Improved Prediction of miRNA-Disease Associations Based on Matrix Completion with Network Regularization


Abstract
1. Introduction
2. Materials and Methods
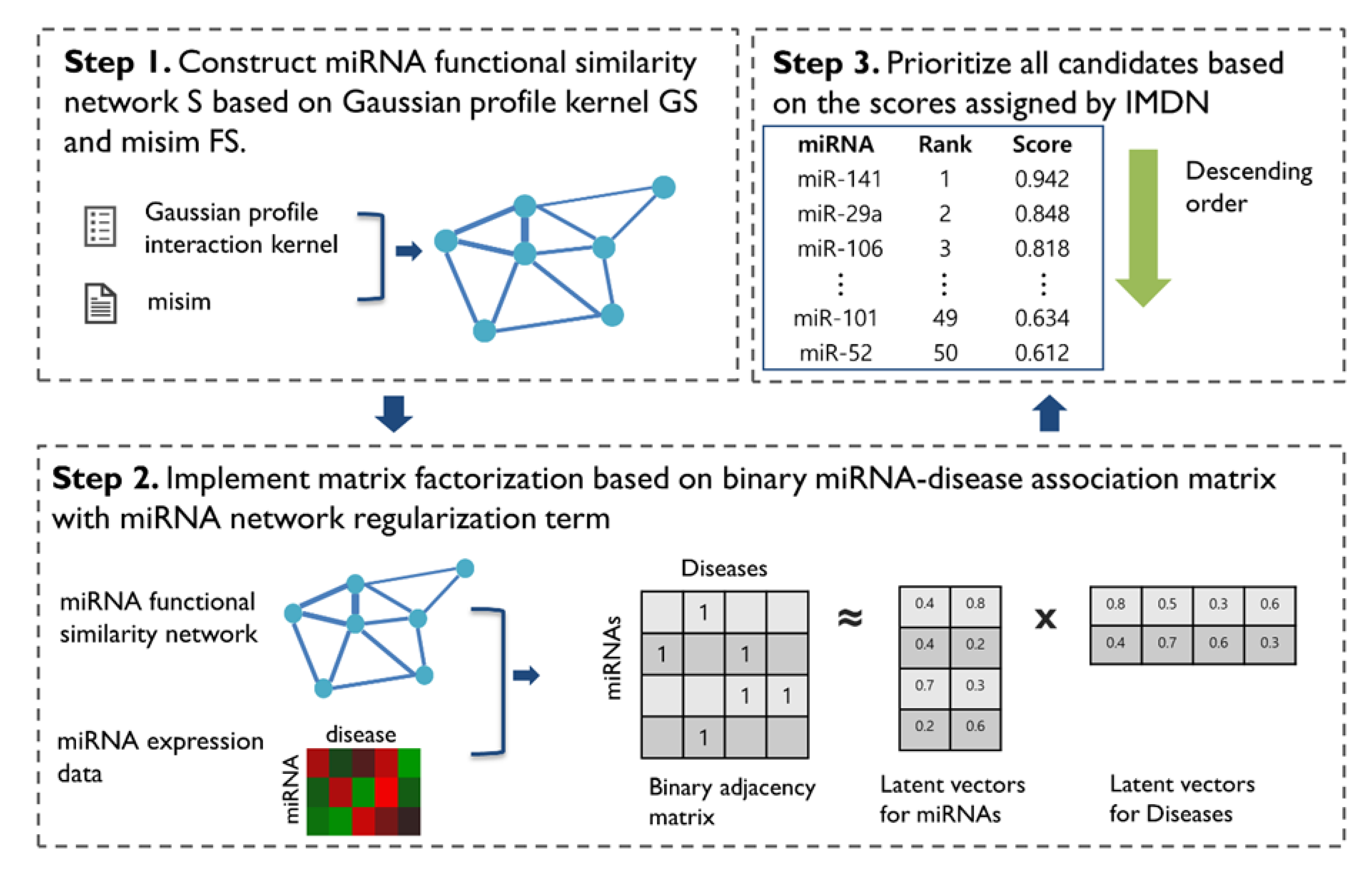
2.1. Methods Overview
2.2. Human miRNA-disease Associations
2.3. miRNA Expression Data
2.4. miRNA Similarity Network
2.4.1. miRNA Functional Similarity
2.4.2. Gaussian Interaction Profile Kernel miRNA Similarity
2.4.3. Integrated miRNA Similarity
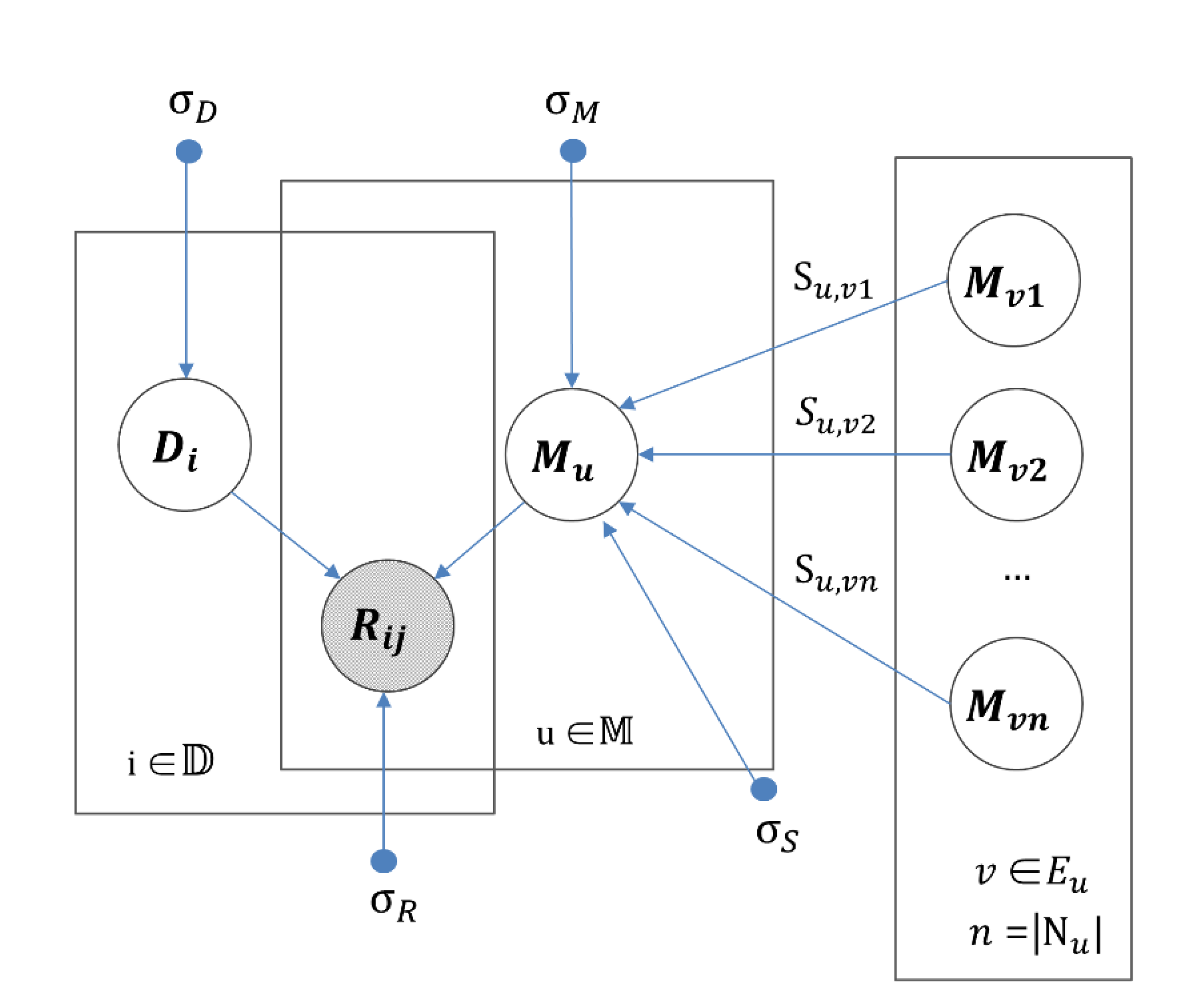
2.5. IMDN
3. Results
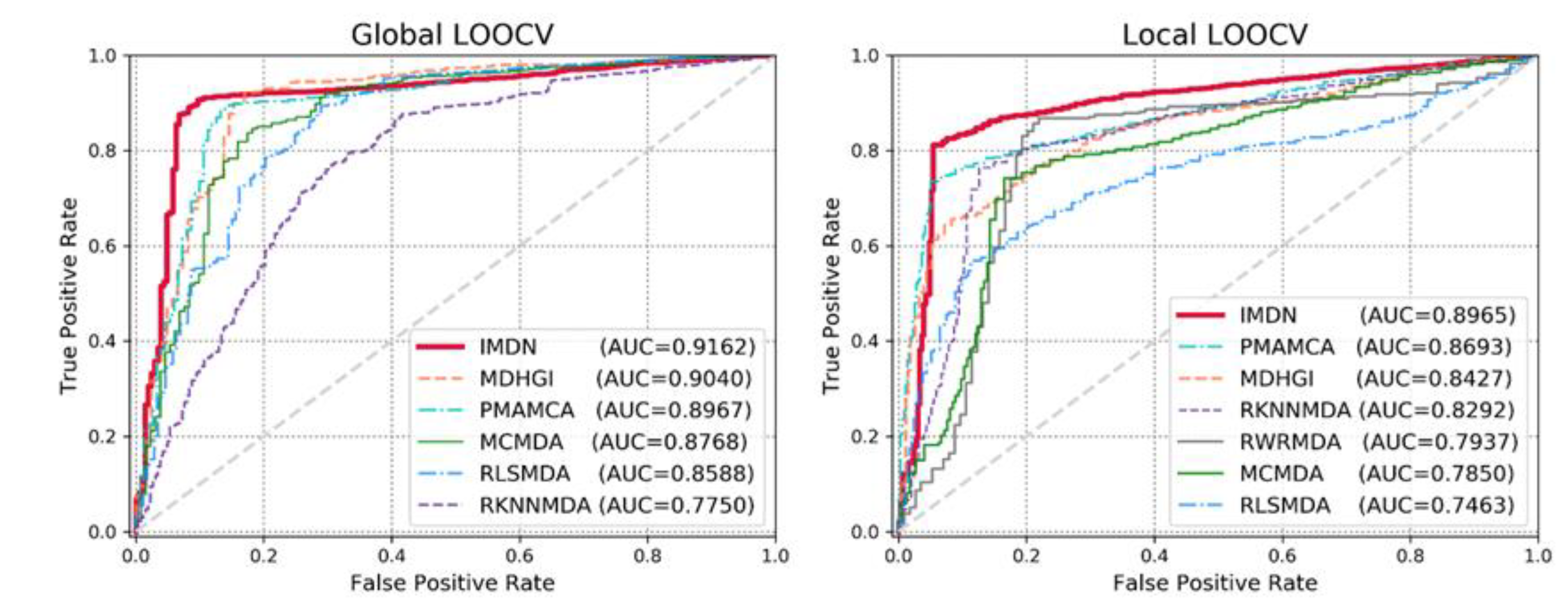
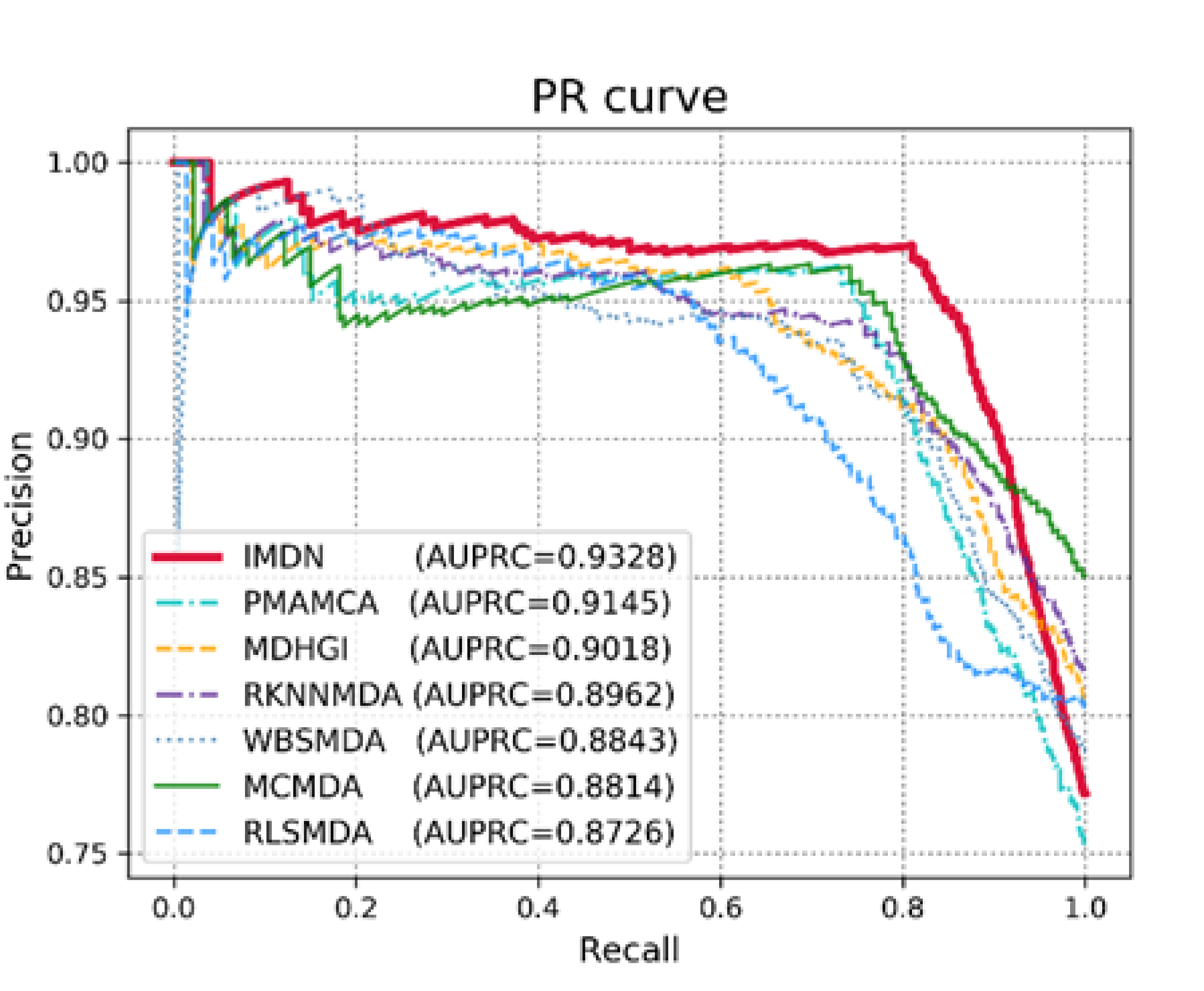
3.1. Performance Evaluation
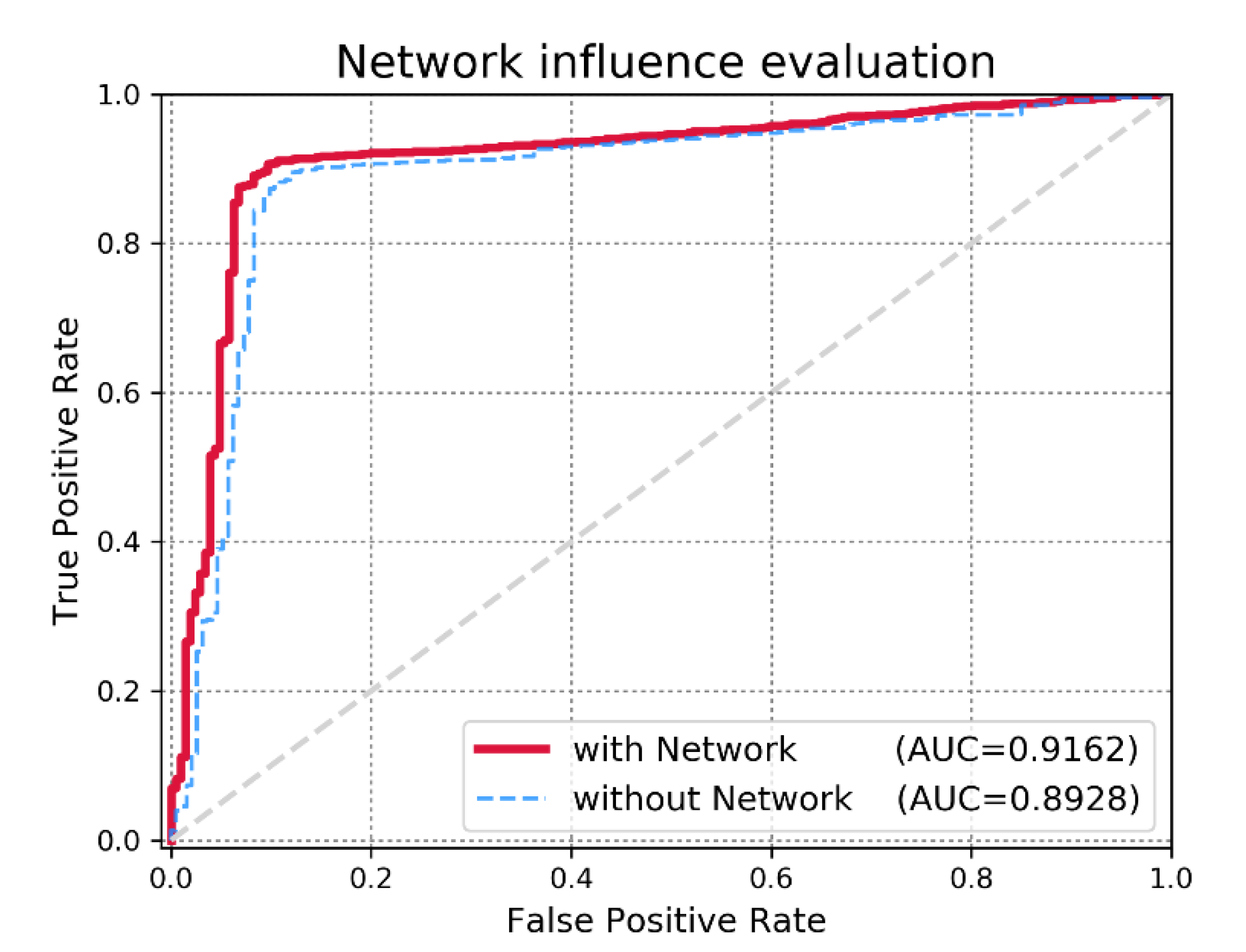
3.2. Effect of miRNA Functional Similarity Network
3.3. Case Studies
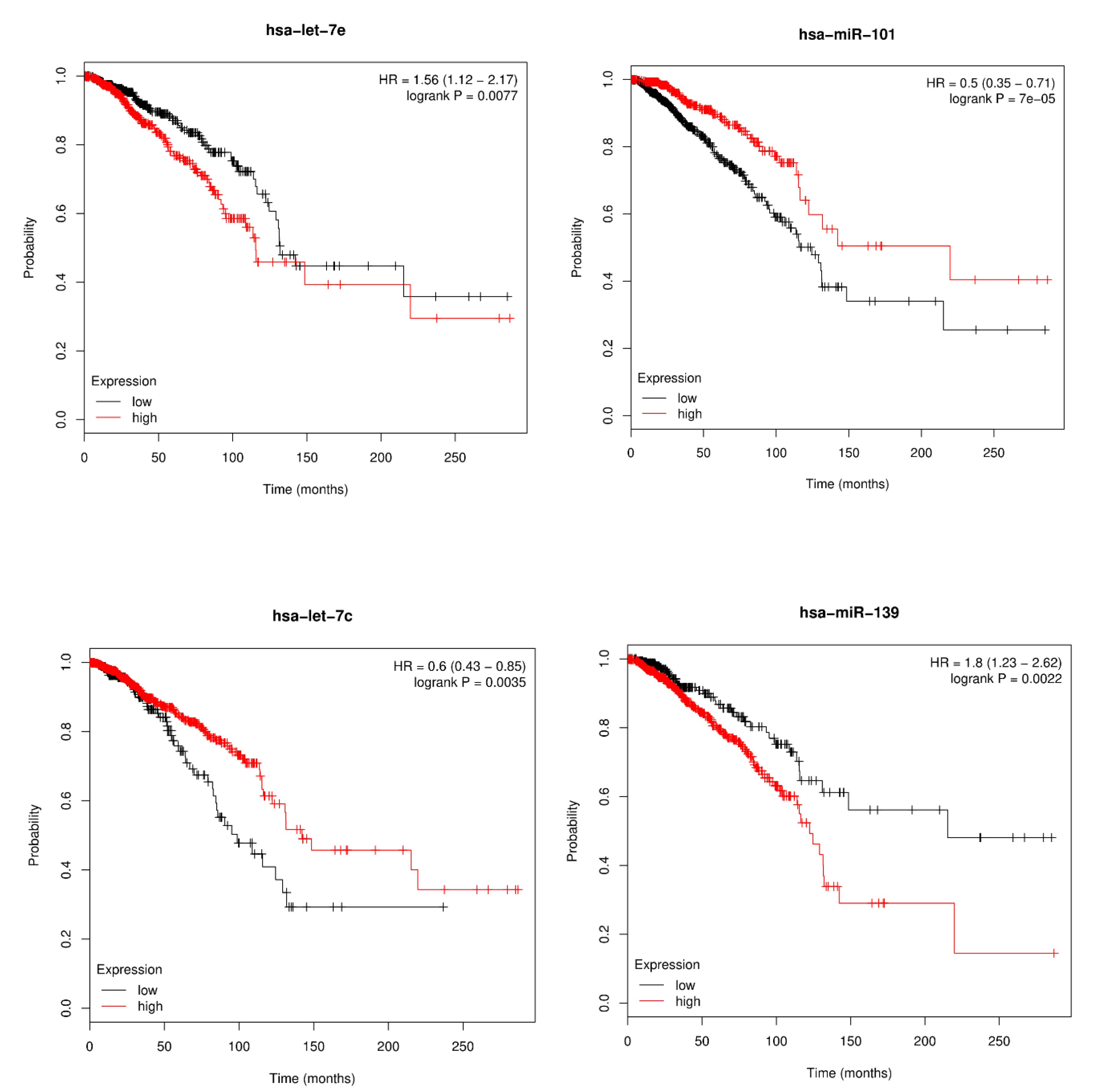
3.4. Survival Analysis
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Bartel, B. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Xu, P.; Guo, M.; Hay, B.A. MicroRNAs and the regulation of cell death. Trends Genet. 2004, 20, 617–624. [Google Scholar] [CrossRef] [PubMed]
- Karp, X.; Ambros, V. DEVELOPMENTAL BIOLOGY: Enhanced: Encountering MicroRNAs in Cell Fate Signaling. Sciety 2005, 310, 1288–1289. [Google Scholar] [CrossRef]
- Cheng, A.M.; Byrom, M.W.; Shelton, J.; Ford, L.P. Antisense inhibition of human miRNAs and indications for an involvement of miRNA in cell growth and apoptosis. Nucleic Acids Res. 2005, 33, 1290–1297. [Google Scholar] [CrossRef]
- Griffiths-Jones, S. miRBase: microRNA Sequences and Annotation. Curr. Protoc. Bioinform. 2010, 29, 1–10. [Google Scholar] [CrossRef]
- Alshalalfa, M.; Alhajj, R. Using context-specific effect of miRNAs to identify functional associations between miRNAs and gene signatures. BMC Bioinform. 2013, 14, S1. [Google Scholar] [CrossRef]
- Zhu, H.C.; Wang, L.M.; Wang, M.; Song, B.; Tan, S.; Teng, J.F.; Duan, D.-X. MicroRNA-195 downregulates Alzheimer’s disease amyloid-b production by targeting BACE1. Brain Res. Bull 2012, 88, 596–601. [Google Scholar]
- Wang, R.; Wang, H.-B.; Hao, C.J.; Cui, Y.; Han, X.-C.; Hu, Y.; Li, F.-F.; Ma, X.; Ma, X. MiR-101 Is Involved in Human Breast Carcinogenesis by Targeting Stathmin1. PLoS ONE 2012, 7, e46173. [Google Scholar] [CrossRef]
- Calin, G.A.; Dumitru, C.D.; Shimizu, M.; Bichi, R.; Zupo, S.; Noch, E.; Aldler, H.; Rattan, S.; Keating, M.; Rai, K.; et al. Nonlinear partial differential equations and applications: Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 2002, 99, 15524–15529. [Google Scholar] [CrossRef]
- Wang, R.; Tian, S.; Wang, H.-B.; Chu, D.-P.; Cao, J.-L.; Xia, H.-F.; Ma, X. MiR-185is involved in human breast carcinogenesis by targetingVegfa. FEBS Lett. 2014, 588, 4438–4447. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Wang, H.; Yang, Z. MiR-122 Inhibits Cell Proliferation and Tumorigenesis of Breast Cancer by Targeting IGF1R. PLoS ONE 2012, 7, e47053. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Hao, Y.; Wang, G.; Juan, L.; Zhang, T.; Teng, M.; Liu, Y.; Wang, Y. Prioritization of disease microRNAs through a human phenome-microRNAome network. BMC Syst. Boil. 2010, 4, S2. [Google Scholar] [CrossRef] [PubMed]
- Mørk, S.; Pletscher-Frankild, S.; Caro, A.P.; Gorodkin, J.; Jensen, L.J. Protein-driven inference of miRNA-disease associations. Bioinformatics 2013, 30, 392–397. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Liu, M.-X.; Yan, G.-Y. RWRMDA: Predicting novel human microRNA–disease associations. Mol. BioSyst. 2012, 8, 2792. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H.; Deng, L.; Liu, Y.; Zhang, Y.; Dai, Q. WBSMDA: Within and Between Score for MiRNA-Disease Association prediction. Sci. Rep. 2016, 6, 21106. [Google Scholar] [CrossRef]
- Chen, X.; Yan, C.C.; Zhang, X.; You, Z.-H.; Huang, Y.-A.; Yan, G.-Y. HGIMDA: Heterogeneous graph inference for miRNA-disease association prediction. Oncotarget 2016, 7, 65257–65269. [Google Scholar] [CrossRef]
- Xuan, P.; Han, K.; Guo, M.; Guo, Y.; Li, J.; Ding, J.; Liu, Y.; Dai, Q.; Li, J.; Teng, Z.; et al. Prediction of microRNAs associated with human diseases based on weighted k most similar neighbors. PLoS ONE 2013, 8, e70204. [Google Scholar]
- Ha, J.; Kim, H.; Yoon, Y.; Park, S. A method of extracting disease-related microRNAs through the propagation algorithm using the environmental factor based global miRNA network. Bio-Medical Mater. Eng. 2015, 26, S1763–S1772. [Google Scholar] [CrossRef]
- Shi, H.; Xu, J.; Zhang, G.; Xu, L.; Li, C.; Wang, L.; Zhao, Z.; Jiang, W.; Guo, Z.; Li, X. Walking the interactome to identify human miRNA-disease associations through the functional link between miRNA targets and disease genes. BMC Syst. Boil. 2013, 7, 101. [Google Scholar] [CrossRef]
- Chen, X.; Yan, G.-Y. Semi-supervised learning for potential human microRNA-disease associations inference. Sci. Rep. 2014, 4, 5501. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yan, C.C.; Zhang, X.; Li, Z.; Deng, L.; Zhang, Y.; Dai, Q. RBMMMDA: Predicting multiple types of disease-microRNA associations. Sci. Rep. 2015, 5, 13877. [Google Scholar] [CrossRef]
- Ha, J.; Park, C.; Park, S. PMAMCA: Prediction of microRNA-disease association utilizing a matrix completion approach. BMC Syst. Boil. 2019, 13, 33. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-Q.; Rong, Z.-H.; Chen, X.; Yan, G.-Y.; You, Z.-H. MCMDA: Matrix completion for MiRNA-disease association prediction. Oncotarget 2017, 8, 21187–21199. [Google Scholar] [CrossRef]
- Xiao, Q.; Luo, J.; Liang, C.; Cai, J.; Ding, P. A graph regularized non-negative matrix factorization method for identifying microRNA-disease associations. Bioinformatics 2017, 34, 239–248. [Google Scholar] [CrossRef]
- Chen, X.; Wu, Q.-F.; Yan, G.-Y. RKNNMDA: Ranking-based KNN for MiRNA-Disease Association prediction. RNA Boil. 2017, 14, 952–962. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, L.; Qu, J.; Guan, N.-N.; Li, J.-Q. Predicting miRNA–disease association based on inductive matrix completion. Bioinformatics 2018, 34, 4256–4265. [Google Scholar] [CrossRef]
- Chen, X.; Yin, J.; Qu, J.; Huang, L. MDHGI: Matrix Decomposition and Heterogeneous Graph Inference for miRNA-disease association prediction. PLoS Comput. Boil. 2018, 14, e1006418. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Qiu, C.; Tu, J.; Geng, B.; Yang, J.; Jiang, T.; Cui, Q. HMDD v2.0: A database for experimentally supported human microRNA and disease associations. Nucleic Acids Res. 2013, 42, D1070–D1074. [Google Scholar] [PubMed]
- Yang, Z.; Ren, F.; Liu, C.; He, S.; Sun, G.; Gao, Q.; Yao, L.; Zhang, Y.; Miao, R.; Cao, Y.; et al. dbDEMC: A database of differentially expressed miRNAs in human cancers. BMC Genom. 2010, 11, S5. [Google Scholar] [CrossRef] [PubMed]
- Jiang, Q.; Wang, Y.; Hao, Y.; Juan, L.; Teng, M.; Zhang, X.; Li, M.; Wang, G.; Liu, Y. miR2Disease: A manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2008, 37, D98–D104. [Google Scholar] [CrossRef] [PubMed]
- Tomczak, K.; Czerwińska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Współczesna Onkol. 2015, 19, A68–A77. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zhang, S.; Wan, Y.; Zhao, Y.; Shi, J.; Zhou, Y.; Cui, Q. MISIM v2.0: A web server for inferring microRNA functional similarity based on microRNA-disease associations. Nucleic Acids Res. 2019, 47, W536–W541. [Google Scholar] [CrossRef] [PubMed]
- Van Laarhoven, T.; Nabuurs, S.B.; Marchiori, E. Gaussian interaction profile kernels for predicting drug–target interaction. Bioinformatics 2011, 27, 3036–3043. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Huang, Y.-A.; You, Z.-H.; Yan, G.-Y.; Wang, X.-S. A novel approach based on KATZ measure to predict associations of human microbiota with non-infectious diseases. Bioinformatics 2017, 33, 733–739. [Google Scholar]
- Jamali, M.; Ester, M. A matrix factorization technique with trust propagation for recommendation in social networks. In Proceedings of the fourth Association for Computing Machinery conference (ACM), Barcelona , Spain, 26 September 2010; 2010; p. 135. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2019, 69, 7. [Google Scholar]
- Feng, J.; Yang, Y.; Zhang, P.; Wang, F.; Ma, Y.; Qin, H.; Wang, Y. miR-150 functions as a tumour suppressor in human colorectal cancer by targeting c-Myb. J. Cell. Mol. Med. 2014, 18, 2125–2134. [Google Scholar] [CrossRef]
- Iino, I.; Kikuchi, H.; Miyazaki, S.; Hiramatsu, Y.; Ohta, M.; Kamiya, K.; Kusama, Y.; Baba, S.; Setou, M.; Konno, H. Effect of miR-122 and its target gene cationic amino acid transporter 1 on colorectal liver metastasis. Cancer Sci. 2013, 104, 624–630. [Google Scholar] [CrossRef]
- Wan, D.; He, S.; Xie, B.; Xu, G.; Gu, W.; Shen, C.; Hu, Y.; Wang, X.; Zhi, Q.; Wang, L. Aberrant expression of miR-199a-3p and its clinical significance in colorectal cancers. Med. Oncol. 2013, 30, 378. [Google Scholar] [CrossRef]
- Li, Y.; Chen, D.; Jin, L.; Liu, J.; Li, Y.; Su, Z.; Qi, Z.; Shi, M.; Jiang, Z.; Yang, S.; et al. Oncogenic microRNA-142-3p is associated with cellular migration, proliferation and apoptosis in renal cell carcinoma. Oncol. Lett. 2015, 11, 1235–1241. [Google Scholar] [CrossRef]
- Li, Y.; Li, Y.; Chen, D.; Jin, L.; Su, Z.; Liu, J.; Duan, H.; Li, X.; Qi, Z.; Shi, M.; et al. miR-30a-5p in the tumorigenesis of renal cell carcinoma: A tumor suppressive microRNA. Mol. Med. Rep. 2016, 13, 4085–4094. [Google Scholar] [CrossRef] [PubMed]
- Harrison, J.S. Leukemia and lymphoma society. Soc. Sci. Electron. Publ. 2013, 21, 3699–3707. [Google Scholar]
- McDuffie, H.H.; Pahwa, P.; Karunanayake, C.; Spinelli, J.J.; A Dosman, J. Clustering of cancer among families of cases with Hodgkin Lymphoma (HL), Multiple Myeloma (MM), Non-Hodgkin’s Lymphoma (NHL), Soft Tissue Sarcoma (STS) and control subjects. BMC Cancer 2009, 9, 70. [Google Scholar] [CrossRef] [PubMed]
- Cimmino, A.; Calin, G.A.; Fabbri, M.; Iorio, M.; Ferracin, M.; Shimizu, M.; Wojcik, S.E.; Aqeilan, R.I.; Zupo, S.; Dono, M.; et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. USA 2005, 102, 13944–13949. [Google Scholar] [CrossRef]
- Husby, S.; Ralfkiaer, U.; Garde, C.; Zandi, R.; Ek, S.; Kolstad, A.; Jerkeman, M.; Laurell, A.; Räty, R.; Pedersen, L.B.; et al. miR-18b overexpression identifies mantle cell lymphoma patients with poor outcome and improves the MIPI-B prognosticator. Blood 2015, 125, 2669–2677. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shi, X.-H.; Li, X.; Zhang, H.; He, R.-Z.; Zhao, Y.; Zhou, M.; Pan, S.-T.; Zhao, C.-L.; Feng, Y.-C.; Wang, M.; et al. A Five-microRNA Signature for Survival Prognosis in Pancreatic Adenocarcinoma based on TCGA Data. Sci. Rep. 2018, 8, 7638. [Google Scholar] [CrossRef]
- Bandyopadhyay, S.; Mallik, S.; Mukhopadhyay, A. A Survey and Comparative Study of Statistical Tests for Identifying Differential Expression from Microarray Data. IEEE/ACM Trans. Comput. Boil. Bioinform. 2013, 11, 95–115. [Google Scholar] [CrossRef]
- Lánczky, A.; Bottai, G.; Munkácsy, G.; Nagy, Á.; Szabó, A.; Santarpia, L.; Gyorffy, B. miRpower: A web-Tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Symbol | Description |
|---|---|
| number of miRNAs | |
| number of diseases | |
| size of latent vector dimension | |
| miRNA-disease association matrix | |
| miRNA latent space | |
| disease latent space | |
| miRNA similarity matrix | |
| miRNA expression weight matrix |
| Rank | Name | Evidence | Rank | Name | Evidence |
|---|---|---|---|---|---|
| 1 | hsa-let-7a-3 | HMDD v2.0 | 26 | hsa-let-7f-2 | HMDD v2.0 |
| 2 | hsa-miR-19a | dbDEMC, HMDD v2.0 | 27 | hsa-miR-205 | dbDEMC, HMDD v2.0 |
| 3 | hsa-let-7f-1 | HMDD v2.0 | 28 | hsa-miR-125a | dbDEMC, HMDD v2.0 |
| 4 | hsa-miR-137 | dbDEMC, HMDD v2.0 | 29 | hsa-miR-106a | dbDEMC, HMDD v2.0 |
| 5 | hsa-let-7a-1 | HMDD v2.0 | 30 | hsa-miR-101-1 | HMDD v2.0 |
| 6 | hsa-miR-24-1 | HMDD v2.0 | 31 | hsa-miR-365a | HMDD v2.0 |
| 7 | hsa-miR-141 | dbDEMC, HMDD v2.0 | 32 | hsa-miR-21 | dbDEMC, HMDD v2.0 |
| 8 | hsa-miR-30c-2 | HMDD v2.0 | 33 | hsa-miR-9-3 | HMDD v2.0 |
| 9 | hsa-miR-128-2 | HMDD v2.0 | 34 | hsa-miR-296 | dbDEMC, HMDD v2.0 |
| 10 | hsa-miR-629 | HMDD v2.0 | 35 | hsa-miR-493 | dbDEMC, HMDD v2.0 |
| 11 | hsa-miR-486 | dbDEMC, HMDD v2.0 | 36 | hsa-miR-142 | HMDD v2.0 |
| 12 | hsa-miR-29b-1 | HMDD v2.0 | 37 | hsa-miR-9-2 | HMDD v2.0 |
| 13 | hsa-miR-92a-1 | dbDEMC, HMDD v2.0 | 38 | hsa-miR-19b-2 | HMDD v2.0 |
| 14 | hsa-miR-132 | dbDEMC, HMDD v2.0 | 39 | hsa-miR-145 | dbDEMC, HMDD v2.0 |
| 15 | hsa-miR-330 | HMDD v2.0 | 40 | hsa-miR-218-2 | HMDD v2.0 |
| 16 | hsa-miR-200c | HMDD v2.0 | 41 | hsa-miR-30a | dbDEMC, HMDD v2.0 |
| 17 | hsa-miR-584 | dbDEMC, HMDD v2.0 | 42 | hsa-miR-16-1 | dbDEMC, HMDD v2.0 |
| 18 | hsa-miR-1-1 | HMDD v2.0 | 43 | hsa-miR-122 | Literature [41] |
| 19 | hsa-miR-365b | HMDD v2.0 | 44 | hsa-miR-125b-2 | HMDD v2.0 |
| 20 | hsa-miR-506 | dbDEMC, HMDD v2.0 | 45 | hsa-miR-127 | dbDEMC, HMDD v2.0 |
| 21 | hsa-miR-199a | Literature [42] | 46 | hsa-miR-150 | Literature [40] |
| 22 | hsa-miR-101-2 | HMDD v2.0 | 47 | hsa-miR-502 | HMDD v2.0 |
| 23 | hsa-miR-22 | dbDEMC, HMDD v2.0 | 48 | hsa-miR-615 | HMDD v2.0 |
| 24 | hsa-miR-9-1 | HMDD v2.0 | 49 | hsa-miR-6815-5p | unconfirmed |
| 25 | hsa-miR-155 | dbDEMC, HMDD v2.0 | 50 | hsa-miR-16-2 | HMDD v2.0 |
| Rank | Name | Evidence | Rank | Name | Evidence |
|---|---|---|---|---|---|
| 1 | hsa-mir-194 | dbDEMC | 26 | hsa-mir-26b | dbDEMC |
| 2 | hsa-mir-204 | dbDEMC | 27 | hsa-mir-29b | dbDEMC, miR2Disease |
| 3 | hsa-mir-124a | dbDEMC | 28 | hsa-mir-30e-3p | dbDEMC |
| 4 | hsa-mir-199a | dbDEMC, miR2Disease | 29 | hsa-mir-143 | dbDEMC |
| 5 | hsa-mir-215 | dbDEMC | 30 | hsa-mir-200a | dbDEMC |
| 6 | hsa-mir-210 | dbDEMC, miR2Disease | 31 | hsa-mir-224 | dbDEMC |
| 7 | hsa-mir-199a* | dbDEMC | 32 | hsa-mir-30a-3p | dbDEMC |
| 8 | hsa-mir-182* | dbDEMC | 33 | hsa-mir-146a | dbDEMC |
| 9 | hsa-mir-30d | dbDEMC | 34 | hsa-mir-20a | dbDEMC, miR2Disease |
| 10 | hsa-mir-15a | dbDEMC, miR2Disease | 35 | hsa-mir-422a | dbDEMC |
| 11 | hsa-mir-136 | dbDEMC | 36 | hsa-mir-130b | dbDEMC |
| 12 | hsa-mir-22 | dbDEMC | 37 | hsa-mir-130a | dbDEMC |
| 13 | hsa-mir-101 | dbDEMC, miR2Disease | 38 | hsa-mir-455 | dbDEMC |
| 14 | hsa-mir-320 | dbDEMC | 39 | hsa-mir-489 | dbDEMC, miR2Disease |
| 15 | hsa-mir-122a | dbDEMC | 40 | hsa-mir-183 | dbDEMC |
| 16 | hsa-mir-30c | dbDEMC | 41 | hsa-mir-30a-5p | dbDEMC |
| 17 | hsa-mir-214 | dbDEMC, miR2Disease | 42 | hsa-mir-30b | dbDEMC |
| 18 | hsa-mir-198 | dbDEMC | 43 | hsa-mir-139 | dbDEMC |
| 19 | hsa-mir-107 | dbDEMC | 44 | hsa-mir-181b | dbDEMC |
| 20 | hsa-mir-192 | dbDEMC | 45 | hsa-mir-30a | Literature [42] |
| 21 | hsa-mir-106a | dbDEMC, miR2Disease | 46 | hsa-mir-187 | dbDEMC |
| 22 | hsa-mir-186 | dbDEMC | 47 | hsa-mir-133b | unconfirmed |
| 23 | hsa-mir-142 | Literature [41] | 48 | hsa-mir-93 | dbDEMC |
| 24 | hsa-mir-191 | dbDEMC, miR2Disease | 49 | hsa-let-7e | unconfirmed |
| 25 | hsa-mir-422b | dbDEMC | 50 | hsa-mir-429 | dbDEMC |
| Rank | Name | Evidence | Rank | Name | Evidence |
|---|---|---|---|---|---|
| 1 | hsa-miR-138-1 | HMDD v2.0 | 26 | hsa-miR-135b | HMDD v2.0, dbDEMC |
| 2 | hsa-miR-139 | HMDD v2.0, dbDEMC | 27 | hsa-miR-19b-1 | HMDD v2.0 |
| 3 | hsa-miR-92a-2 | HMDD v2.0 | 28 | hsa-miR-101-2 | HMDD v2.0 |
| 4 | hsa-miR-124-1 | HMDD v2.0 | 29 | hsa-miR-181a-2 | HMDD v2.0 |
| 5 | hsa-miR-218-2 | HMDD v2.0 | 30 | hsa-miR-499a | HMDD v2.0 |
| 6 | hsa-miR-20b | HMDD v2.0, dbDEMC | 31 | hsa-miR-122 | HMDD v2.0, dbDEMC |
| 7 | hsa-miR-29c | HMDD v2.0, dbDEMC | 32 | hsa-miR-135a-2 | HMDD v2.0 |
| 8 | hsa-miR-16-1 | HMDD v2.0 | 33 | hsa-miR-150 | HMDD v2.0, dbDEMC |
| 9 | hsa-miR-200b | HMDD v2.0, dbDEMC | 34 | hsa-miR-92a-1 | HMDD v2.0 |
| 10 | hsa-miR-181a-1 | HMDD v2.0 | 35 | hsa-miR-550a-2 | HMDD v2.0 |
| 11 | hsa-miR-550a-1 | HMDD v2.0 | 36 | hsa-miR-155 | HMDD v2.0, dbDEMC |
| 12 | hsa-miR-125a | HMDD v2.0, dbDEMC | 37 | hsa-miR-15a | HMDD v2.0, dbDEMC |
| 13 | hsa-miR-24-1 | HMDD v2.0 | 38 | hsa-miR-92b | HMDD v2.0, dbDEMC |
| 14 | hsa-miR-17 | HMDD v2.0, dbDEMC | 39 | hsa-miR-16-2 | HMDD v2.0 |
| 15 | hsa-miR-133b | HMDD v2.0, dbDEMC | 40 | hsa-miR-138-2 | HMDD v2.0 |
| 16 | hsa-miR-218-1 | HMDD v2.0 | 41 | hsa-miR-18a | HMDD v2.0, dbDEMC |
| 17 | hsa-miR-382 | unconfirmed | 42 | hsa-miR-203 | HMDD v2.0, dbDEMC |
| 18 | hsa-miR-363 | HMDD v2.0, dbDEMC | 43 | hsa-miR-518b | HMDD v2.0, dbDEMC |
| 19 | hsa-miR-19b-2 | HMDD v2.0 | 44 | hsa-miR-26a-1 | HMDD v2.0 |
| 20 | hsa-miR-146a | HMDD v2.0, dbDEMC | 45 | hsa-miR-429 | unconfirmed |
| 21 | hsa-miR-184 | HMDD v2.0, dbDEMC | 46 | hsa-miR-126 | HMDD v2.0, dbDEMC |
| 22 | hsa-miR-511 | unconfirmed | 47 | hsa-miR-135a-1 | HMDD v2.0 |
| 23 | hsa-miR-101-1 | HMDD v2.0 | 48 | hsa-miR-147 | unconfirmed |
| 24 | hsa-miR-26a-2 | HMDD v2.0 | 49 | hsa-miR-210 | HMDD v2.0, dbDEMC |
| 25 | hsa-miR-21 | HMDD v2.0, dbDEMC | 50 | hsa-mir-320a | unconfirmed |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, J.; Park, C.; Park, C.; Park, S. Improved Prediction of miRNA-Disease Associations Based on Matrix Completion with Network Regularization. Cells 2020, 9, 881. https://doi.org/10.3390/cells9040881
Ha J, Park C, Park C, Park S. Improved Prediction of miRNA-Disease Associations Based on Matrix Completion with Network Regularization. Cells. 2020; 9(4):881. https://doi.org/10.3390/cells9040881
Chicago/Turabian StyleHa, Jihwan, Chihyun Park, Chanyoung Park, and Sanghyun Park. 2020. "Improved Prediction of miRNA-Disease Associations Based on Matrix Completion with Network Regularization" Cells 9, no. 4: 881. https://doi.org/10.3390/cells9040881
APA StyleHa, J., Park, C., Park, C., & Park, S. (2020). Improved Prediction of miRNA-Disease Associations Based on Matrix Completion with Network Regularization. Cells, 9(4), 881. https://doi.org/10.3390/cells9040881