Suppression of CaMKIIβ Inhibits ANO1-Mediated Glioblastoma Progression


 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Cell Culture and Transfection
2.3. Production and Infection With Lentivirus
2.4. Construction of Expression Vectors
2.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR) and Quantittative Real Time PCR (qPCR)
2.6. Immunocytochemistry
2.7. Surface Biotinylation Assay
2.8. Electrophysiological Recording for Cell
2.9. Migration Assay
2.10. Cell Invasion Assay
2.11. Cell Proliferation Assay
2.12. Statistical Analysis
3. Results
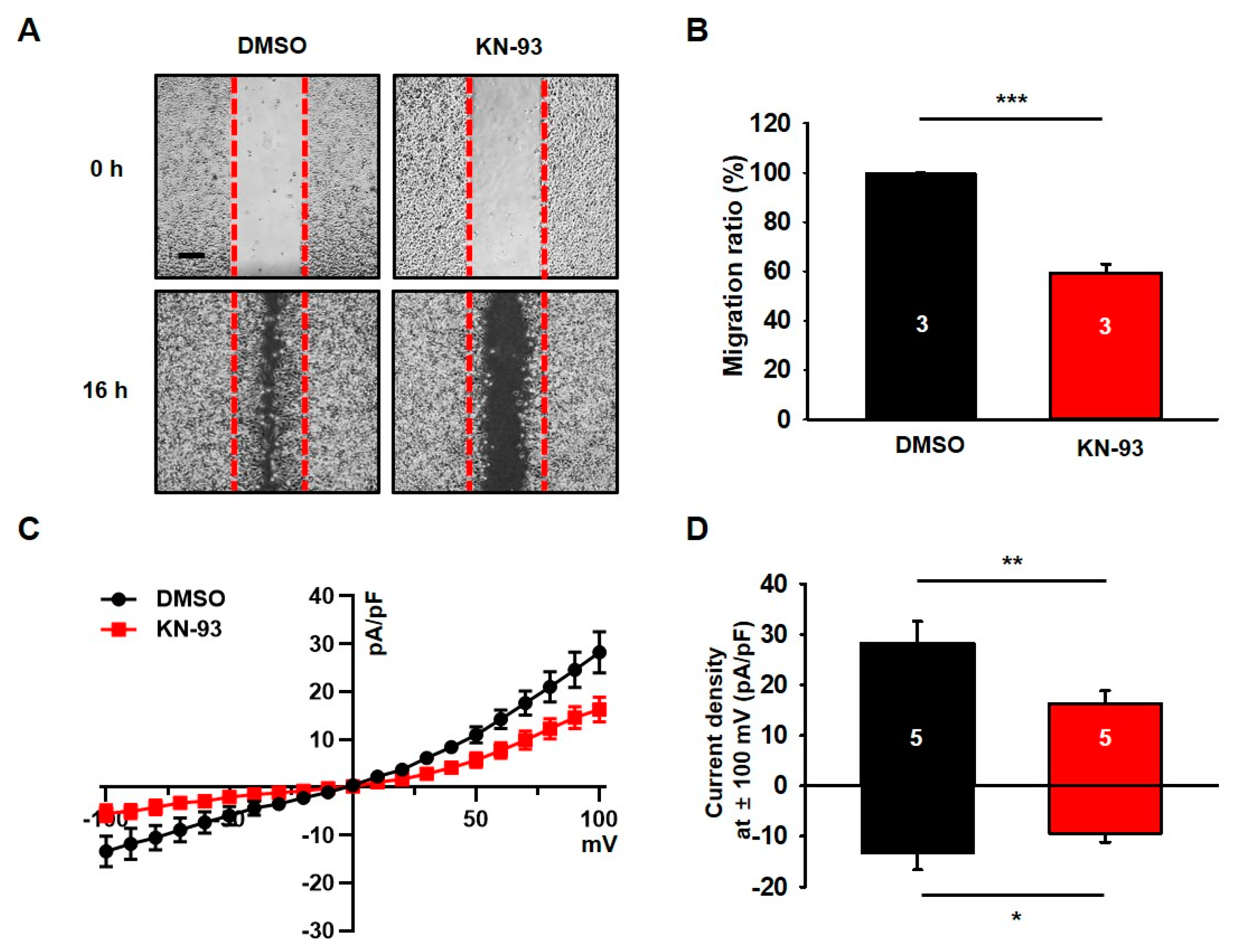
3.1. KN-93, a Selective CaMKII Blocker, Reduces Migration and Chloride Currents in U251 Cells
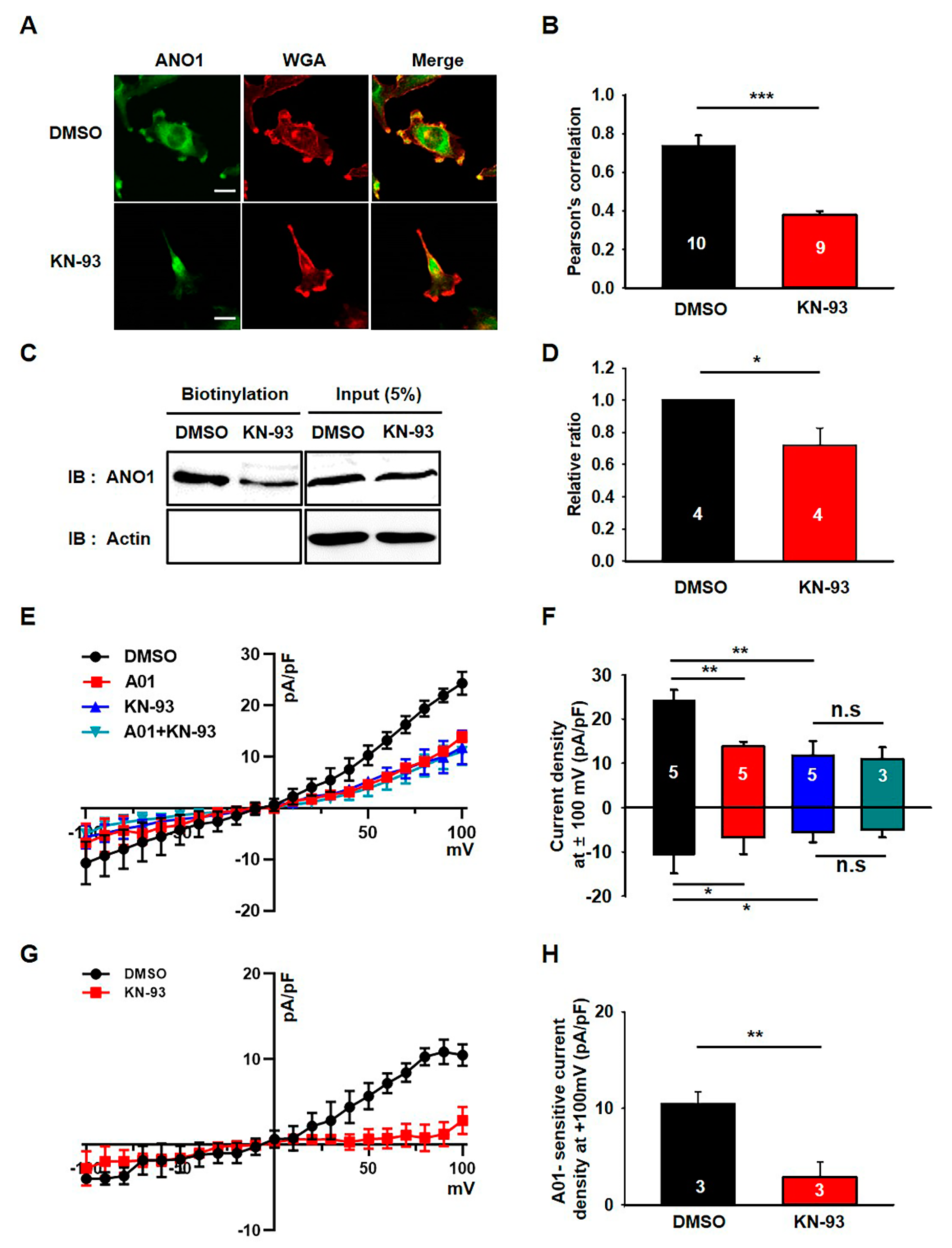
3.2. KN-93 Reduces the Surface Expression and Activity of ANO1 in U251 Cells
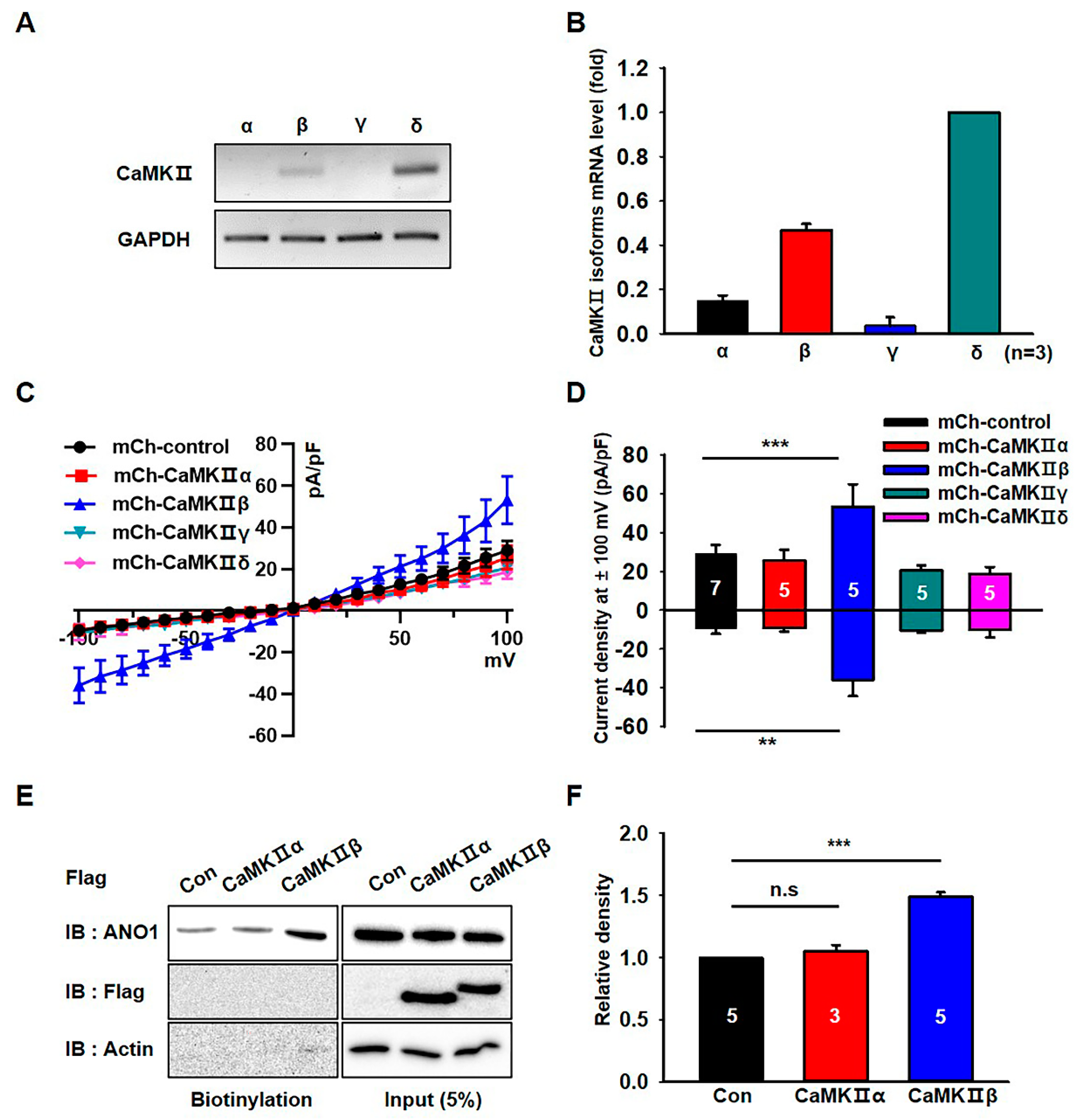
3.3. CaMKIIβ Specifically Increases the Surface Expression and Activity of ANO1 in U251 Cells
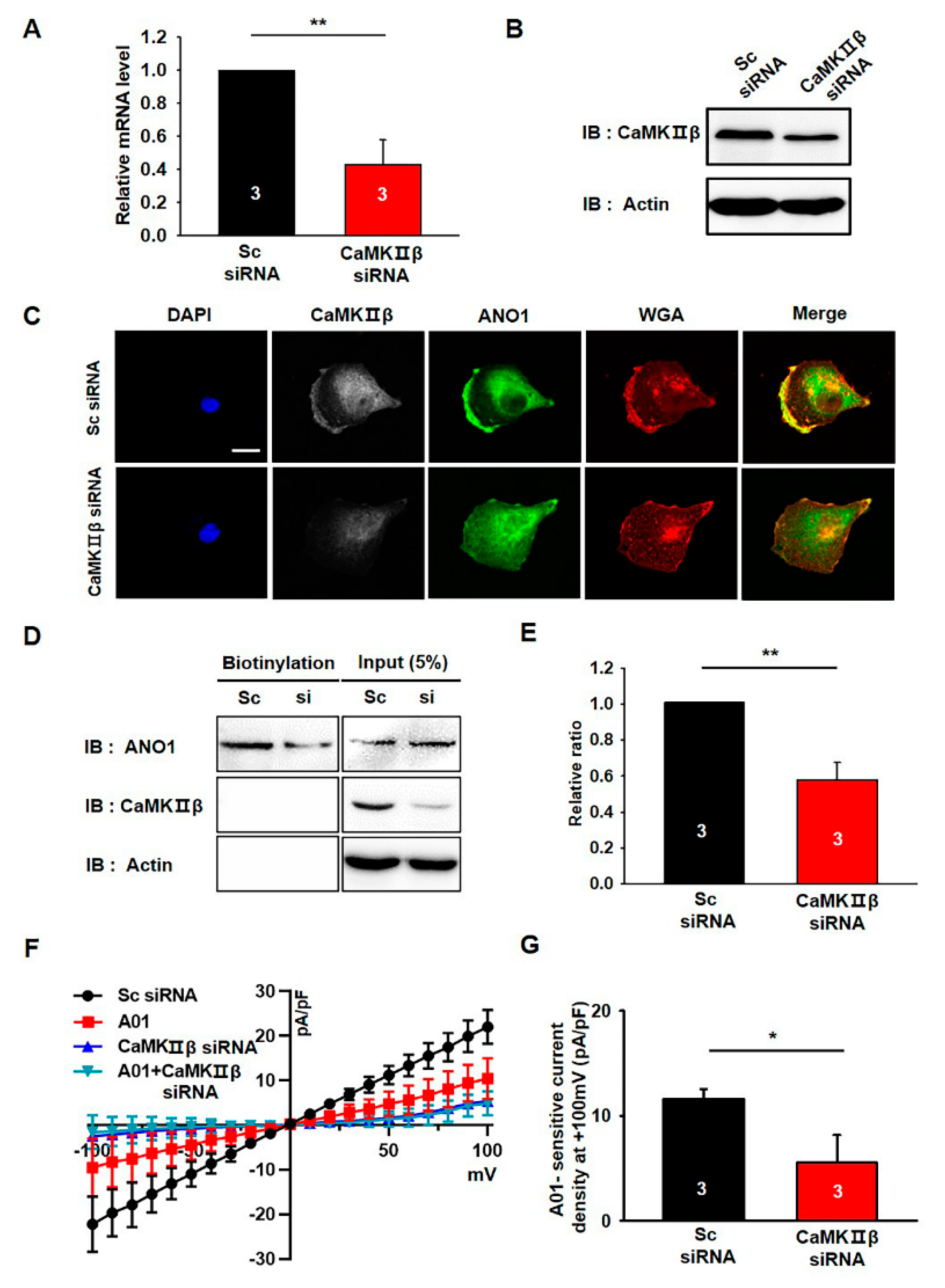
3.4. CaMKIIβ Knockdown Reduces the Surface Expression of ANO1 in U251 Cells
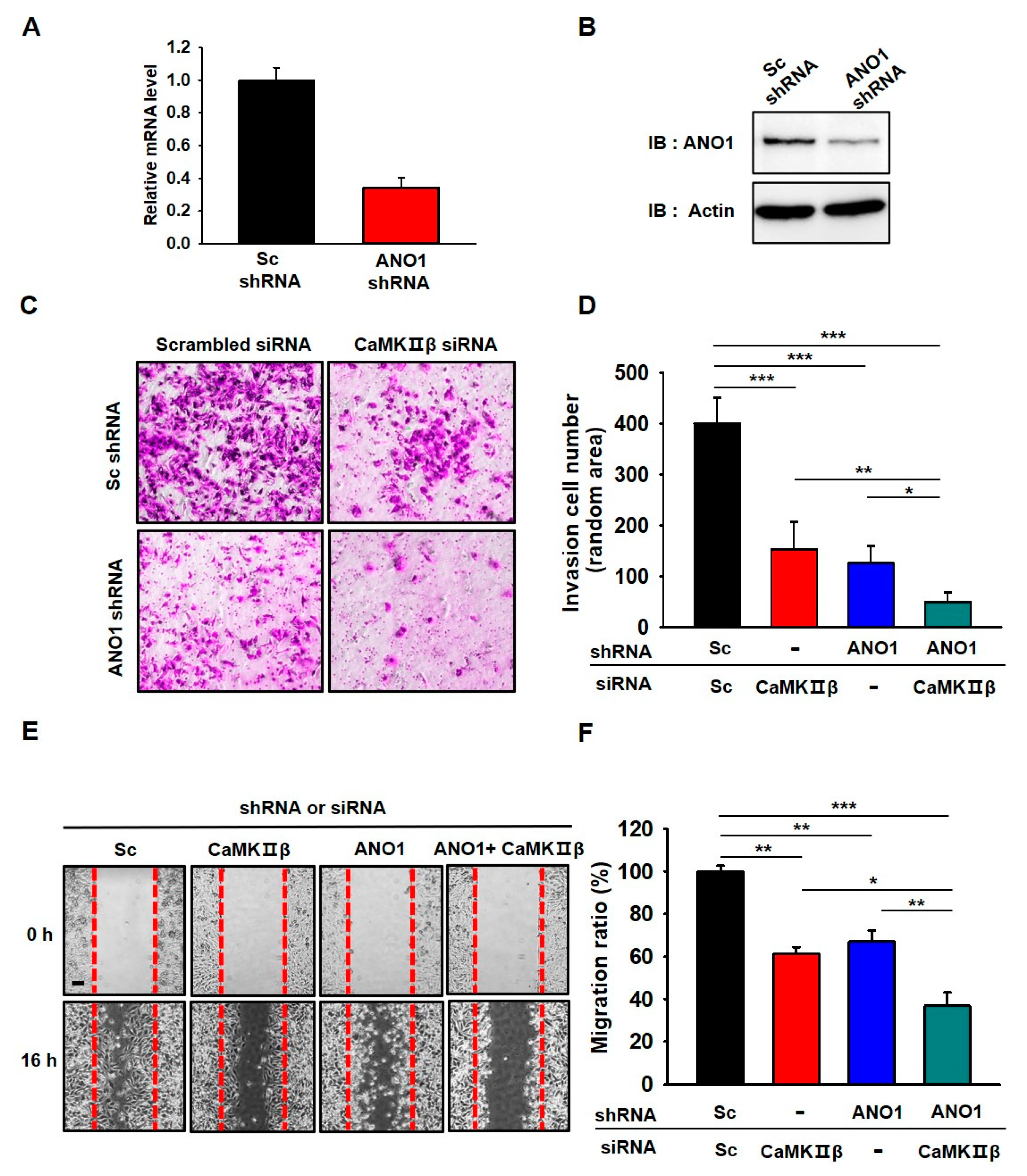
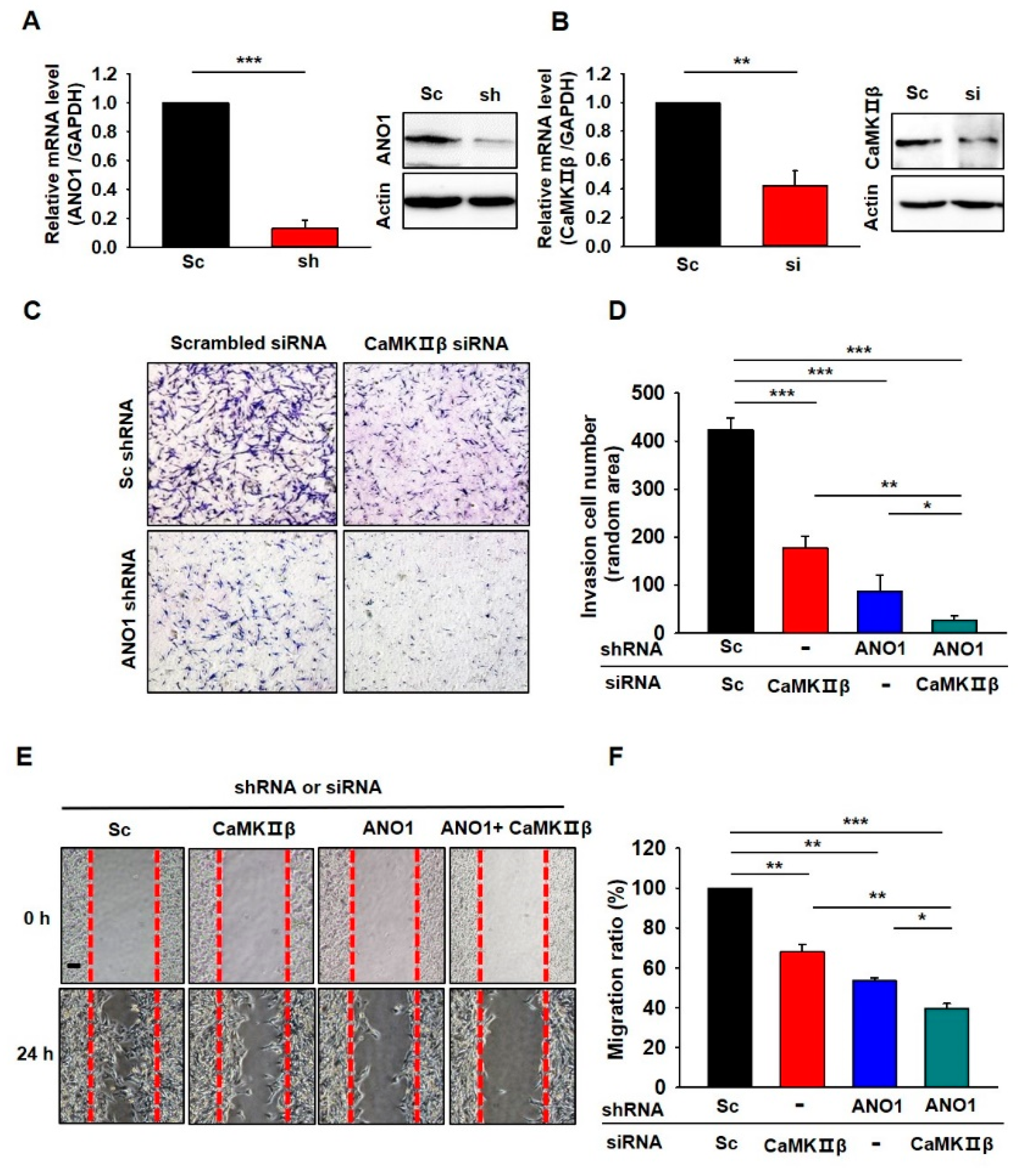
3.5. Deficiency of ANO1 or CaMKIIβ Suppresses Invasion and Migration of Glioblastoma Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Molenaar, R.J. Ion channels in glioblastoma. ISRN Neurol. 2011, 2011, 590249. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019, 21, v1–v100. [Google Scholar] [CrossRef] [PubMed]
- Farin, A.; Suzuki, S.O.; Weiker, M.; Goldman, J.E.; Bruce, J.N.; Canoll, P. Transplanted glioma cells migrate and proliferate on host brain vasculature: A dynamic analysis. Glia 2006, 53, 799–808. [Google Scholar] [CrossRef]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.; Porto-Carreiro, I.; Pereira, C.M.; Balça-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell. Neurosci. 2014, 8, 418. [Google Scholar] [CrossRef]
- Sontheimer, H. An unexpected role for ion channels in brain tumor metastasis. Exp. Boil. Med. 2008, 233, 779–791. [Google Scholar] [CrossRef]
- Habela, C.W.; Olsen, M.L.; Sontheimer, H. ClC3 is a critical regulator of the cell cycle in normal and malignant glial cells. J. Neurosci. 2008, 28, 9205–9217. [Google Scholar] [CrossRef]
- Ransom, C.B.; Liu, X.; Sontheimer, H. BK channels in human glioma cells have enhanced calcium sensitivity. Glia 2002, 38, 281–291. [Google Scholar] [CrossRef]
- Ishiuchi, S.; Tsuzuki, K.; Yoshida, Y.; Yamada, N.; Hagimura, N.; Okado, H.; Miwa, A.; Kurihara, H.; Nakazato, Y.; Tamura, M.; et al. Blockage of Ca 2+-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat. Med. 2002, 8, 971–978. [Google Scholar] [CrossRef]
- Lee, Y.S.; Lee, J.K.; Bae, Y.; Lee, B.S.; Kim, E.; Cho, C.H.; Ryoo, K.; Yoo, J.; Kim, C.H.; Yi, G.S.; et al. Suppression of 14-3-3γ-mediated surface expression of ANO1 inhibits cancer progression of glioblastoma cells. Sci. Rep. 2016, 6, 26413. [Google Scholar] [CrossRef]
- Lee, Y.S.; Bae, Y.; Park, N.; Yoo, J.C.; Cho, C.H.; Ryoo, K.; Hwang, E.M.; Park, J.Y. Surface expression of the Anoctamin-1 (ANO1) channel is suppressed by protein-protein interactions with beta-COP. Biochem. Biophys. Res. Commun. 2016, 475, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.D.; Cho, H.; Koo, J.Y.; Tak, M.H.; Cho, Y.; Shim, W.S.; Park, S.P.; Lee, J.; Lee, B.; Kim, B.M.; et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature 2008, 455, 1210–1215. [Google Scholar] [CrossRef] [PubMed]
- Caputo, A.; Caci, E.; Ferrera, L.; Pedemonte, N.; Barsanti, C.; Sondo, E.; Pfeffer, U.; Ravazzolo, R.; Zegarra-Moran, O.; Galietta, L.J. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science 2008, 322, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, B.C.; Cheng, T.; Jan, Y.N.; Jan, L.Y. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell 2008, 134, 1019–1029. [Google Scholar] [CrossRef]
- Catalán, M.A.; Kondo, Y.; Peña-Munzenmayer, G.; Jaramillo, Y.; Liu, F.; Choi, S.; Crandall, E.; Borok, Z.; Flodby, P.; Shull, G.E.; et al. A fluid secretion pathway unmasked by acinar-specific Tmem16A gene ablation in the adult mouse salivary gland. Proc. Natl. Acad. Sci. USA 2015, 112, 2263–2268. [Google Scholar]
- Huang, F.; Rock, J.R.; Harfe, B.D.; Cheng, T.; Huang, X.; Jan, Y.N.; Jan, L.Y. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc. Natl. Acad. Sci. USA 2009, 106, 21413–21418. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, H.; Wu, M.; Yang, H.; Kudo, M.; Peters, C.J.; Woodruff, P.G.; Solberg, O.D.; Donne, M.L.; Huang, X.; et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. USA 2012, 109, 16354–16359. [Google Scholar] [CrossRef]
- Cho, C.H.; Lee, S.; Kim, A.; Yarishkin, O.; Ryoo, K.; Lee, Y.S.; Jung, H.G.; Yang, E.; Lee, D.Y.; Lee, B.J.; et al. TMEM16A expression in cholinergic neurons of the medial habenula mediates anxiety-related behaviors. EMBO. Rep. 2020, 21. [Google Scholar] [CrossRef]
- Hanzu, F.A.; Gasa, R.; Bulur, N.; Lybaert, P.; Gomis, R.; Malaisse, W.J.; Beauwens, R.; Sener, A. Expression of TMEM16A and SLC4A4 in human pancreatic islets. Cell. Physiol. Biochem. 2012, 29, 61–64. [Google Scholar] [CrossRef]
- Britschgi, A.; Bill, A.; Brinkhaus, H.; Rothwell, C.; Clay, I.; Duss, S.; Rebhan, M.; Raman, P.; Guy, C.T.; Wetzel, K.; et al. Calcium-activated chloride channel ANO1 promotes breast cancer progression by activating EGFR and CAMK signaling. Proc Natl Acad Sci. USA 2013, 110, E1026–E1034. [Google Scholar] [CrossRef]
- Liu, W.; Lu, M.; Liu, B.; Huang, Y.; Wang, K. Inhibition of Ca2+-activated Cl− channel ANO1/TMEM16A expression suppresses tumor growth and invasiveness in human prostate carcinoma. Cancer. Lett. 2012, 326, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Liu, Y.; Ren, Y.; Kang, L.; Zhang, L. Transmembrane protein with unknown function 16A overexpression promotes glioma formation through the nuclear factor-κB signaling pathway. Mol. Med. Rep. 2014, 9, 1068–1074. [Google Scholar] [CrossRef] [PubMed]
- Bae, J.S.; Park, J.Y.; Park, S.H.; Ha, S.H.; An, A.R.; Noh, S.J.; Kwon, K.S.; Jung, S.H.; Park, H.S.; Kang, M.J.; et al. Expression of ANO1/DOG1 is associated with shorter survival and progression of breast carcinomas. Oncotarget 2018, 9, 607–621. [Google Scholar] [CrossRef] [PubMed]
- Bill, A.; Hall, M.L.; Borawski, J.; Hodgson, C.; Jenkins, J.; Piechon, P.; Popa, O.; Rothwell, C.; Tranter, P.; Tria, S.; et al. Small Molecule-facilitated Degradation of ANO1 Protein A NEW TARGETING APPROACH FOR ANTICANCER THERAPEUTICS. J. Biol. Chem. 2014, 289, 11029–11041. [Google Scholar] [CrossRef]
- Brzozowski, J.; Skelding, K. The multi-functional calcium/calmodulin stimulated protein kinase (CaMK) family: Emerging targets for anti-cancer therapeutic intervention. Pharmaceuticals 2019, 12, 8. [Google Scholar] [CrossRef]
- Wang, Y.-y.; Zhao, R.; Zhe, H. The emerging role of CaMKII in cancer. Oncotarget 2015, 6, 11725–11734. [Google Scholar] [CrossRef]
- Lisman, J.; Yasuda, R.; Raghavachari, S. Mechanisms of CaMKII action in long-term potentiation. Nat. Rev. Neurosci. 2012, 13, 169–182. [Google Scholar] [CrossRef]
- Erickson, J. Mechanisms of CaMKII activation in the heart. Front. Pharmacol. 2014, 5, 59. [Google Scholar] [CrossRef]
- Lin, C.X.; Lv, X.F.; Yuan, F.; Li, X.Y.; Ma, M.M.; Liu, C.Z.; Zhou, J.G.; Wang, G.L.; Guan, Y.Y. Ca2+/calmodulin-dependent protein kinase II γ-dependent serine727 phosphorylation is required for TMEM16A Ca2+-activated Cl− channel regulation in cerebrovascular cells. Circ. J. 2018, 82, 903–913. [Google Scholar] [CrossRef]
- Ayon, R.J.; Hawn, M.B.; Aoun, J.; Wiwchar, M.; Forrest, A.S.; Cunningham, F.; Singer, C.A.; Valencik, M.L.; Greenwood, I.A.; Leblanc, N. Molecular mechanism of TMEM16A regulation: Role of CaMKII and PP1/PP2A. Am. J. Physiol. Cell. Physiol. 2019, 317, C1093–C1106. [Google Scholar] [CrossRef]
- Yin, J.; Oh, Y.T.; Kim, J.Y.; Kim, S.S.; Choi, E.; Kim, T.H.; Hong, J.H.; Chang, N.; Cho, H.J.; Sa, J.K.; et al. Transglutaminase 2 inhibition reverses mesenchymal transdifferentiation of glioma stem cells by regulating C/EBPβ signaling. Cancer. Res. 2017, 77, 4973–4984. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.J.; Lee, S.; Jung, H.J. A curcumin derivative hydrazinobenzoylcurcumin suppresses stem-like features of glioblastoma cells by targeting Ca2+/calmodulin-dependent protein kinase II. J. Cell. Biochem. 2019, 120, 6741–6752. [Google Scholar] [CrossRef]
- Ransom, C.B.; O’Neal, J.T.; Sontheimer, H. Volume-activated chloride currents contribute to the resting conductance and invasive migration of human glioma cells. J. Neurosci. 2001, 21, 7674–7683. [Google Scholar] [CrossRef] [PubMed]
- Chai, S.; Qian, Y.; Tang, J.; Liang, Z.; Zhang, M.; Si, J.; Li, X.; Huang, W.; Xu, R.; Wang, K. Corrigendum to’Ca2+/calmodulin-dependent protein kinase IIγ, a critical mediator of the NF-κB network, is a novel therapeutic target in non-small cell lung cancer. Cancer. Lett. 2016, 380, 609–610. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Guo, S.; Liu, Z.; Wu, L.; Li, M.; Yang, J.; Chen, R.; Liu, X.; Xu, H.; Cai, S.; et al. CAMK2N1 inhibits prostate cancer progression through androgen receptor-dependent signaling. Oncotarget 2014, 5, 10293–10306. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, N.; Liu, X.; Zheng, Y.; Cao, X. A novel endogenous human CaMKII inhibitory protein suppresses tumor growth by inducing cell cycle arrest via p27 stabilization. J. Biol. Chem. 2008, 283, 11565–11574. [Google Scholar] [CrossRef]
- Cuddapah, V.A.; Sontheimer, H. Molecular interaction and functional regulation of ClC-3 by Ca2+/calmodulin-dependent protein kinase II (CaMKII) in human malignant glioma. J. Biol. Chem. 2010, 285, 11188–11196. [Google Scholar] [CrossRef]
- Kang, S.S.; Han, K.S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1, 4, 5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer. Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef]
- Lyons, S.A.; Chung, W.J.; Weaver, A.K.; Ogunrinu, T.; Sontheimer, H. Autocrine glutamate signaling promotes glioma cell invasion. Cancer. Res. 2007, 67, 9463–9471. [Google Scholar] [CrossRef]
- Rondé, P.; Giannone, G.; Gerasymova, I.; Stoeckel, H.; Takeda, K.; Haiech, J. Mechanism of calcium oscillations in migrating human astrocytoma cells. Biochim. Biophys. Acta 2000, 1498, 273–280. [Google Scholar] [CrossRef]
- Bill, A.; Gutierrez, A.; Kulkarni, S.; Kemp, C.; Bonenfant, D.; Voshol, H.; Duvvuri, U.; Gaither, L.A. ANO1/TMEM16A interacts with EGFR and correlates with sensitivity to EGFR-targeting therapy in head and neck cancer. Oncotarget 2015, 6, 9173–9188. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | Size | |
|---|---|---|---|
| CaMKIIα | Forward Primer Reverse Primer | 5′- AGGCTTCAATTCCCAGCTCT -3′ 5′- TGTGTCAGCCAATGAAAGGC -3′ | 137 |
| CaMKIIβ | Forward Primer Reverse Primer | 5′- CCAGAAGCTGGAGAGAGAGG-3′ 5′- AAGACCAGGTAGTGGAAGCC -3′ | 105 |
| CaMKIIδ | Forward Primer Reverse Primer | 5′- GATTTCTGCTGACAGTGCGT-3′ 5′- GAGTAGGACACACTGCCCTT-3′ | 100 |
| CaMKIIγ | Forward Primer Reverse Primer | 5′-CTTCTCTCACAGGAGCCACA-3′ 5′-TCTCCTGCTGACCTGGAAAG-3′ | 104 |
| GAPDH | Forward Primer Reverse Primer | 5′-CCATGGAGAAGGCTGG-3′ 5′-CAAAGTTGTCAGGATGACC-3′ | 195 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sim, K.M.; Lee, Y.-S.; Kim, H.J.; Cho, C.-H.; Yi, G.-S.; Park, M.-J.; Hwang, E.M.; Park, J.-Y. Suppression of CaMKIIβ Inhibits ANO1-Mediated Glioblastoma Progression. Cells 2020, 9, 1079. https://doi.org/10.3390/cells9051079
Sim KM, Lee Y-S, Kim HJ, Cho C-H, Yi G-S, Park M-J, Hwang EM, Park J-Y. Suppression of CaMKIIβ Inhibits ANO1-Mediated Glioblastoma Progression. Cells. 2020; 9(5):1079. https://doi.org/10.3390/cells9051079
Chicago/Turabian StyleSim, Kyoung Mi, Young-Sun Lee, Hee Jin Kim, Chang-Hoon Cho, Gwan-Su Yi, Myung-Jin Park, Eun Mi Hwang, and Jae-Yong Park. 2020. "Suppression of CaMKIIβ Inhibits ANO1-Mediated Glioblastoma Progression" Cells 9, no. 5: 1079. https://doi.org/10.3390/cells9051079
APA StyleSim, K. M., Lee, Y.-S., Kim, H. J., Cho, C.-H., Yi, G.-S., Park, M.-J., Hwang, E. M., & Park, J.-Y. (2020). Suppression of CaMKIIβ Inhibits ANO1-Mediated Glioblastoma Progression. Cells, 9(5), 1079. https://doi.org/10.3390/cells9051079