J. Epidemiol. 2017, 185, 681–687. [Google Scholar] [CrossRef]
- Bello, A.E.; Perkins, E.L.; Jay, R.; Efthimiou, P. Recommendations for optimizing methotrexate treatment for patients with rheumatoid arthritis. Open Access Rheumatol. Res. Rev. 2017, 9, 67–79. [Google Scholar] [CrossRef]
- Eberhardt, K.; Fex, E. Clinical course and remission rate in patients with early rheumatoid arthritis: Relationship to outcome after 5 years. Br. J. Rheumatol. 1998, 37, 1324–1329. [Google Scholar] [CrossRef]
- Van der Woude, D.; Young, A.; Jayakumar, K.; Mertens, B.J.; Toes, R.E.; van der Heijde, D.; Huizinga, T.W.; van der Helm-van Mil, A.H. Prevalence of and predictive factors for sustained disease-modifying antirheumatic drug-free remission in rheumatoid arthritis: Results from two large early arthritis cohorts. Arthritis Rheum. 2009, 60, 2262–2271. [Google Scholar] [CrossRef]
- Hider, S.L.; Buckley, C.; Silman, A.J.; Symmons, D.P.; Bruce, I.N. Factors influencing response to disease modifying antirheumatic drugs in patients with rheumatoid arthritis. J. Rheumatol. 2005, 32, 11–16. [Google Scholar] [PubMed]
- Ling, S.F.; Bluett, J. Pharmacogenetics of methotrexate response in rheumatoid arthritis: An update. Pharmacogenomics 2020, 21, 3–6. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Hirose, J.; Yonemura, K. Contribution of HLA-DRB1*04 alleles and anti-cyclic citrullinated antibodies to development of resistance to disease-modifying antirheumatic drugs in early rheumatoid arthritis. Clin. Rheumatol. 2010, 29, 1357–1366. [Google Scholar] [CrossRef] [PubMed]
- Ferraccioli, G.F.; Gremese, E.; Tomietto, P.; Favret, G.; Damato, R.; Di Poi, E. Analysis of improvements, full responses, remission and toxicity in rheumatoid patients treated with step-up combination therapy (methotrexate, cyclosporin A, sulphasalazine) or monotherapy for three years. Rheumatology 2002, 41, 892–898. [Google Scholar] [CrossRef][Green Version]
- Ali, A.A.; Moatter, T.; Baig, J.A.; Iqbal, A.; Hussain, A.; Iqbal, M.P. Polymorphism of HLA-DR and HLA-DQ in rheumatoid arthritis patients and clinical response to methotrexate—A hospital-based study. JPMA J. Pak. Med Assoc. 2006, 56, 452–456. [Google Scholar]
- Qiu, Q.; Huang, J.; Shu, X.; Fan, H.; Zhou, Y.; Xiao, C. Polymorphisms and Pharmacogenomics for the Clinical Efficacy of Methotrexate in Patients with Rheumatoid Arthritis: A Systematic Review and Meta-analysis. Sci. Rep. 2017, 7, 44015. [Google Scholar] [CrossRef]
- Gonzalez-Gay, M.A.; Hajeer, A.H.; Garcia-Porrua, C.; Dababneh, A.; Thomson, W.; Ollier, W.E.; Mattey, D.L. Patients chosen for treatment with cyclosporine because of severe rheumatoid arthritis are more likely to carry HLA-DRB1 shared epitope alleles, and have earlier disease onset. J. Rheumatol. 2002, 29, 271–275. [Google Scholar]
- Hyrich, K.L.; Watson, K.D.; Silman, A.J.; Symmons, D.P.M. Predictors of response to anti-TNF-α therapy among patients with rheumatoid arthritis: Results from the British Society for Rheumatology Biologics Register. Rheumatology 2006, 45, 1558–1565. [Google Scholar] [CrossRef]
- Criswell, L.A.; Lum, R.F.; Turner, K.N.; Woehl, B.; Zhu, Y.; Wang, J.; Tiwari, H.K.; Edberg, J.C.; Kimberly, R.P.; Moreland, L.W.; et al. The influence of genetic variation in the HLA-DRB1 and LTA-TNF regions on the response to treatment of early rheumatoid arthritis with methotrexate or etanercept. Arthritis Rheum. 2004, 50, 2750–2756. [Google Scholar] [CrossRef]
- Murdaca, G.; Spano, F.; Contatore, M.; Guastalla, A.; Magnani, O.; Puppo, F. Pharmacogenetics of etanercept: Role of TNF-alpha gene polymorphisms in improving its efficacy. Expert Opin. Drug Metab. Toxicol. 2014, 10, 1703–1710. [Google Scholar] [CrossRef]
- Skapenko, A.; Smolen, J.S.; Kavanaugh, A.; Arora, V.; Kupper, H.; Schulze-Koops, H. Genetic markers associated with clinical and radiographic response in adalimumab plus methotrexate- or methotrexate-treated rheumatoid arthritis patients in OPTIMA. Clin. Exp. Rheumatol. 2019, 37, 783–790. [Google Scholar] [PubMed]
- Liu, M.; Degner, J.; Davis, J.W.; Idler, K.B.; Nader, A.; Mostafa, N.M.; Waring, J.F. Identification of HLA-DRB1 association to adalimumab immunogenicity. PLoS ONE 2018, 13, e0195325. [Google Scholar] [CrossRef] [PubMed]
- Rigby, W.; Buckner, J.; Bridges, L.; Nys, M.; Gao, S.; Polinsky, M.; Johnsen, A.; Ray, N.; Bykerk, V. LB0008 the effect of hla-drb1 risk alleles on the clinicalefficacy of abatacept and adalimumab in seropositive biologic-naïve patientswith early, moderate-to-severe ra: Data from a head-to-head single-blindedtrial. Ann. Rheum. Dis. 2019, 78, 263–264. [Google Scholar] [CrossRef]
- Oryoji, K.; Yoshida, K.; Kashiwado, Y.; Tanaka, K.; Mizuki, S.I.; Tsukamoto, H.; Kamada, K.; Akashi, K. Shared epitope positivity is related to efficacy of abatacept in rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1234–1236. [Google Scholar] [CrossRef]
- Phillips, E.J.; Sukasem, C.; Whirl-Carrillo, M.; Muller, D.J.; Dunnenberger, H.M.; Chantratita, W.; Goldspiel, B.; Chen, Y.T.; Carleton, B.C.; George, A.L., Jr.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for HLA Genotype and Use of Carbamazepine and Oxcarbazepine: 2017 Update. Clin. Pharmacol. Ther. 2018, 103, 574–581. [Google Scholar] [CrossRef]
- Saito, Y.; Stamp, L.K.; Caudle, K.E.; Hershfield, M.S.; McDonagh, E.M.; Callaghan, J.T.; Tassaneeyakul, W.; Mushiroda, T.; Kamatani, N.; Goldspiel, B.R.; et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guidelines for human leukocyte antigen B (HLA-B) genotype and allopurinol dosing: 2015 update. Clin. Pharmacol. Ther. 2016, 99, 36–37. [Google Scholar] [CrossRef]
- Khanna, D.; Fitzgerald, J.D.; Khanna, P.P.; Bae, S.; Singh, M.K.; Neogi, T.; Pillinger, M.H.; Merill, J.; Lee, S.; Prakash, S.; et al. 2012 American College of Rheumatology guidelines for management of gout. Part 1: Systematic nonpharmacologic and pharmacologic therapeutic approaches to hyperuricemia. Arthritis Care Res. 2012, 64, 1431–1446. [Google Scholar] [CrossRef]
- Martin, M.A.; Hoffman, J.M.; Freimuth, R.R.; Klein, T.E.; Dong, B.J.; Pirmohamed, M.; Hicks, J.K.; Wilkinson, M.R.; Haas, D.W.; Kroetz, D.L.; et al. Clinical Pharmacogenetics Implementation Consortium Guidelines for HLA-B Genotype and Abacavir Dosing: 2014 update. Clin. Pharmacol. Ther. 2014, 95, 499–500. [Google Scholar] [CrossRef]
- Bauer, D.C.; Zadoorian, A.; Wilson, L.O.W.; Melbourne Genomics Health, A.; Thorne, N.P. Evaluation of computational programs to predict HLA genotypes from genomic sequencing data. Brief. Bioinform. 2018, 19, 179–187. [Google Scholar] [CrossRef]
- Ka, S.; Lee, S.; Hong, J.; Cho, Y.; Sung, J.; Kim, H.N.; Kim, H.L.; Jung, J. HLAscan: Genotyping of the HLA region using next-generation sequencing data. BMC Bioinform. 2017, 18, 258. [Google Scholar] [CrossRef]
- Teare, M.D.; Santibanez Koref, M.F. Linkage analysis and the study of Mendelian disease in the era of whole exome and genome sequencing. Brief. Funct. Genom. 2014, 13, 378–383. [Google Scholar] [CrossRef] [PubMed]
- Goswami, R.S.; Harada, S. An Overview of Molecular Genetic Diagnosis Techniques. Curr. Protoc. Hum. Genet. 2020, 105, e97. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Diogo, D.; Greenberg, J.D.; Mouassess, F.; Achkar, W.A.; Fulton, R.S.; Denny, J.C.; Gupta, N.; Mirel, D.; Gabriel, S.; et al. Integration of sequence data from a Consanguineous family with genetic data from an outbred population identifies PLB1 as a candidate rheumatoid arthritis risk gene. PLoS ONE 2014, 9, e87645. [Google Scholar] [CrossRef] [PubMed]
- Jordan, C.T.; Cao, L.; Roberson, E.D.; Pierson, K.C.; Yang, C.F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef] [PubMed]
- Al-Mayouf, S.M.; Sunker, A.; Abdwani, R.; Abrawi, S.A.; Almurshedi, F.; Alhashmi, N.; Al Sonbul, A.; Sewairi, W.; Qari, A.; Abdallah, E.; et al. Loss-of-function variant in DNASE1L3 causes a familial form of systemic lupus erythematosus. Nat. Genet. 2011, 43, 1186–1188. [Google Scholar] [CrossRef]
- Kiezun, A.; Garimella, K.; Do, R.; Stitziel, N.O.; Neale, B.M.; McLaren, P.J.; Gupta, N.; Sklar, P.; Sullivan, P.F.; Moran, J.L.; et al. Exome sequencing and the genetic basis of complex traits. Nat. Genet. 2012, 44, 623–630. [Google Scholar] [CrossRef]
- Murdaca, G.; Gulli, R.; Spano, F.; Mandich, P.; Puppo, F. Pharmacogenetics and future therapeutic scenarios: What affects the prediction of response to treatment with etanercept? Drug Dev. Res. 2014, 75 (Suppl. S1), S7–S10. [Google Scholar] [CrossRef]
- Maxwell, J.R.; Potter, C.; Hyrich, K.L.; BRAGGSS; Genomics Study, S; Barton, A.; Worthington, J.; Isaacs, J.D.; Morgan, A.W.; Wilson, A.G. Association of the tumour necrosis factor-308 variant with differential response to anti-TNF agents in the treatment of rheumatoid arthritis. Hum. Mol. Genet. 2008, 17, 3532–3538. [Google Scholar] [CrossRef]
- Toonen, E.J.; Coenen, M.J.; Kievit, W.; Fransen, J.; Eijsbouts, A.M.; Scheffer, H.; Radstake, T.R.; Creemers, M.C.; de Rooij, D.J.; van Riel, P.L.; et al. The tumour necrosis factor receptor superfamily member 1b 676T>G polymorphism in relation to response to infliximab and adalimumab treatment and disease severity in rheumatoid arthritis. Ann. Rheum. Dis. 2008, 67, 1174–1177. [Google Scholar] [CrossRef]
- Sode, J.; Vogel, U.; Bank, S.; Andersen, P.S.; Hetland, M.L.; Locht, H.; Heegaard, N.H.H.; Andersen, V. Confirmation of an IRAK3 polymorphism as a genetic marker predicting response to anti-TNF treatment in rheumatoid arthritis. Pharm. J. 2018, 18, 81–86. [Google Scholar] [CrossRef]
- Bank, S.; Andersen, P.S.; Burisch, J.; Pedersen, N.; Roug, S.; Galsgaard, J.; Turino, S.Y.; Brodersen, J.B.; Rashid, S.; Rasmussen, B.K.; et al. Genetically determined high activity of IL-12 and IL-18 in ulcerative colitis and TLR5 in Crohns disease were associated with non-response to anti-TNF therapy. Pharm. J. 2018, 18, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, M.; Paradowska-Gorycka, A.; Dabrowska-Zamojcin, E.; Czerewaty, M.; Sluczanowska-Glabowska, S.; Pawlik, A. The effect of gene polymorphisms on patient responses to rheumatoid arthritis therapy. Expert Opin. Drug Metab. Toxicol. 2016, 12, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Plant, D.; Wilson, A.G.; Barton, A. Genetic and epigenetic predictors of responsiveness to treatment in RA. Nat. Rev. Rheumatol. 2014, 10, 329–337. [Google Scholar] [CrossRef] [PubMed]
- Iwaszko, M.; Swierkot, J.; Dratwa, M.; Wysoczanska, B.; Korman, L.; Bugaj, B.; Kolossa, K.; Jeka, S.; Wiland, P.; Bogunia-Kubik, K. Association of MICA-129Met/Val polymorphism with clinical outcome of anti-TNF therapy and MICA serum levels in patients with rheumatoid arthritis. TPharm. J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Guis, S.; Balandraud, N.; Bouvenot, J.; Auger, I.; Toussirot, E.; Wendling, D.; Mattei, J.P.; Nogueira, L.; Mugnier, B.; Legeron, P.; et al. Influence of -308 A/G polymorphism in the tumor necrosis factor alpha gene on etanercept treatment in rheumatoid arthritis. Arthritis Rheum. 2007, 57, 1426–1430. [Google Scholar] [CrossRef] [PubMed]
- Seitz, M.; Wirthmuller, U.; Moller, B.; Villiger, P.M. The -308 tumour necrosis factor-alpha gene polymorphism predicts therapeutic response to TNFalpha-blockers in rheumatoid arthritis and spondyloarthritis patients. Rheumatology 2007, 46, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Padyukov, L.; Lampa, J.; Heimburger, M.; Ernestam, S.; Cederholm, T.; Lundkvist, I.; Andersson, P.; Hermansson, Y.; Harju, A.; Klareskog, L.; et al. Genetic markers for the efficacy of tumour necrosis factor blocking therapy in rheumatoid arthritis. Ann. Rheum. Dis. 2003, 62, 526–529. [Google Scholar] [CrossRef]
- Yarwood, A.; Viatte, S.; Okada, Y.; Plenge, R.; Yamamoto, K.; Braggss, R.; Barton, A.; Symmons, D.; Raychaudhuri, S.; Klareskog, L.; et al. Loci associated with N-glycosylation of human IgG are not associated with rheumatoid arthritis: A Mendelian randomisation study. Ann. Rheum. Dis. 2016, 75, 317–320. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Allele | Ala 74 | Val 11 | Lys 71 | SE | DERAA |
|---|---|---|---|---|---|
| *0101 | + | - | - | + | - |
| *0102 | + | - | - | + | - |
| *0103 | + | - | - | - | + |
| *1102 | + | - | - | - | + |
| *1301 | + | - | - | - | + |
| *1302 | + | - | - | - | + |
| *0401 | + | + | + | + | - |
| *0404 | - | + | - | + | - |
| *0405 | - | + | - | + | - |
| *0408 | - | + | - | + | - |
| *1001 | - | + | - | + | - |
| *0402 | + | + | - | - | + |
| *0403 | - | + | - | - | - |
| *0407 | - | + | - | - | - |
| *1101 | + | - | - | - | - |
| *1103 | + | - | - | - | - |
| *1501 | + | - | - | - | - |
| *1502 | + | - | - | - | - |
| *1104 | + | - | - | - | - |
| *1201 | + | - | - | - | - |
| *1601 | + | - | - | - | - |
| *0301 | - | - | + | - | - |
| *1303 | + | - | + | - | - |
| HLA-DRB1 Allele | Influence on RA Risk | P4 Pocket Charge | Preference for Cit over Arg Binding | Amino Acid Specificity | Binding Affinity of Selected RA Epitopes | Reference | ||
|---|---|---|---|---|---|---|---|---|
| Vim-64,69,71Cit (59–71) | Vim-71Cit (66–78) | CII1240Cit | ||||||
| *0401 | risk allele | positive | P4; other unknown | no | low | high | high | [38,83] |
| *0402 | protective | negative | Preference for Arg in P4; other unknown | E71K at P4 pocket | unknown | high | low | [83,87] |
| *0404 | risk allele | positive | P4, P7 | G86V at P1 pocket | low | high | low | [38,88] |
| *0405 | risk allele | positive | P1, P4, P6, P9 | D57S at P9 pocket | moderate | moderate | low | [38,88] |
| *0301 | protective in Asians | positive | Preference for Arg in P6, P9 | unknown | unknown | no binding | moderate | [87,88] |
| Allele/Genotype | Treatment Response | f | Number of Patients (Male/Female) | Number of Patients Positive for Respective Variant | Number of Patients Anti-CCP-Positive at Diagnosis (%) | Additional Demographic Data | Reference |
|---|---|---|---|---|---|---|---|
| HLA-DRB1*0405 | Inadequate response to csDMARDs | 0.0003 | 124 (29/95) | 64 | 85.5 | Japanese population; mean disease duration 4.2 months; current/former smokers 19.3% | [143] |
| HLA-DRB1*0401/*0404 | favorable response to CsA | 0.016 | 54 (12/42) | 4 | unknown | Spanish population, Mean disease duration 12.1 years | [147] |
| HLA-DRB1*0401 | favorable primary response to TNF-α inhibitors | 0.007 | 1846 (432/1414) | 1188 | 83 | Data not shown | [123] |
| HLA-DRB1*03 | high risk of secondary failure to ADA | 0.006 | 634 | 37 | unknown | Data not shown | [152] |
| HLA-DRB1*01 | low risk of secondary failure to ADA | 0.012 | 365 | Data not shown | unknown | Data not shown | [152] |
| HLA-DRB1*07 | low risk of secondary unresponsiveness to ADA | 0.018 | 365 | Data not shown | unknown | Data not shown | [152] |
| HLA-DRB1 SE | higher efficacy response with ABA vs ADA at week 24 | Estimate of difference (95% CI) for DAS28 (CRP): 27.4 | 80 | 61 | unknown | Mean disease duration 5.5 months | [153] |
| HLA-DRB1 SE | favorable response to ABA at week 24 | <0.0001 | 72 (49/23) | 47 | 89 | Japanese population; mean disease duration 10.4 years | [154] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wysocki, T.; Olesińska, M.; Paradowska-Gorycka, A. Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis–From Research to Clinical Practice. Cells 2020, 9, 1127. https://doi.org/10.3390/cells9051127
Wysocki T, Olesińska M, Paradowska-Gorycka A. Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis–From Research to Clinical Practice. Cells. 2020; 9(5):1127. https://doi.org/10.3390/cells9051127
Chicago/Turabian StyleWysocki, Tomasz, Marzena Olesińska, and Agnieszka Paradowska-Gorycka. 2020. "Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis–From Research to Clinical Practice" Cells 9, no. 5: 1127. https://doi.org/10.3390/cells9051127
APA StyleWysocki, T., Olesińska, M., & Paradowska-Gorycka, A. (2020). Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis–From Research to Clinical Practice. Cells, 9(5), 1127. https://doi.org/10.3390/cells9051127