Progressive Photoreceptor Dysfunction and Age-Related Macular Degeneration-Like Features in rp1l1 Mutant Zebrafish

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Zebrafish Care
2.2. gDNA Extraction
2.3. CRISPR/Cas9 Genome Editing
2.4. Finclips and Genotyping
2.5. qPCR
2.6. Electroretinography
2.7. Optical Coherence Tomography
2.8. Electron Microscopy
2.9. Paraffin Processing and Hematoxylin and Eosin Staining
2.10. Oil Red O Staining on Cryopreserved Tissue
2.11. Statistical Analysis
3. Results
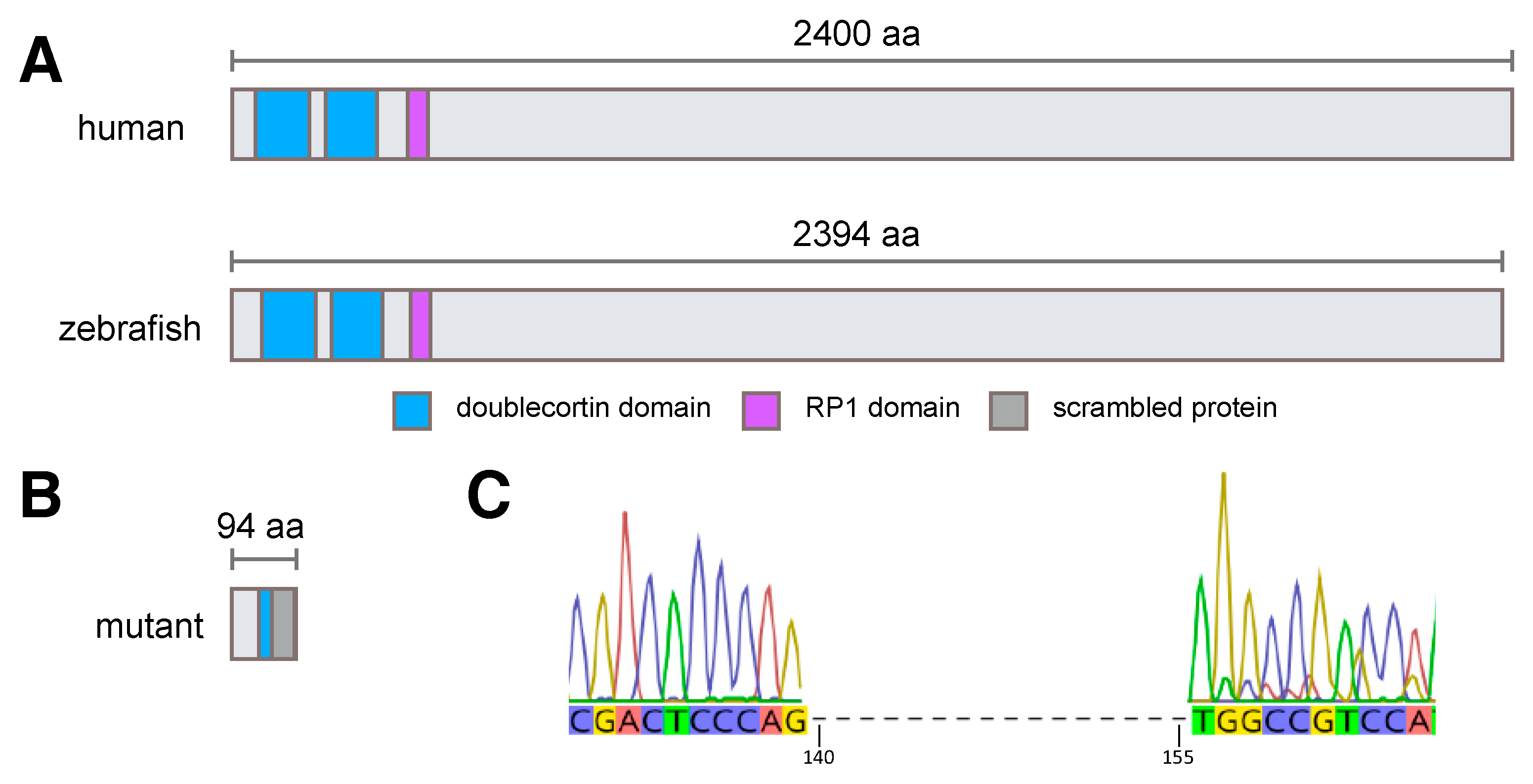
3.1. Generation of rp1l1 Mutant Zebrafish
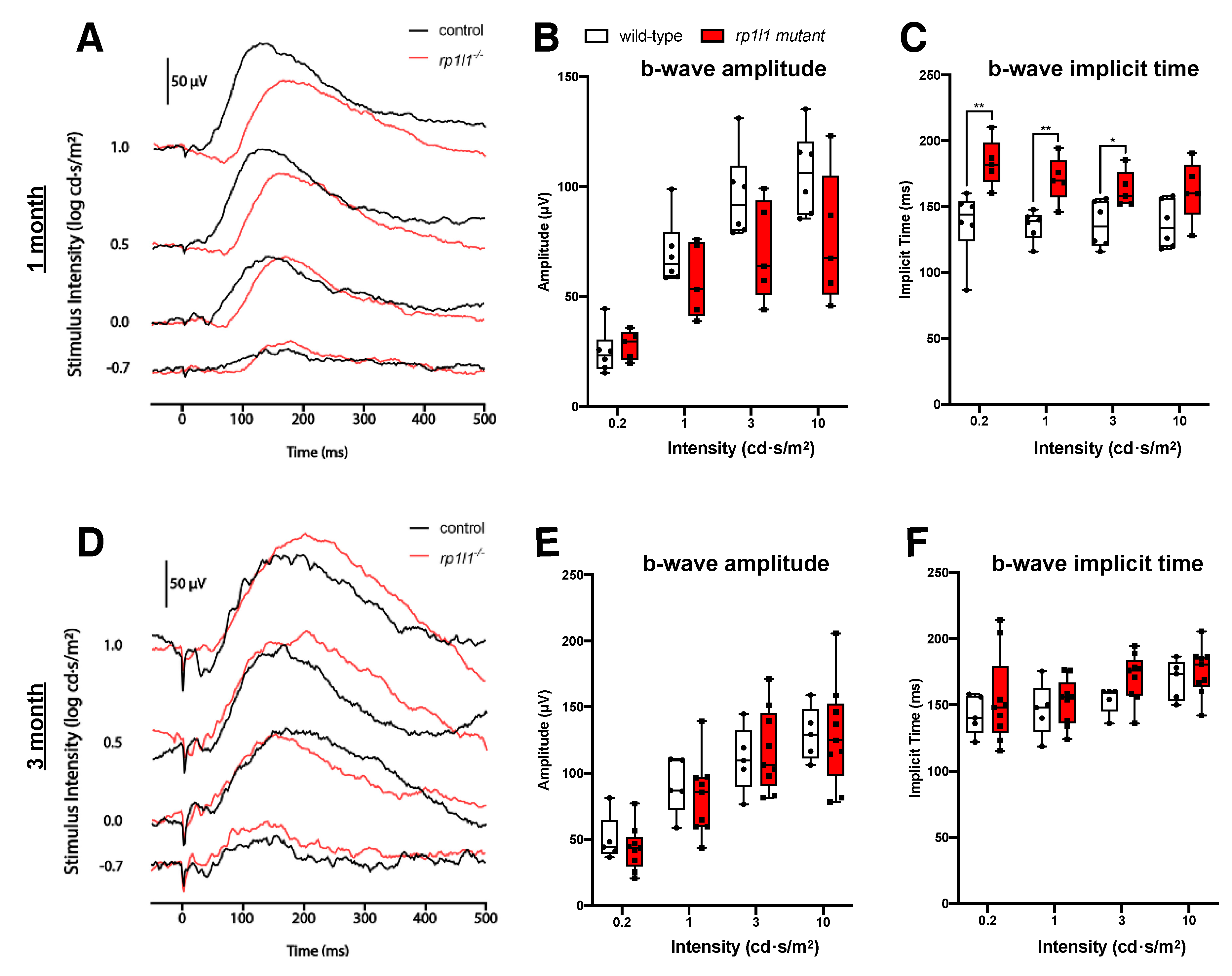
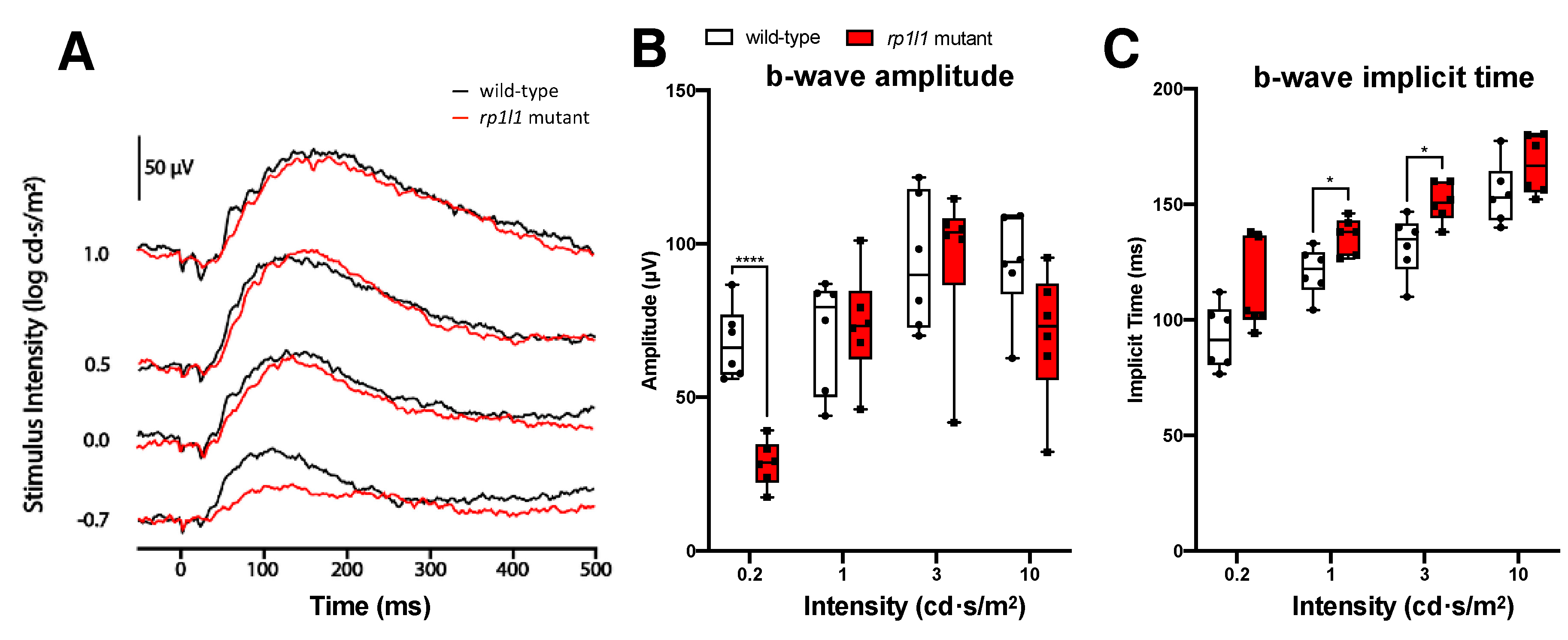
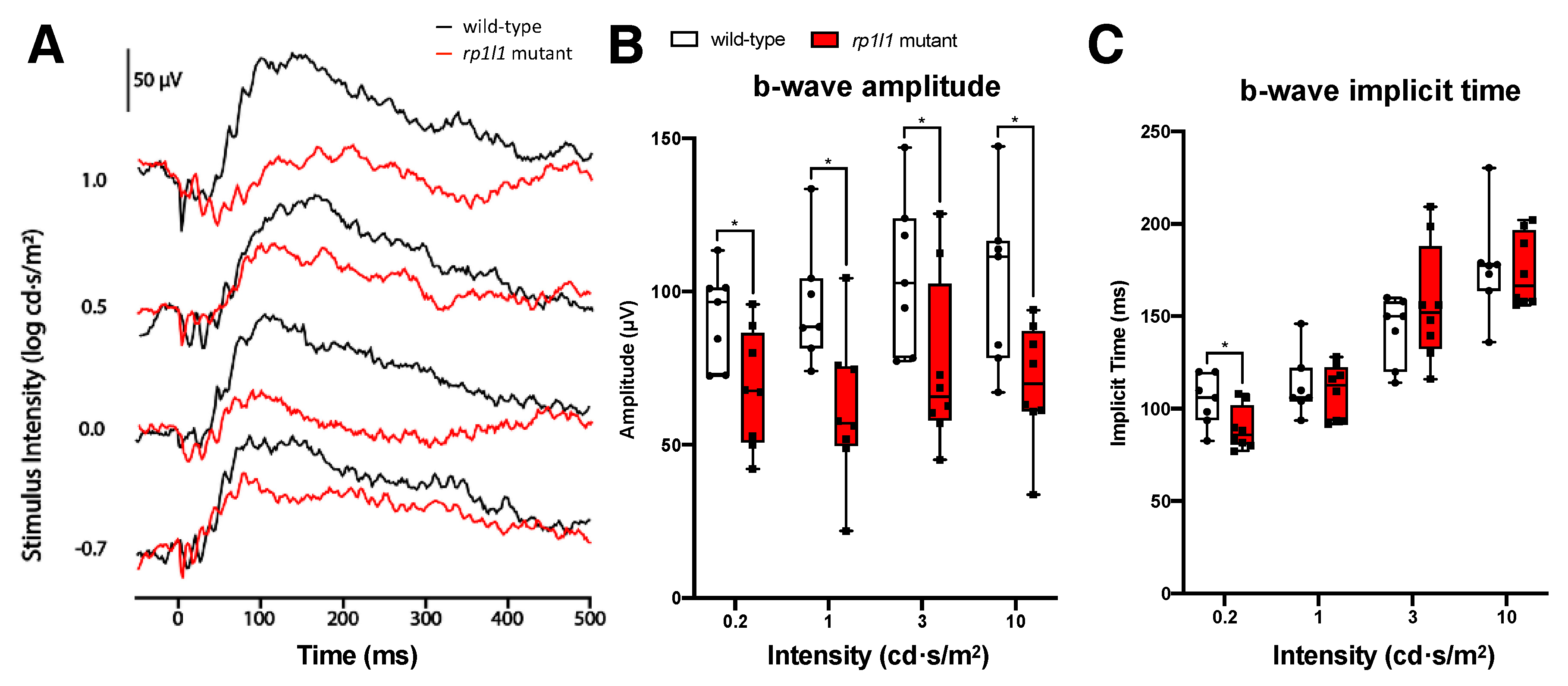
3.2. rp1l1 Mutants Have Progressive Photoreceptor Dysfunction
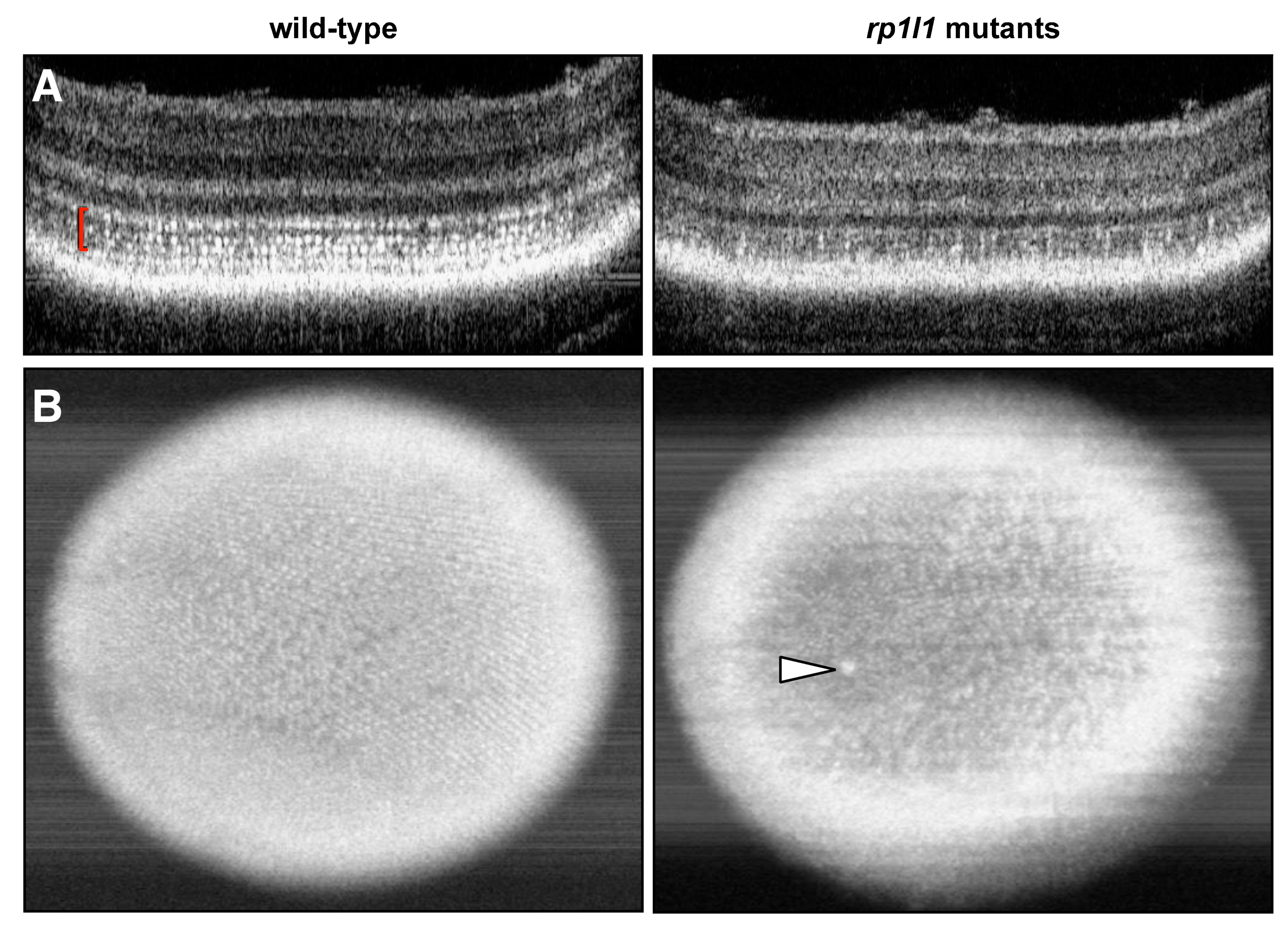
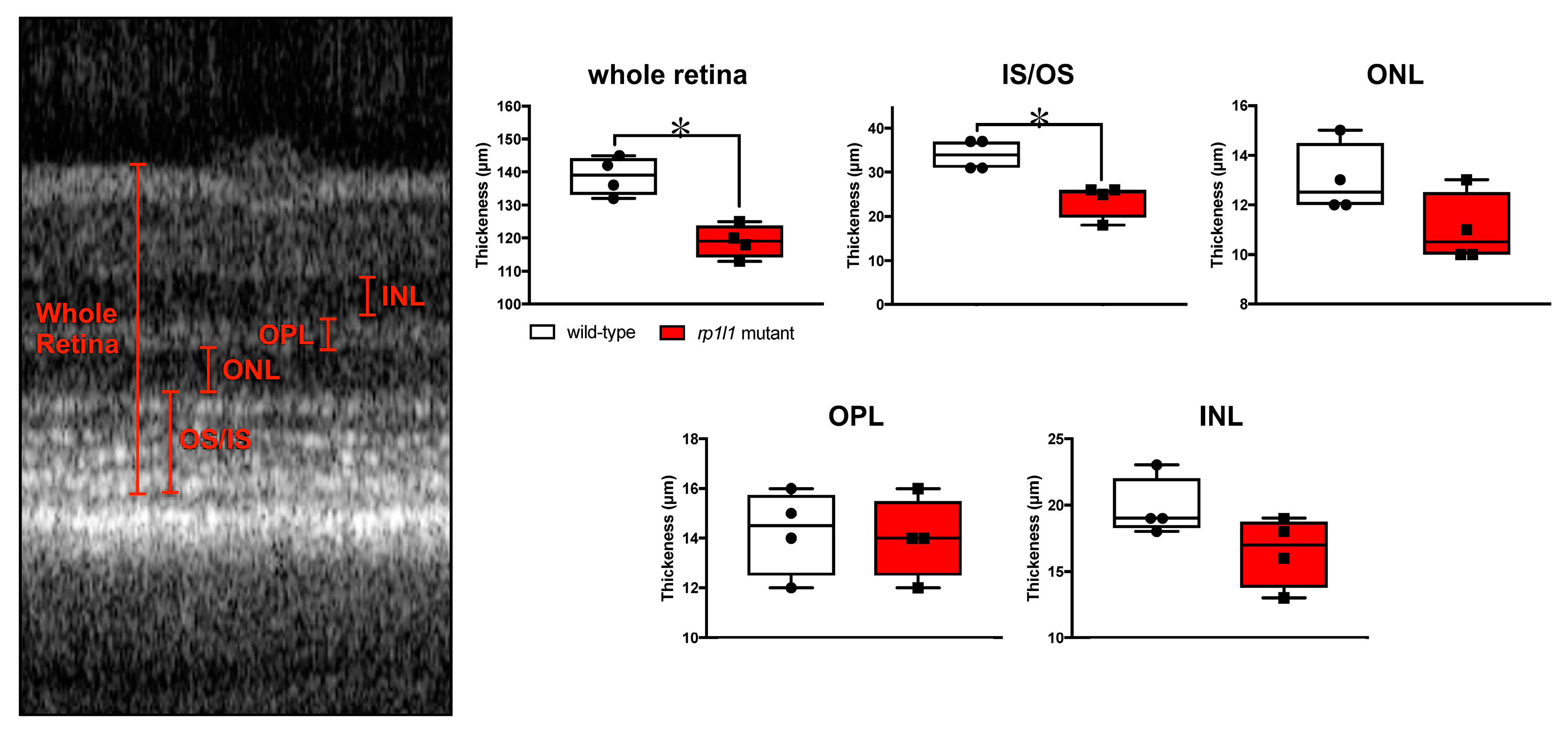
3.3. Live Imaging Reveals Abnormalities in Outer Retinas of rp1l1 Mutant Zebrafish
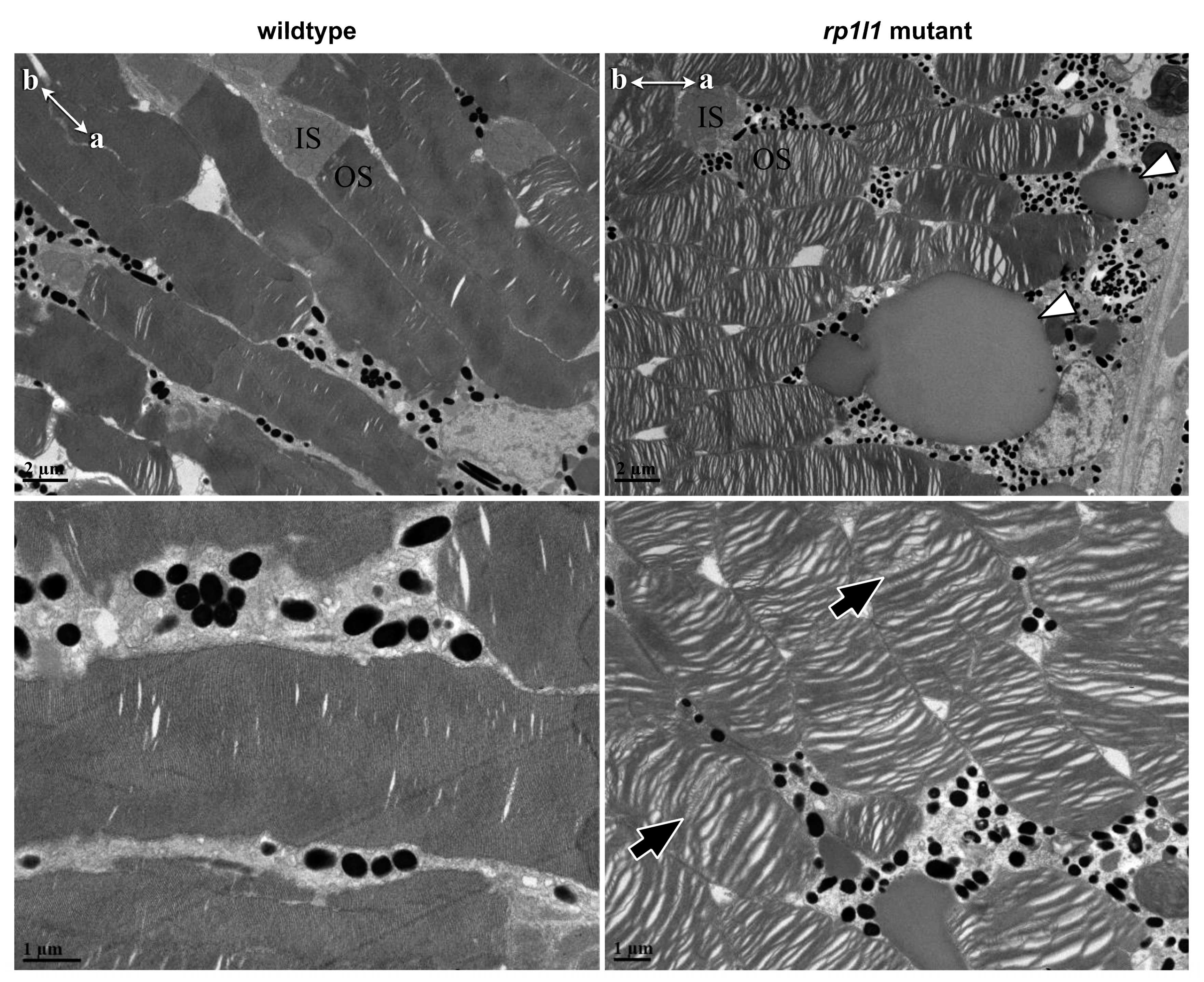
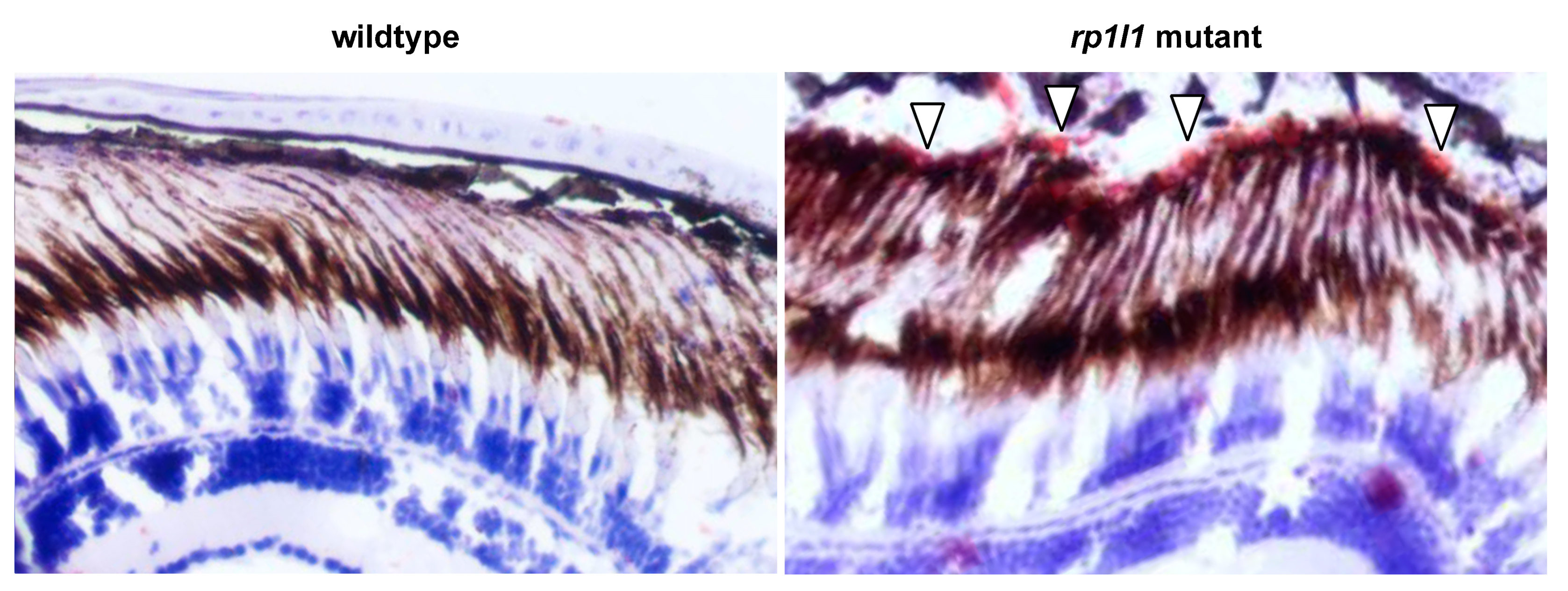
3.4. rp1l1 is Required for Normal Organization of Photoreceptor Outer Segments
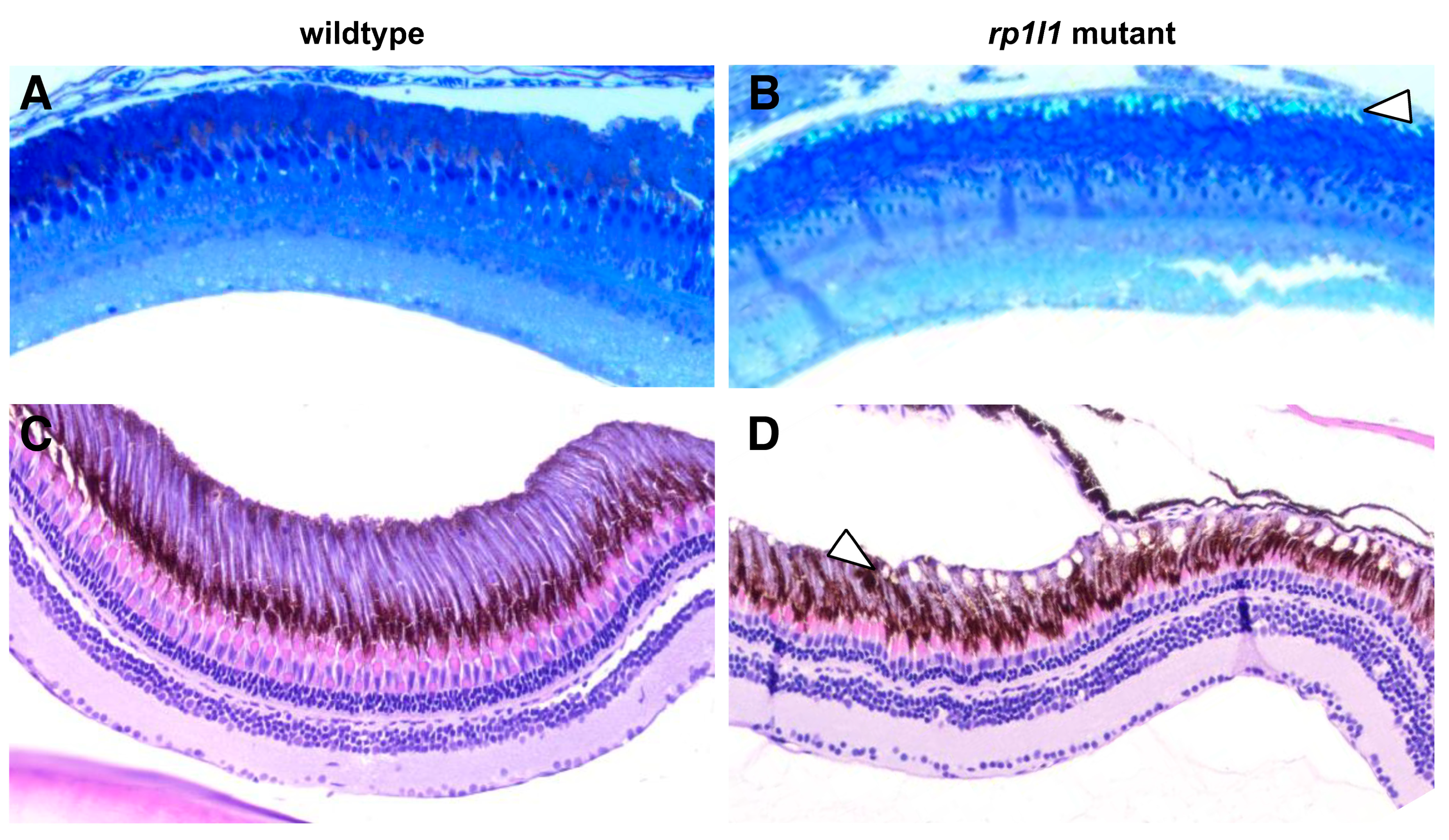
3.5. Diseased Zebrafish Retinas Have Lipid-Rich Subretinal Drusenoid Deposits
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bachmann-Gagescu, R.; Neuhauss, S.C. The photoreceptor cilium and its diseases. Curr. Opin. Genet. Dev. 2019, 56, 22–33. [Google Scholar] [CrossRef]
- Bujakowska, K.M.; Liu, Q.; Pierce, E.A. Photoreceptor cilia and retinal ciliopathies. Cold Spring Harb. Perspect. Biol. 2017, 9, a028274. [Google Scholar] [CrossRef] [PubMed]
- National Eye Institute (National Institutes of Health) AMD Data and Statistics. Available online: https://www.nei.nih.gov/learn-about-eye-health/resources-for-health-educators/eye-health-data-and-statistics/age-related-macular-degeneration-amd-data-and-statistics (accessed on 1 June 2020).
- Miyake, Y.; Horiguchi, M.; Tomita, N.; Kondo, M.; Tanikawa, A.; Takahashi, H.; Suzuki, S.; Terasaki, H. Occult Macular Dystrophy. Am. J. Ophthalmol. 1996, 122, 644–653. [Google Scholar] [CrossRef]
- Miyake, Y.; Tsunoda, K. Occult macular dystrophy. Jpn. J. Ophthalmol. 2015, 59, 71–80. [Google Scholar] [CrossRef]
- Miyake, Y.; Ichikawa, K.; Shiose, Y.; Kawase, Y. Hereditary macular dystrophy without visible fundus abnormality. Am. J. Ophthalmol. 1989, 108, 292–299. [Google Scholar] [CrossRef]
- Brockhurst, R.J.; Sandberg, M.A. Optical coherence tomography findings in occult macular dystrophy. Am. J. Ophthalmol. 2007, 143, 516–518. [Google Scholar] [CrossRef]
- Noel, N.C.L.; MacDonald, I.M. RP1L1 and inherited photoreceptor disease: A review. Surv. Ophthalmol. 2020, 65, 725–739. [Google Scholar] [CrossRef]
- Akahori, M.; Tsunoda, K.; Miyake, Y.; Fukuda, Y.; Ishiura, H.; Tsuji, S.; Usui, T.; Hatase, T.; Nakamura, M.; Ohde, H.; et al. Dominant mutations in RP1L1 are responsible for occult macular dystrophy. Am. J. Hum. Genet. 2010, 87, 424–429. [Google Scholar] [CrossRef]
- Conte, I.; Lestingi, M.; den Hollander, A.; Alfano, G.; Ziviello, C.; Pugliese, M.; Circolo, D.; Caccioppoli, C.; Ciccodicola, A.; Banfi, S. Identification and characterisation of the retinitis pigmentosa 1-like 1 gene (RP1L1): A novel candidate for retinal degenerations. Eur. J. Hum. Genet. 2003, 11, 155–162. [Google Scholar] [CrossRef]
- Bowne, S.J.; Daiger, S.P.; Malone, K.A.; Heckenlively, J.R.; Kennan, A.; Humphries, P.; Hughbanks-Wheaton, D.; Birch, D.G.; Liu, Q.; Pierce, E.A.; et al. Characterization of RP1L1, a highly polymorphic paralog of the retinitis pigmentosa 1 (RP1) gene. Mol. Vis. 2003, 9, 129–137. [Google Scholar]
- Yamashita, T.; Liu, J.; Gao, J.; LeNoue, S.; Wang, C.; Kaminoh, J.; Bowne, S.J.; Sullivan, L.S.; Daiger, S.P.; Zhang, K.; et al. Essential and synergistic roles of RP1 and RP1L1 in rod photoreceptor axoneme and retinitis pigmentosa. J. Neurosci. 2009, 29, 9748–9760. [Google Scholar] [CrossRef] [PubMed]
- Davidson, A.E.; Sergouniotis, P.I.; Mackay, D.S.; Wright, G.A.; Waseem, N.H.; Michaelides, M.; Holder, G.E.; Robson, A.G.; Moore, A.T.; Plagnol, V.; et al. RP1L1 variants are associated with a spectrum of inherited retinal diseases including retinitis pigmentosa and occult macular dystrophy. Hum. Mutat. 2013, 34, 506–514. [Google Scholar] [CrossRef] [PubMed]
- Oishi, M.; Oishi, A.; Gotoh, N.; Ogino, K.; Higasa, K.; Iida, K.; Makiyama, Y.; Morooka, S.; Matsuda, F.; Yoshimura, N. Comprehensive molecular diagnosis of a large cohort of Japanese retinitis pigmentosa and usher syndrome patients by next-generation sequencing. Investig. Ophthalmol. Vis. Sci. 2014, 55, 7369–7375. [Google Scholar] [CrossRef]
- Dias, M.F.; Joo, K.; Kemp, J.A.; Fialho, S.L.; da Silva Cunha, A.; Woo, S.J.; Kwon, Y.J. Molecular genetics and emerging therapies for retinitis pigmentosa: Basic research and clinical perspectives. Prog. Retin. Eye Res. 2018, 63, 107–131. [Google Scholar] [CrossRef]
- Narayan, D.S.; Wood, J.P.M.; Chidlow, G.; Casson, R.J. A review of the mechanisms of cone degeneration in retinitis pigmentosa. Acta Ophthalmol. 2016, 94, 748–754. [Google Scholar] [CrossRef] [PubMed]
- Meeker, N.D.; Hutchinson, S.A.; Ho, L.; Trede, N.S. Method for isolation of PCR-ready genomic DNA from zebrafish tissues. Biotechniques 2007, 43, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, J.A.; Valen, E.; Thyme, S.B.; Huang, P.; Akhmetova, L.; Pauli, A.; Montague, T.G.; Zimmerman, S.; Richter, C.; Schier, A.F. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS ONE 2014, 9, e98186. [Google Scholar] [CrossRef]
- Wilkinson, R.N.; Elworthy, S.; Ingham, P.W.; van Eeden, F.J.M. A method for high-throughput PCR-based genotyping of larval zebrafish tail biopsies. Biotechniques 2013, 55, 314–316. [Google Scholar] [CrossRef]
- Nadolski, N.J.; Wong, C.X.L.; Hocking, J.C. Electroretinogram analysis of zebrafish retinal function across development. Doc. Ophthalmol. 2020. [Google Scholar] [CrossRef]
- Fernández, E.J.; Hermann, B.; Považay, B.; Unterhuber, A.; Sattmann, H.; Hofer, B.; Ahnelt, P.K.; Drexler, W. Ultrahigh resolution optical coherence tomography and pancorrection for cellular imaging of the living human retina. Opt. Express 2008, 16, 11083–11094. [Google Scholar] [CrossRef]
- Dimopoulos, I.S.; Hoang, S.C.; Radziwon, A.; Binczyk, N.M.; Seabra, M.C.; MacLaren, R.E.; Somani, R.; Tennant, M.T.S.; MacDonald, I.M. Two-year results after AAV2-mediated gene therapy for choroideremia: The Alberta experience. Am. J. Ophthalmol. 2018, 193, 130–142. [Google Scholar] [CrossRef]
- Coscas, G.; de Benedetto, U.; Coscas, F.; Li Calzi, C.I.; Vismara, S.; Roudot-Thoraval, F.; Bandello, E.; Souied, E. Hyperreflective dots: A new spectral-domain optical coherence tomography entity for follow-up and prognosis in exudative age-related macular degeneration. Ophthalmologica 2013, 229, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Vujosevic, S.; Bini, S.; Midena, G.; Berton, M.; Pilotto, E.; Midena, E. Hyperreflective intraretinal spots in diabetics without and with nonproliferative diabetic retinopathy: An in vivo study using spectral domain OCT. J. Diabetes Res. 2013, 2013, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Castanos, M.V.; Zhou, D.B.; Linderman, R.E.; Allison, R.; Milman, T.; Carroll, J.; Migacz, J.; Rosen, R.B.; Chui, T.Y.P. Imaging of macrophage-like cells in living human retina using clinical OCT. Invest. Ophthalmol. Vis. Sci. 2020, 61, 48. [Google Scholar] [CrossRef] [PubMed]
- Okunuki, Y.; Mukai, R.; Pearsall, E.A.; Klokman, G.; Husain, D.; Park, D.H.; Korobkina, E.; Weiner, H.L.; Butovsky, O.; Ksander, B.R.; et al. Microglia inhibit photoreceptor cell death and regulate immune cell infiltration in response to retinal detachment. Proc. Natl. Acad. Sci. USA 2018, 115, E6264–E6273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Zabel, M.K.; Wang, X.; Ma, W.; Shah, P.; Fariss, R.N.; Qian, H.; Parkhurst, C.N.; Gan, W.; Wong, W.T. Microglial phagocytosis of living photoreceptors contributes to inherited retinal degeneration. EMBO Mol. Med. 2015, 7, 1179–1197. [Google Scholar] [CrossRef]
- Liu, Y.P.; Bosch, D.G.M.; Siemiatkowska, A.M.; Rendtorff, N.D.; Boonstra, F.N.; Möller, C.; Tranebjærg, L.; Katsanis, N.; Cremers, F.P.M. Putative digenic inheritance of heterozygous RP1L1 and C2orf71 null mutations in syndromic retinal dystrophy. Ophthalmic Genet. 2017, 38, 127–132. [Google Scholar] [CrossRef]
- Allison, W.T.; Barthel, L.K.; Skebo, K.M.; Takechi, M.; Kawamura, S.; Raymond, P.A. Ontogeny of cone photoreceptor mosaics in zebrafish. J. Comp. Neurol. 2010, 518, 4182–4195. [Google Scholar] [CrossRef] [PubMed]
- Doerre, G.; Malicki, J. A mutation of early photoreceptor development, mikre oko, reveals cell-cell interactions involved in the survival and differentiation of zebrafish photoreceptors. J. Neurosci. 2001, 21, 6745–6757. [Google Scholar] [CrossRef]
- Doerre, G.; Malicki, J. Genetic analysis of photoreceptor cell development in the zebrafish retina. Mech. Dev. 2002, 110, 125–138. [Google Scholar] [CrossRef]
- Fadool, J.M. Development of a rod photoreceptor mosaic revealed in transgenic zebrafish. Dev. Biol. 2003, 258, 277–290. [Google Scholar] [CrossRef]
- Liu, Q.; Zuo, J.; Pierce, E.A. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J. Neurosci. 2004, 24, 6427–6436. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.; Lyubarsky, A.; Skalet, J.H.; Pugh, E.N.; Pierce, E.A. RP1 is required for the correct stacking of outer segment discs. Investig. Opthalmol. Vis. Sci. 2003, 44, 4171–4183. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Liu, Q.; Zhou, J.; Daiger, S.P.; Farber, D.B.; Heckenlively, J.R.; Smith, J.E.; Sullivan, L.S.; Zuo, J.; Milam, A.H.; Pierce, E.A. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Investig. Ophthalmol. Vis. Sci. 2002, 43, 22–32. [Google Scholar]
- Pierce, E.A.; Quinn, T.; Meehan, T.; McGee, T.L.; Berson, E.L.; Dryja, T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat. Genet. 1999, 22, 248–254. [Google Scholar] [CrossRef]
- Albarry, M.A.; Hashmi, J.A.; Alreheli, A.Q.; Alia, M.; Khan, B.; Ramzan, K.; Basit, S. Novel homozygous loss-of-function mutations in RP1 and RP1L1 genes in retinitis pigmentosa patients. Ophthalmic Genet. 2019, 40, 507–513. [Google Scholar] [CrossRef]
- Verbakel, S.K.; Van Huet, R.A.C.; Den Hollander, A.I.; Geerlings, M.J.; Kersten, E.; Klevering, B.J.; Klaver, C.C.W.; Plomp, A.S.; Wesseling, N.L.; Bergen, A.A.B.; et al. Macular dystrophy and cone-rod dystrophy caused by mutations in the RP1 gene: Extending the RP1 disease spectrum. Investig. Ophthalmol. Vis. Sci. 2019, 60, 1192–1203. [Google Scholar] [CrossRef]
- Huisingh, C.; McGwin, G.; Neely, D.; Zarubina, A.; Clark, M.; Zhang, Y.; Curcio, C.A.; Owsley, C. The association between subretinal drusenoid deposits in older adults in normal macular health and incident age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 739–745. [Google Scholar] [CrossRef][Green Version]
- Rudolf, M.; Malek, G.; Messinger, J.D.; Clark, M.E.; Wang, L.; Curcio, C.A. Sub-retinal drusenoid deposits in human retina: Organization and composition. Exp. Eye Res. 2008, 87, 402–408. [Google Scholar] [CrossRef]
- Saade, C.; Smith, R.T. Reticular macular lesions: A review of the phenotypic hallmarks and their clinical significance. Clin. Exp. Ophthalmol. 2014, 42, 865–874. [Google Scholar] [CrossRef]
- Cheng, S.Y.; Cipi, J.; Ma, S.; Hafler, B.P.; Kanadia, R.N.; Brush, R.S.; Agbaga, M.P.; Punzo, C. Altered photoreceptor metabolism in mouse causes late stage age-related macular degeneration-like pathologies. Proc. Natl. Acad. Sci. USA 2020, 117, 13094–13104. [Google Scholar] [CrossRef]
- Imamura, Y.; Noda, S.; Hashizume, K.; Shinoda, K.; Yamaguchi, M.; Uchiyama, S.; Shimizu, T.; Mizushima, Y.; Shirasawa, T.; Tsubota, K. Drusen, choroidal neovascularization, and retinal pigment epithelium dysfunction in SOD1-deficient mice: A model of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2006, 103, 11282–11287. [Google Scholar] [CrossRef]
- Malek, G.; Johnson, L.V.; Mace, B.E.; Saloupis, P.; Schmechel, D.E.; Rickman, D.W.; Toth, C.A.; Sullivan, P.M.; Rickman, C.B. Apolipoprotein E allele-dependent pathogenesis: A model for age-related retinal degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 11900–11905. [Google Scholar] [CrossRef]
- Do, K.V.; Kautzmann, M.A.I.; Jun, B.; Gordon, W.C.; Nshimiyimana, R.; Yang, R.; Petasis, N.A.; Bazan, N.G. Elovanoids counteract oligomeric β-amyloid-induced gene expression and protect photoreceptors. Proc. Natl. Acad. Sci. USA 2019, 116, 24317–24325. [Google Scholar] [CrossRef] [PubMed]
- Ban, N.; Lee, T.J.; Sene, A.; Choudhary, M.; Lekwuwa, M.; Dong, Z.; Santeford, A.; Lin, J.B.; Malek, G.; Ory, D.S.; et al. Impaired monocyte cholesterol clearance initiates age-related retinal degeneration and vision loss. JCI Insight 2018, 3, e120824. [Google Scholar] [CrossRef] [PubMed]
- Luhmann, U.F.O.; Robbie, S.; Munro, P.M.G.; Barker, S.E.; Duran, Y.; Luong, V.; Fitzke, F.W.; Bainbridge, J.W.B.; Ali, R.R.; MacLaren, R.E. The drusen-like phenotype in aging Ccl2 knockout mice is caused by an accelerated accumulation of swollen autofluorescent subretinal macrophages. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5934–5943. [Google Scholar] [CrossRef] [PubMed]
- Vessey, K.A.; Greferath, U.; Jobling, A.I.; Phipps, J.A.; Ho, T.; Waugh, M.; Fletcher, E.L. Ccl2/Cx3cr1 knockout mice have inner retinal dysfunction but are not an accelerated model of AMD. Investig. Ophthalmol. Vis. Sci. 2012, 53, 7833–7846. [Google Scholar] [CrossRef]
- Aleman, T.S.; Cideciyan, A.V.; Aguirre, G.K.; Huang, W.C.; Mullins, C.L.; Roman, A.J.; Sumaroka, A.; Olivares, M.B.; Tsai, F.F.; Schwartz, S.B.; et al. Human CRB1-associated retinal degeneration: Comparison with the rd8 Crb1-mutant mouse model. Investig. Ophthalmol. Vis. Sci. 2011, 52, 6898–6910. [Google Scholar] [CrossRef]
- Chang, B.; Hurd, R.; Wang, J.; Nishina, P. Survey of common eye diseases in laboratory mouse strains. Investig. Ophthalmol. Vis. Sci. 2013, 54, 4974–4981. [Google Scholar] [CrossRef]
- Bergen, A.A.; Arya, S.; Koster, C.; Pilgrim, M.G.; Wiatrek-Moumoulidis, D.; van der Spek, P.J.; Hauck, S.M.; Boon, C.J.F.; Emri, E.; Stewart, A.J.; et al. On the origin of proteins in human drusen: The meet, greet and stick hypothesis. Prog. Retin. Eye Res. 2019, 70, 55–84. [Google Scholar] [CrossRef]
- Spaide, R.F. Outer retinal atrophy after regression of subretinal drusenoid deposits as a newly recognized form of late age-related macular degeneration. Retina 2013, 33, 1800–1808. [Google Scholar] [CrossRef] [PubMed]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Noel, N.C.L.; Nadolski, N.J.; Hocking, J.C.; MacDonald, I.M.; Allison, W.T. Progressive Photoreceptor Dysfunction and Age-Related Macular Degeneration-Like Features in rp1l1 Mutant Zebrafish. Cells 2020, 9, 2214. https://doi.org/10.3390/cells9102214
Noel NCL, Nadolski NJ, Hocking JC, MacDonald IM, Allison WT. Progressive Photoreceptor Dysfunction and Age-Related Macular Degeneration-Like Features in rp1l1 Mutant Zebrafish. Cells. 2020; 9(10):2214. https://doi.org/10.3390/cells9102214
Chicago/Turabian StyleNoel, Nicole C. L., Nathan J. Nadolski, Jennifer C. Hocking, Ian M. MacDonald, and W. Ted Allison. 2020. "Progressive Photoreceptor Dysfunction and Age-Related Macular Degeneration-Like Features in rp1l1 Mutant Zebrafish" Cells 9, no. 10: 2214. https://doi.org/10.3390/cells9102214
APA StyleNoel, N. C. L., Nadolski, N. J., Hocking, J. C., MacDonald, I. M., & Allison, W. T. (2020). Progressive Photoreceptor Dysfunction and Age-Related Macular Degeneration-Like Features in rp1l1 Mutant Zebrafish. Cells, 9(10), 2214. https://doi.org/10.3390/cells9102214