A G-Protein Coupled Receptor and Macular Degeneration


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
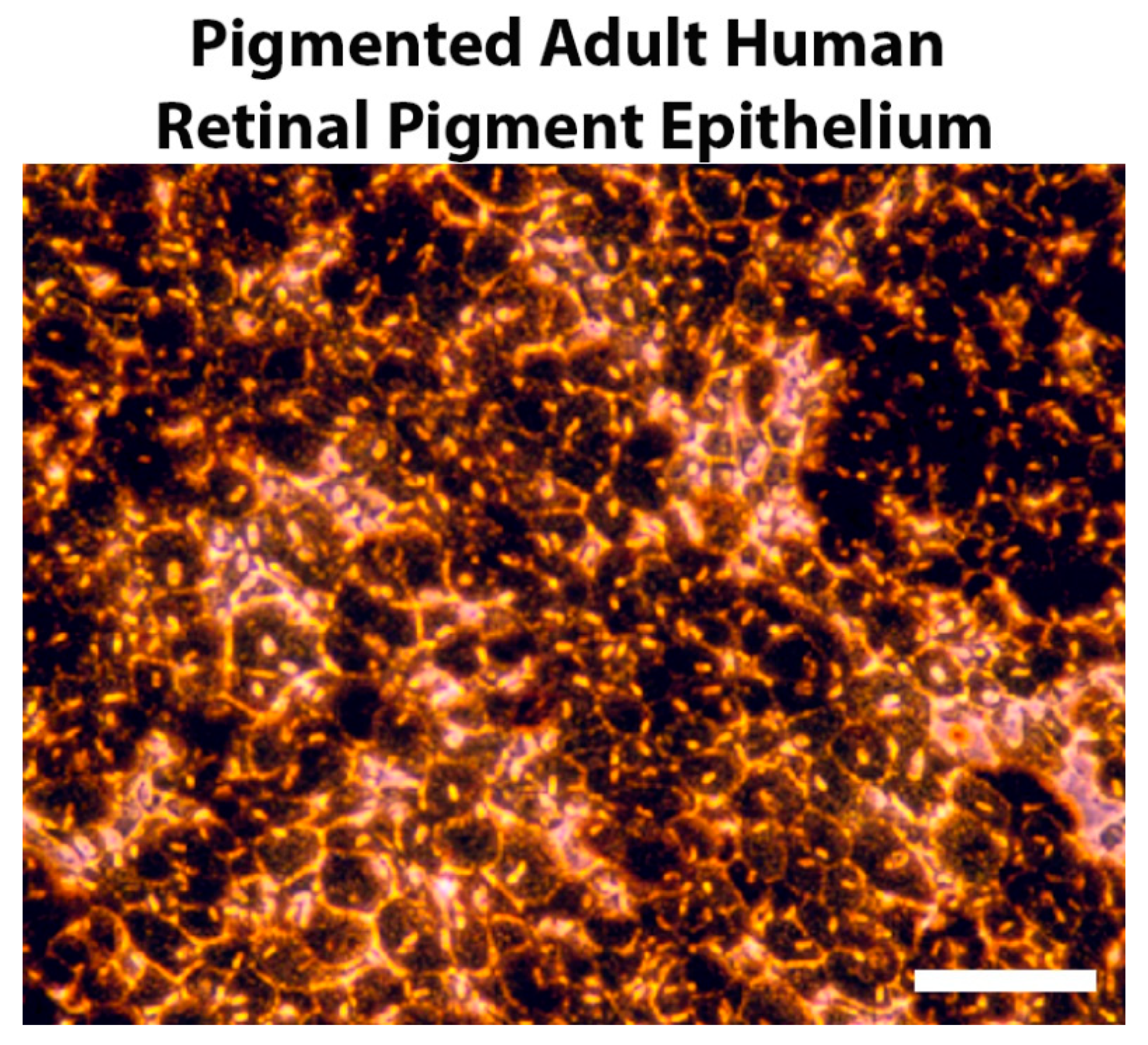
2. Pigmentation and Retinal Development
3. GPR143 and Pigmentation
4. Downstream Effectors of GPR143
4.1. Pigment Epithelium-Derived Factor
4.2. Vascular Endothelial Growth Factor
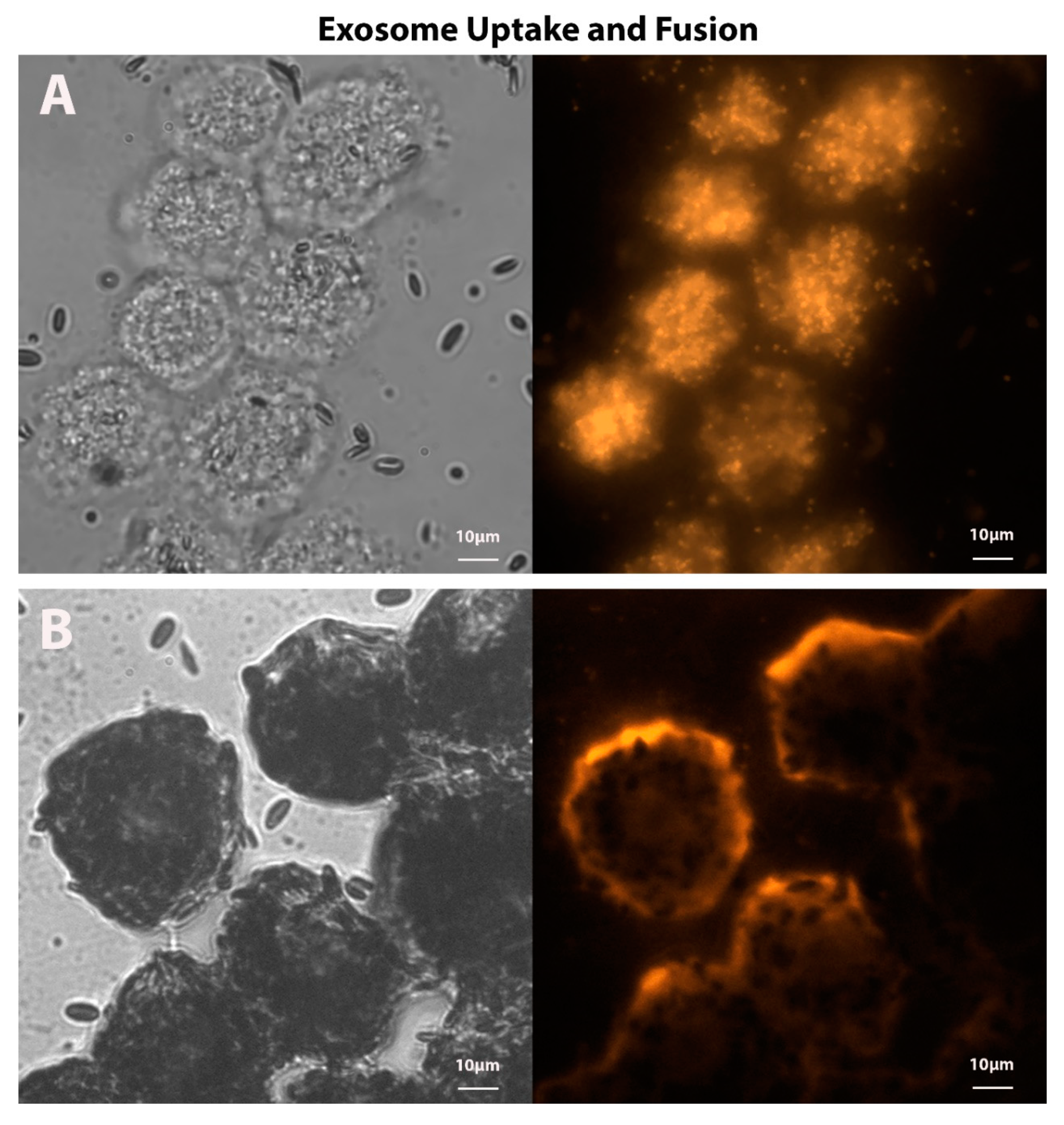
4.3. Exosomes
4.4. GPR143 Signaling
5. RPE Pigmentation in Aging
6. L-DOPA and AMD
Exosomes, GPR143, and AMD
- AMD is caused, or accompanied by diminished choroidal perfusion and RPE hypoxia.
- Hypoxia causes cells to release exosomes.
- Exosomes stimulate angiogenesis.
- Retinal neovascularization is a primary cause of vision loss in AMD.
- The ligand of GPR143 is L-DOPA.
- L-DOPA stops RPE exosome release.
- L-DOPA systemically treats wet AMD.
7. Conclusions
- The absence of GPR143 signaling/disruption of the pigmentation pathway, as observed in ocular albinism (OA).
- Race and age-related decrease of GPR143 signaling, as observed in the prevalence of age-related macular degeneration (AMD) in whites compared to other races.
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Strauss, O. The retinal pigment epithelium in visual function. Physiol. Rev. 2005, 85, 845–881. [Google Scholar] [CrossRef] [PubMed]
- Fronk, A.H.; Vargis, E. Methods for culturing retinal pigment epithelial cells: A review of current protocols and future recommendations. J. Tissue Eng. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.S.; Burke, J.M. Separation of phenotypically distinct subpopulations of cultured human retinal pigment epithelial cells. Exp. Cell Res. 1994, 213, 85–92. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.Y.; Peisch, R.D. Melanin concentration in normal human retinal pigment epithelium. Investig. Ophthalmol. Vis. Sci. 1986, 27, 1063–1067. [Google Scholar]
- Weiter, J.J.; Delori, F.C.; Wing, G.L.; Fitch, K.A. Retinal pigment epithelial lipofuscin and melanin and choroidal melanin in human eyes. Invest. Ophthalmol. Vis. Sci. 1986, 27, 145–152. [Google Scholar]
- Fraga, S.; Serrão, M.P.; Soares-da-Silva, P. The L-3,4-dihydroxyphenylalanine transporter in human and rat epithelial intestinal cells is a type 2 hetero amino acid exchanger. Eur. J. Pharmacol. 2002, 441, 127–135. [Google Scholar] [CrossRef]
- Satoh, M.; Kuzuu, A.; Doi, H.; Masaka, T.; Sakakibara, R. Monitoring Of Blood L-DOPA And L-DOPA Metabolite Concentrations And Adverse Events In Patients With Advanced Parkinsons Disease Receiving L-DOPA And Amantadine Combination Therapy: A Clinical Study. Res. Rev. J. Pharm. Pharm. Sci. 2015, 4, 88–96. [Google Scholar]
- Doudet, D.J.; Cornfeldt, M.L.; Honey, C.R.; Schweikert, A.W.; Allen, R.C. PET imaging of implanted human retinal pigment epithelial cells in the MPTP-induced primate model of Parkinson’s disease. Exp. Neurol. 2004, 189, 361–368. [Google Scholar] [CrossRef]
- Dutton, J.; Copeland, L.G.; Playfer, J.R.; Roberts, N.B. Measuring L-dopa in plasma and urine to monitor therapy of elderly patients with Parkinson disease treated with L-dopa and a dopa decarboxylase inhibitor. Clin. Chem. 1993, 39, 629–634. [Google Scholar] [CrossRef]
- Barbeau, A. L-dopa therapy in Parkinson’s disease: A critical review of nine years’ experience. Can. Med. Assoc. J. 1969, 101, 59–68. [Google Scholar]
- Gracies, J.-M.; Olanow, W. Current And Experimental Therapeutics Of Parkinson’s Disease. Neuropsychopharmacology 2002, 124, 1795–1816. [Google Scholar]
- Bok, D. The retinal pigment epithelium: A versatile partner in vision. J. Cell Sci. Suppl. 1993, 17, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Boulton, M.; Dayhaw-Barker, P. The role of the retinal pigment epithelium: Topographical variation and ageing changes. Eye 2001, 15, 384–389. [Google Scholar] [CrossRef] [PubMed]
- Bharti, K.; Nguyen, M.T.T.; Skuntz, S.; Bertuzzi, S.; Arnheiter, H. The other pigment cell: Specification and development of the pigmented epithelium of the vertebrate eye. Pigment Cell Res. 2006, 19, 380–394. [Google Scholar] [CrossRef] [PubMed]
- Surace, E.M.; Angeletti, B.; Ballabio, A.; Marigo, V. Expression pattern of the ocular albinism type 1 (Oa1) gene in the murine retinal pigment epithelium. Invest. Ophthalmol. Vis. Sci. 2000, 41, 4333–4337. [Google Scholar]
- Roffler-Tarlov, S.; Liu, J.H.; Naumova, E.N.; Bernal-Ayala, M.M.; Mason, C.A. L-Dopa and the albino riddle: Content of L-Dopa in the developing retina of pigmented and albino mice. PLoS ONE 2013, 8. [Google Scholar] [CrossRef]
- Creel, D.J.; Summers, C.G.; King, R.A. Visual anomalies associated with albinism. Ophthalmic Paediatr. Genet. 1990, 11, 193–200. [Google Scholar] [CrossRef]
- McAllister, J.T.; Dubis, A.M.; Tait, D.M.; Ostler, S.; Rha, J.; Stepien, K.E.; Gail Summers, C.; Carroll, J.; Summers, C.G.; Carroll, J. Arrested development: High-resolution imaging of foveal morphology in albinism. Vis. Res. 2010, 50, 810–817. [Google Scholar] [CrossRef]
- Mohammad, S.; Gottlob, I.; Kumar, A.; Thomas, M.; Degg, C.; Sheth, V.; Proudlock, F.A. The functional significance of foveal abnormalities in albinism measured using spectral-domain optical coherence tomography. Ophthalmology 2011, 118, 1645–1652. [Google Scholar] [CrossRef]
- McKay, B.S. Pigmentation and vision: Is GPR143 in control? J. Neurosci. Res. 2019, 97, 77–87. [Google Scholar] [CrossRef]
- Lopez, V.M.; Decatur, C.L.; Stamer, W.D.; Lynch, R.M.; McKay, B.S. L-DOPA is an endogenous ligand for OA1. PLoS Biol. 2008, 6, 1861–1869. [Google Scholar] [CrossRef] [PubMed]
- Jin, N.G.; Chuang, A.Z.; Masson, P.J.; Ribelayga, C.P. Rod electrical coupling is controlled by a circadian clock and dopamine in mouse retina. J. Physiol. 2015, 593, 1597–1631. [Google Scholar] [CrossRef] [PubMed]
- Rajan, P.D.; Kekuda, R.; Chancy, C.D.; Huang, W.; Ganapathy, V.; Smith, S.B. Expression of the extraneuronal monoamine transporter in RPE and neural retina. Curr. Eye Res. 2000, 20, 195–204. [Google Scholar] [CrossRef]
- Jackson, C.R.; Chaurasia, S.S.; Hwang, C.K.; Iuvone, P.M. Dopamine D4 receptor activation controls circadian timing of the adenylyl cyclase 1/cyclic AMP signaling system in mouse retina. Eur. J. Neurosci. 2011, 34, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Falk, T.; Congrove, N.R.; Zhang, S.; McCourt, A.D.; Sherman, S.J.; McKay, B.S. PEDF and VEGF-A output from human retinal pigment epithelial cells grown on novel microcarriers. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Locke, C.J.; Congrove, N.R.; Dismuke, W.M.; Bowen, T.J.; Stamer, W.D.; McKay, B.S. Controlled exosome release from the retinal pigment epithelium insitu. Exp. Eye Res. 2014, 129, 1–4. [Google Scholar] [CrossRef]
- Falk, T.; Gonzalez, R.T.; Sherman, S.J. The Yin and Yang of VEGF and PEDF: Multifaceted neurotrophic factors and their potential in the treatment of Parkinson’s disease. Int. J. Mol. Sci. 2010, 11, 2875–2900. [Google Scholar] [CrossRef]
- Tombran-Tink, J.; Barnstable, C.J. PEDF: A multifaceted neurotrophic factor. Nat. Rev. Neurosci. 2003, 4, 628–636. [Google Scholar] [CrossRef]
- Hardy, K.M.; Hoffman, E.A.; Gonzalez, P.; McKay, B.S.; Stamer, W.D. Extracellular trafficking of myocilin in human trabecular meshwork cells. J. Biol. Chem. 2005, 280, 28917–28926. [Google Scholar] [CrossRef]
- Tombran-Tink, J.; Shivaram, S.M.; Chader, G.J.; Johnson, L.V.; Bok, D. Expression, secretion, and age-related downregulation of pigment epithelium-derived factor, a serpin with neurotrophic activity. J. Neurosci. 1995, 15, 4992–5003. [Google Scholar] [CrossRef]
- Steele, F.R.; Chader, G.J.; Johnson, L.V.; Tombran-Tink, J. Pigment epithelium-derived factor: Neurotrophic activity and identification as a member of the serine protease inhibitor gene family. Proc. Natl. Acad. Sci. USA 1993, 90, 1526–1530. [Google Scholar] [CrossRef] [PubMed]
- Tombran-Tink, J.; Chader, G.G.; Johnson, L.V. PEDF: A pigment epithelium-derived factor with potent neuronal differentiative activity. Exp. Eye Res. 1991, 53, 411–414. [Google Scholar] [CrossRef]
- Huang, Q.; Wang, S.; Sorenson, C.M.; Sheibani, N. PEDF-deficient mice exhibit an enhanced rate of retinal vascular expansion and are more sensitive to hyperoxia-mediated vessel obliteration. Exp. Eye Res. 2008, 87, 226–241. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Cheng, R.; Benyajati, S.; Ma, J. PEDF and its roles in physiological and pathological conditions: Implication in diabetic and hypoxia-induced angiogenic diseases. Clin. Sci. 2015, 128, 805–823. [Google Scholar] [CrossRef] [PubMed]
- McKay, B.S.; Goodman, B.; Falk, T.; Sherman, S.J. Retinal pigment epithelial cell transplantation could provide trophic support in Parkinson’s disease: Results from an in vitro model system. Exp. Neurol. 2006, 201, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Rosenfeld, P.J.; Rich, R.M.; Lalwani, G.A. Ranibizumab: Phase III Clinical Trial Results. Ophthalmol. Clin. N. Am. 2006, 19, 361–372. [Google Scholar]
- Rofagha, S.; Bhisitkul, R.B.; Boyer, D.S.; Sadda, S.R.; Zhang, K. Seven-year outcomes in ranibizumab-treated patients in ANCHOR, MARINA, and HORIZON: A multicenter cohort study (SEVEN-UP). Ophthalmology 2013, 120, 2292–2299. [Google Scholar] [CrossRef]
- Mitchell, P.; Korobelnik, J.F.; Lanzetta, P.; Holz, F.G.; Prünte, C.; Schmidt-Erfurth, U.; Tano, Y.; Wolf, S. Ranibizumab (Lucentis) in neovascular age-related macular degeneration: Evidence from clinical trials. Br. J. Ophthalmol. 2010, 94, 2–13. [Google Scholar] [CrossRef]
- Schorey, J.S.; Bhatnagar, S. Exosome function: From tumor immunology to pathogen biology. Traffic 2008, 9, 871–881. [Google Scholar] [CrossRef]
- Pant, S.; Hilton, H.; Burczynski, M.E. The multifaceted exosome: Biogenesis, role in normal and aberrant cellular function, and frontiers for pharmacological and biomarker opportunities. Biochem. Pharmacol. 2012, 83, 1484–1494. [Google Scholar] [CrossRef]
- Bang, C.; Thum, T. Exosomes: New players in cell-cell communication. Int. J. Biochem. Cell Biol. 2012, 44, 2060–2064. [Google Scholar] [CrossRef] [PubMed]
- Klingeborn, M.; Dismuke, W.M.; Skiba, N.P.; Kelly, U.; Stamer, W.D.; Bowes Rickman, C. Directional Exosome Proteomes Reflect Polarity-Specific Functions in Retinal Pigmented Epithelium Monolayers. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed]
- Klingeborn, M.; Stamer, W.D.; Bowes Rickman, C. Polarized exosome release from the retinal pigmented epithelium. Adv. Exp. Med. Biol. 2018, 1074, 539–544. [Google Scholar]
- Vautrot, V.; Chanteloup, G.; Elmallah, M.; Cordonnier, M.; Aubin, F.; Garrido, C.; Gobbo, J. Exosomal miRNA: Small Molecules, Big Impact in Colorectal Cancer. J. Oncol. 2019, 2019. [Google Scholar] [CrossRef] [PubMed]
- Sahebi, R.; Langari, H.; Fathinezhad, Z.; Bahari Sani, Z.; Avan, A.; Ghayour Mobarhan, M.; Rezayi, M. Exosomes: New insights into cancer mechanisms. J. Cell. Biochem. 2020, 121, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Ohyashiki, J.H.; Umezu, T.; Ohyashiki, K. Exosomes promote bone marrow angiogenesis in hematologic neoplasia: The role of hypoxia. Curr. Opin. Hematol. 2016, 23, 268–273. [Google Scholar] [CrossRef]
- Zhang, J.; Li, S.; Li, L.; Li, M.; Guo, C.; Yao, J.; Mi, S. Exosome and exosomal microRNA: Trafficking, sorting, and function. Genom. Proteom. Bioinforma. 2015, 13, 17–24. [Google Scholar] [CrossRef]
- d’Addio, M.; Pizzigoni, A.; Bassi, M.T.; Baschirotto, C.; Valetti, C.; Incerti, B.; Clementi, M.; De Luca, M.; Ballabio, A.; Schiaffino, M. V Defective intracellular transport and processing of OA1 is a major cause of ocular albinism type 1. Hum. Mol. Genet. 2000, 9, 3011–3018. [Google Scholar] [CrossRef]
- O’Donnell, F.E., Jr.; Hambrick, G.W., Jr.; Green, W.R.; Iliff, W.J.; Stone, D.L. X-linked ocular albinism. An oculocutaneous macromelanosomal disorder. Arch. Ophthalmol. 1976, 94, 1883–1892. [Google Scholar]
- Schiaffino, M.V.; Baschirotto, C.; Pellegrini, G.; Montalti, S.; Tacchetti, C.; De Luca, M.; Ballabio, A. The ocular albinism type 1 gene product is a membrane glycoprotein localized to melanosomes. Proc. Natl. Acad. Sci. USA 1996, 93, 9055–9060. [Google Scholar] [CrossRef]
- Burgoyne, T.; Jolly, R.; Martin-Martin, B.; Seabra, M.C.; Piccirillo, R.; Schiaffino, M.V.; Futter, C.E. Expression of OA1 limits the fusion of a subset of MVBs with lysosomes-a mechanism potentially involved in the initial biogenesis of melanosomes. J. Cell Sci. 2013, 126, 5143–5152. [Google Scholar] [CrossRef]
- McKay, B.S.; Congrove, N.R.; Johnson, A.A.; Dismuke, W.M.; Bowen, T.J.; Stamer, W.D. A role for myocilin in receptor-mediated endocytosis. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Perkumas, K.M.; Hoffman, E.A.; McKay, B.S.; Allingham, R.R.; Stamer, W.D. Myocilin-associated exosomes in human ocular samples. Exp. Eye Res. 2007, 84, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Chou, C.F.; Klein, B.E.K.; Zhang, X.; Meuer, S.M.; Saaddine, J.B. Prevalence of age-related macular degeneration in the US population. Arch. Ophthalmol. 2011, 129, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Li, X.; Kuo, J.Z.; Klein, B.E.K.; Cotch, M.F.; Wong, T.Y.; Taylor, K.D.; Rotter, J.I. Associations of candidate genes to age-related macular degeneration among racial/ethnic groups in the multi-ethnic study of atherosclerosis. Am. J. Ophthalmol. 2013, 156, 1010–1020. [Google Scholar] [CrossRef]
- Arias, E.; Heronm, M.; Xu, J. United States life tables, 2014. Natl. Vital Stat. Rep. 2017, 66, 1–64. [Google Scholar]
- Anderson, D.H.; Fisher, S.K.; Steinberg, R.H. Mammalian cones: Disc shedding, phagocytosis, and renewal. Invest. Ophthalmol. Vis. Sci. 1978, 17, 117–133. [Google Scholar]
- Adler, R.; Curcio, C.; Hicks, D.; Price, D.; Wong, F. Cell death in age-related macular degeneration. Mol. Vis. 1999, 5, 31. [Google Scholar]
- Bergen, A.A.; Arya, S.; Koster, C.; Pilgrim, M.G.; Wiatrek-Moumoulidis, D.; van der Spek, P.J.; Hauck, S.M.; Boon, C.J.F.; Emri, E.; Stewart, A.J.; et al. On the origin of proteins in human drusen: The meet, greet and stick hypothesis. Prog. Retin. Eye Res. 2019, 70, 55–84. [Google Scholar] [CrossRef]
- Jager, R.D.; Mieler, W.F.; Miller, J.W. Age-related macular degeneration. N. Engl. J. Med. 2008, 358, 2606–2617. [Google Scholar] [CrossRef]
- Bastide, M.F.; Meissner, W.G.; Picconi, B.; Fasano, S.; Fernagut, P.O.; Feyder, M.; Francardo, V.; Alcacer, C.; Ding, Y.; Brambilla, R.; et al. Pathophysiology of L-dopa-induced motor and non-motor complications in Parkinson’s disease. Prog. Neurobiol. 2015, 132, 96–168. [Google Scholar] [CrossRef] [PubMed]
- Brilliant, M.H.; Vaziri, K.; Connor, T.B.; Schwartz, S.G.; Carroll, J.J.; McCarty, C.A.; Schrodi, S.J.; Hebbring, S.J.; Kishor, K.S.; Flynn, H.W.; et al. Mining Retrospective Data for Virtual Prospective Drug Repurposing: L-DOPA and Age-related Macular Degeneration. Am. J. Med. 2016, 129, 292–298. [Google Scholar] [CrossRef]
- Tomany, S.C.; Wang, J.J.; Van Leeuwen, R.; Klein, R.; Mitchell, P.; Vingerling, J.R.; Klein, B.E.; Smith, W.; De Jong, P.T. Risk factors for incident age-related macular degeneration: Pooled findings from 3 continents. Ophthalmology 2004, 111, 1280–1287. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Scott, J.; Griffiths, H.; Self, J.E.; Lotery, A. Oral levodopa rescues retinal morphology and visual function in a murine model of human albinism. Pigment Cell Melanoma Res. 2019, 32, 657–671. [Google Scholar] [CrossRef] [PubMed]
- Summers, C.G.; Connett, J.E.; Holleschau, A.M.; Anderson, J.L.; De Becker, I.; McKay, B.S.; Brilliant, M.H. Does levodopa improve vision in albinism? Results of a randomized, controlled clinical trial. Clin. Exp. Ophthalmol. 2014, 42, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Baba, K.; Debruyne, J.P.; Tosini, G. Dopamine 2 Receptor Activation Entrains Circadian Clocks in Mouse Retinal Pigment Epithelium. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Figueroa, A.G.; McKay, B.S. A G-Protein Coupled Receptor and Macular Degeneration. Cells 2020, 9, 910. https://doi.org/10.3390/cells9040910
Figueroa AG, McKay BS. A G-Protein Coupled Receptor and Macular Degeneration. Cells. 2020; 9(4):910. https://doi.org/10.3390/cells9040910
Chicago/Turabian StyleFigueroa, Anna G., and Brian S. McKay. 2020. "A G-Protein Coupled Receptor and Macular Degeneration" Cells 9, no. 4: 910. https://doi.org/10.3390/cells9040910
APA StyleFigueroa, A. G., & McKay, B. S. (2020). A G-Protein Coupled Receptor and Macular Degeneration. Cells, 9(4), 910. https://doi.org/10.3390/cells9040910