Human AlphoidtetO Artificial Chromosome as a Gene Therapy Vector for the Developing Hemophilia A Model in Mice

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Vector Construction
2.3. Cre-Recombinase Mediated Insertion of the FVIII Construct into the AlphoidtetO-HAC in Hamster CHO Cells
2.4. Lentivirus Preparations
2.5. Western Blot
2.6. PCR
2.7. Preparation of Metaphase Spreads
2.8. Fluorescence In Situ Hybridization (FISH) with the PNA Probes
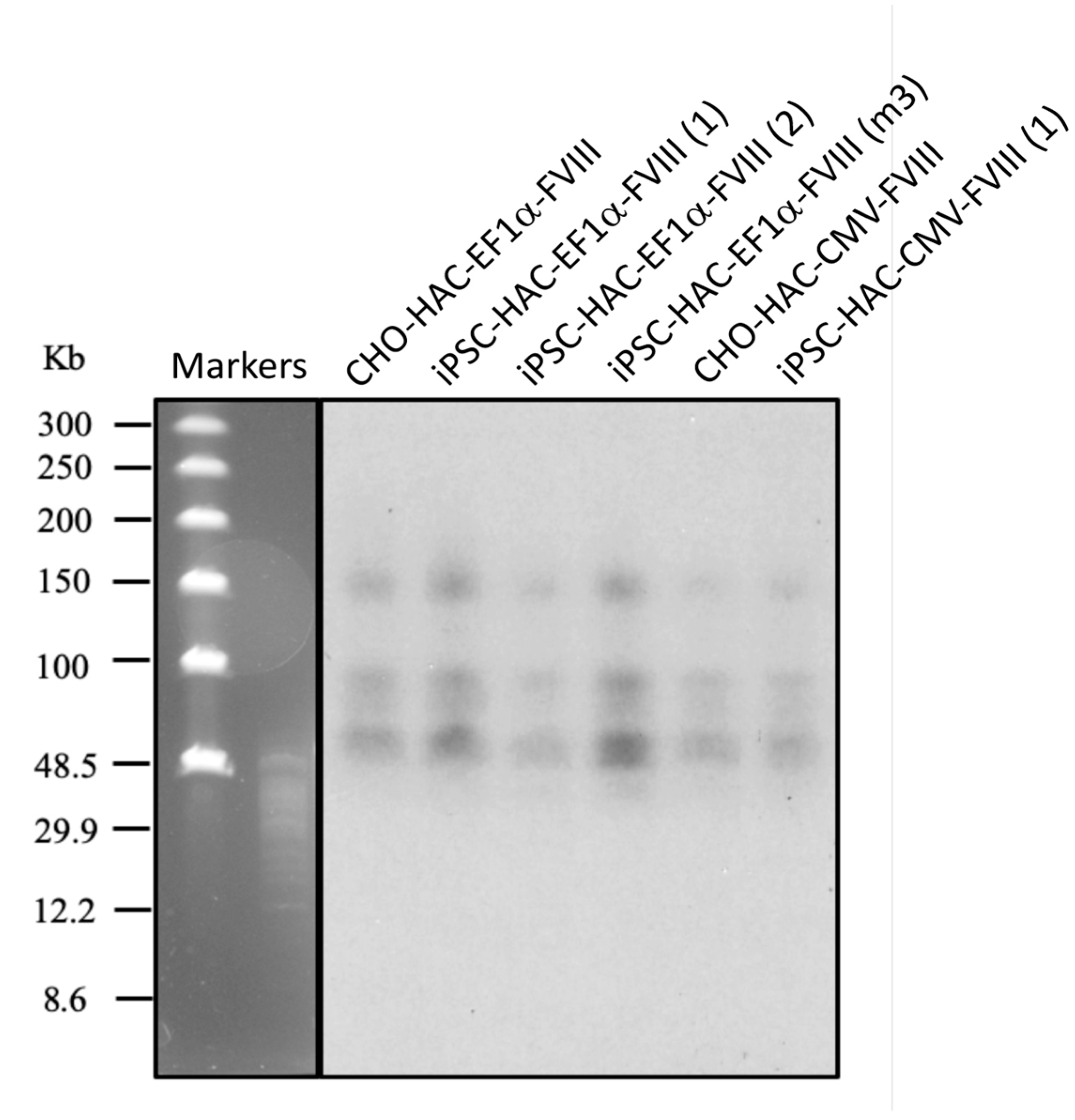
2.9. Southern-Blot Analysis
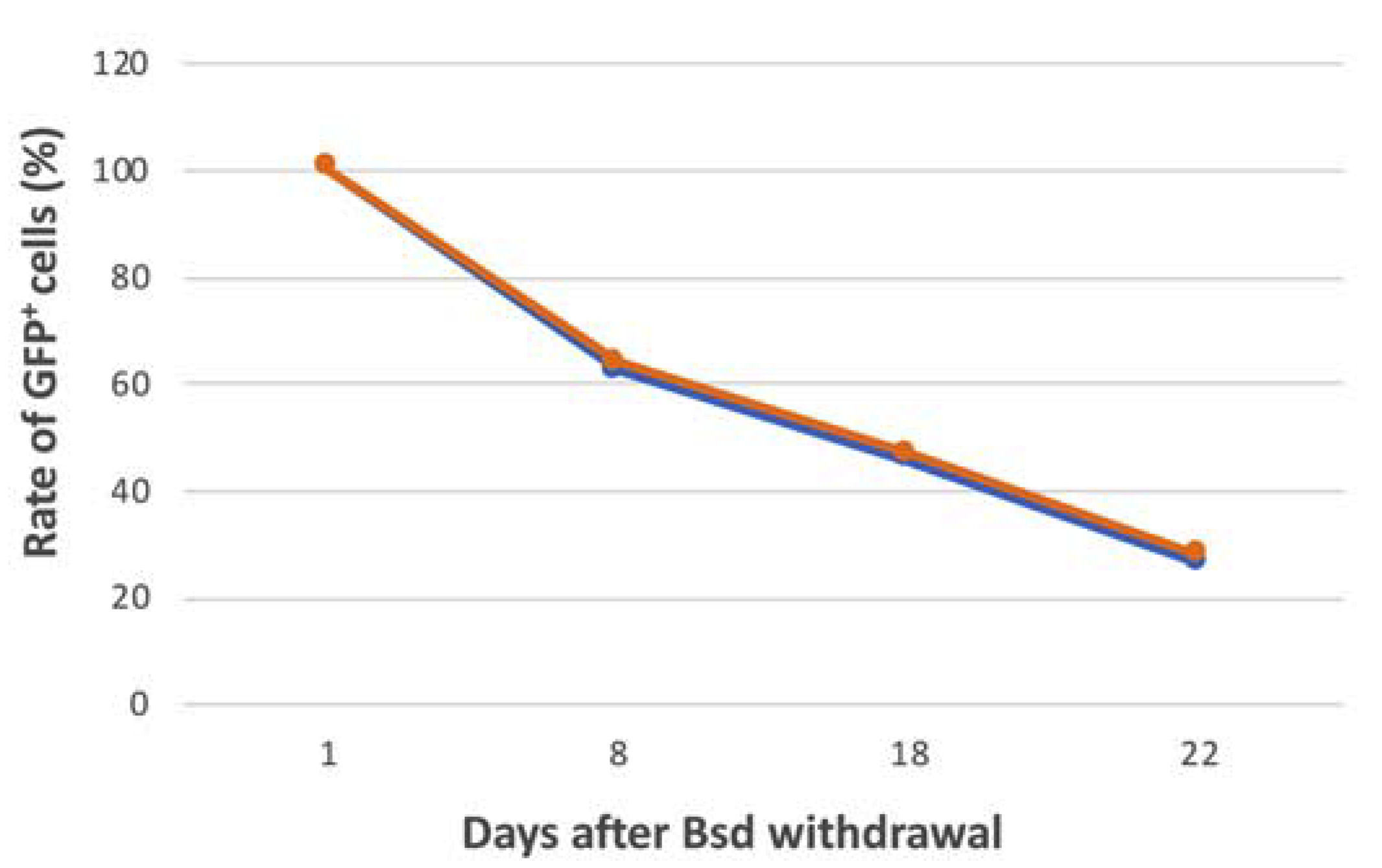
2.10. Analysis of Mitotic Stability of the FVIII-alphoidtetO-HAC in iPSCs
2.11. Murine iPSCs Generation
2.12. Teratoma Formation and Histological Analysis
2.13. Microcell-Mediated Chromosome Transfer (MMCT) Method
2.14. Fluorescence-Activated Cell Sorting (FACS) Assay of iPSCs
2.15. Immunocytochemistry
2.16. Ethical Statement
3. Results and Discussion
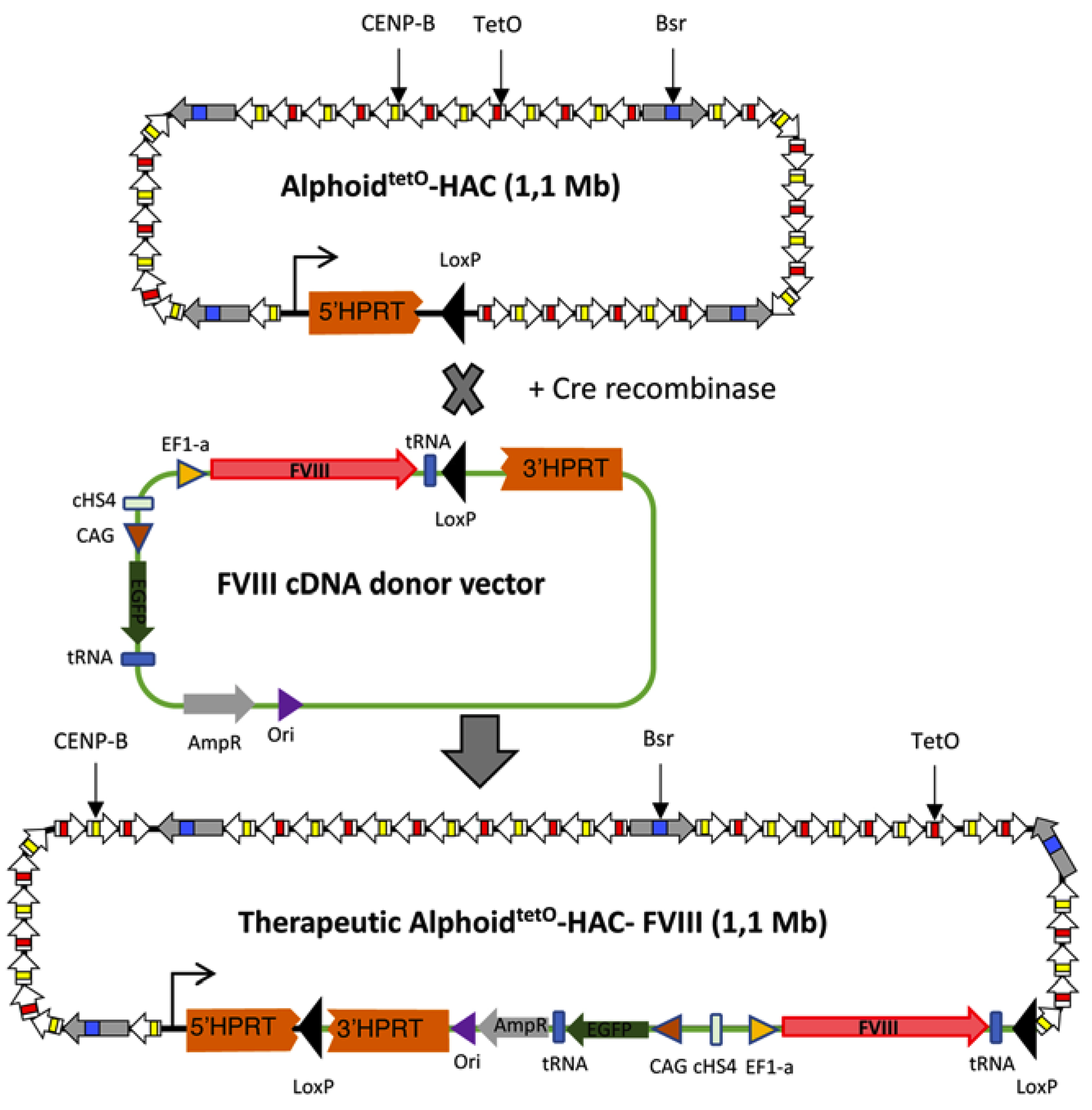
3.1. FVIII Targeting Vector Design
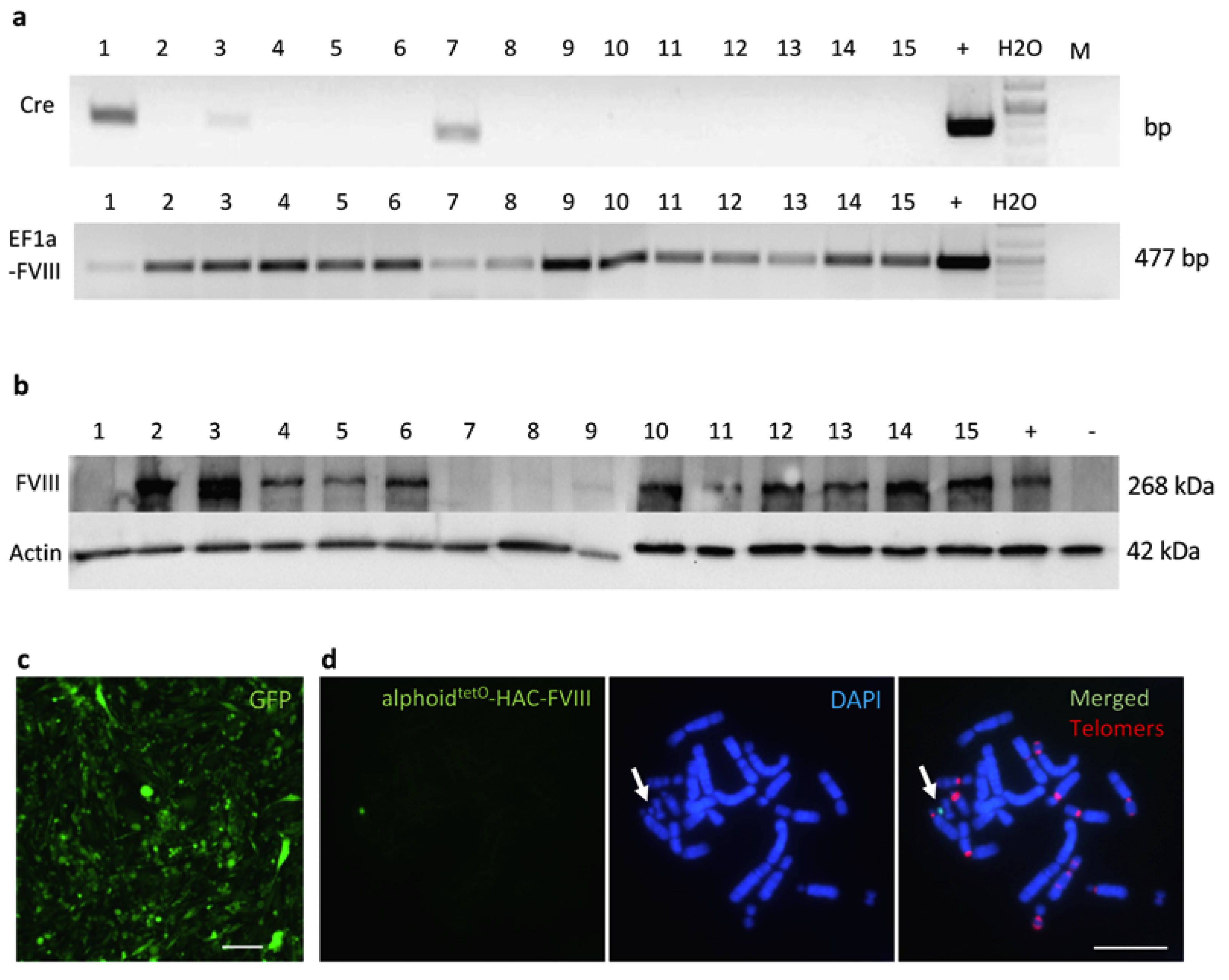
3.2. Recombinase-Mediated Insertion of the Targeting Vector into AlphoidtetO-HAC
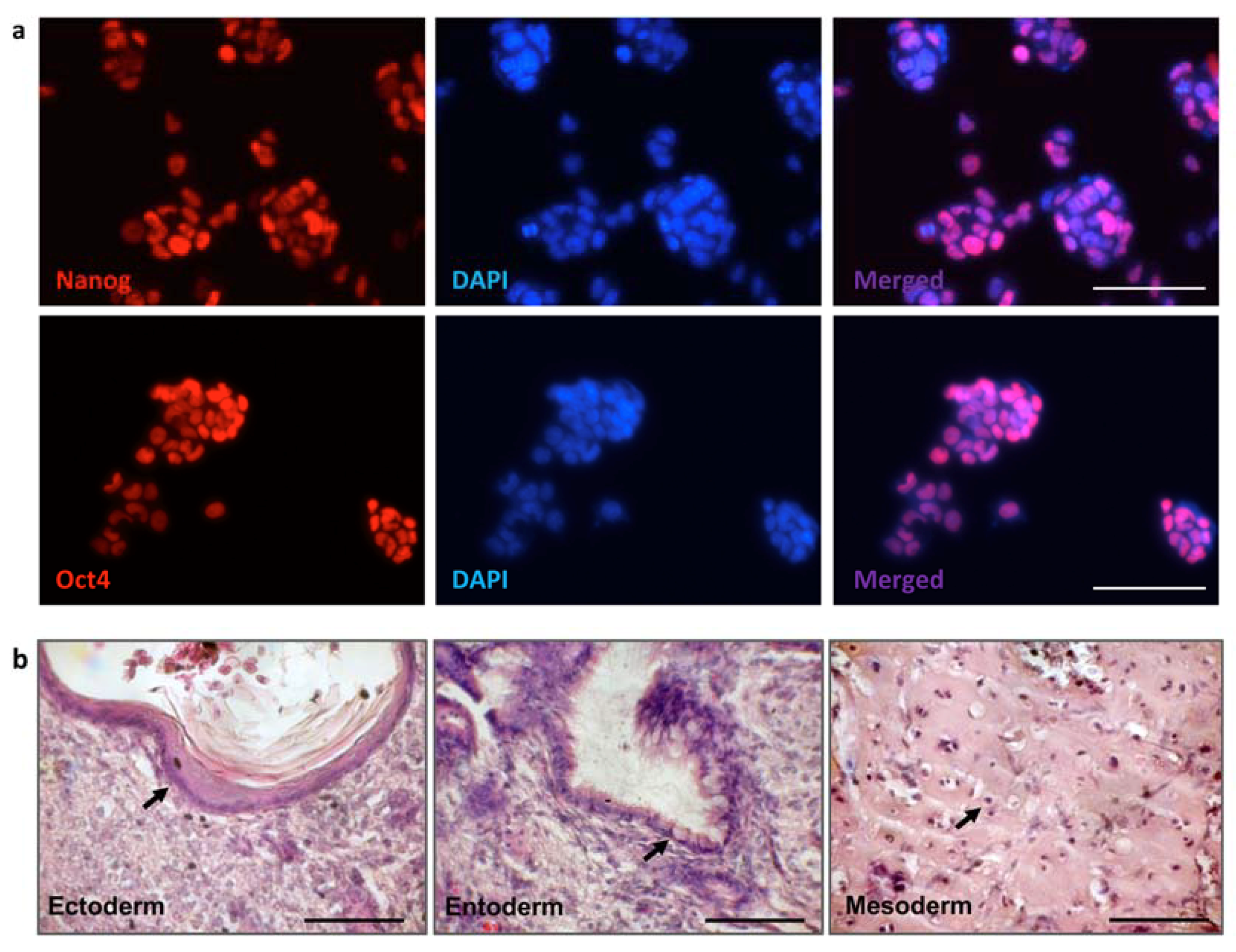
3.3. iPSC Generation from Fibroblasts of FVIII-Deficient Mice
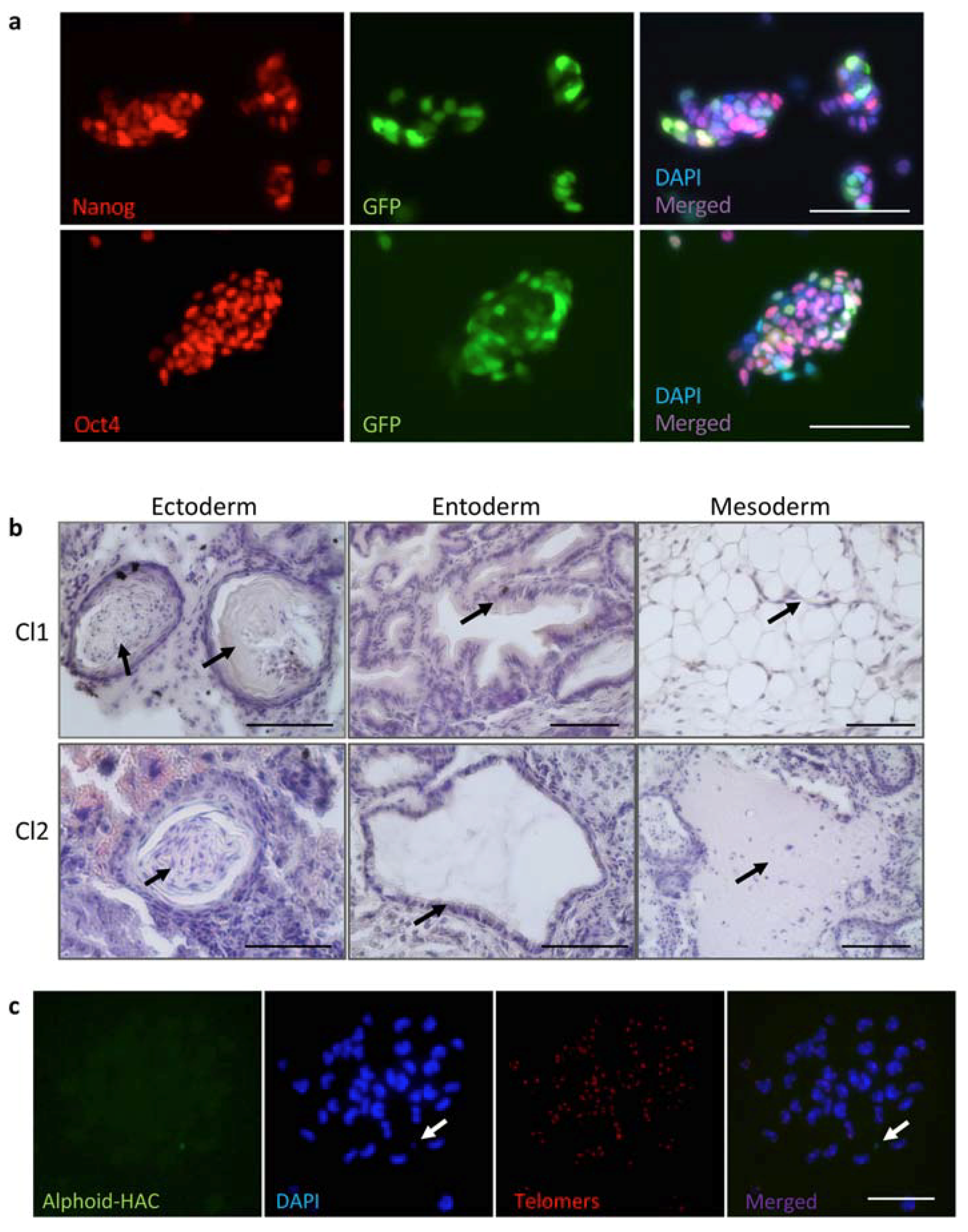
3.4. MMCT of the FVIII-Therapeutic HAC from CHO Cells in Mouse FVIIIY/– iPSCs
3.5. Characterization of iPSC Clones Bearing the AlphoidtetO-HAC-EF1α-FVIII
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kouprina, N.; Ebersole, T.; Koriabine, M.; Pak, E.; Rogozin, I.B.; Katoh, M.; Oshimura, M.; Ogi, K.; Peredelchuk, M.; Solomon, G.; et al. Cloning of human centromeres by transformation-associated recombination in yeast and generation of functional human artificial chromosomes. Nucleic Acids Res. 2003, 31, 922–934. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M.; Ayabe, F.; Norikane, S.; Okada, T.; Masumoto, H.; Horike, S.; Shirayoshi, Y.; Oshimura, M. Construction of a novel human artificial chromosome vector for gene delivery. Biochem. Biophys. Res. Commun. 2004, 321, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Oshimura, M.; Uno, N.; Kazuki, Y.; Katoh, M.; Inoue, T. A pathway from chromosome transfer to engineering resulting in human and mouse artificial chromosomes for a variety of applications to bio-medical challenges. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2015, 23, 111–133. [Google Scholar] [CrossRef] [PubMed]
- Hoshiya, H.; Kazuki, Y.; Abe, S.; Takiguchi, M.; Kajitani, N.; Watanabe, Y.; Yoshino, T.; Shirayoshi, Y.; Higaki, K.; Messina, G.; et al. A highly stable and nonintegrated human artificial chromosome (HAC) containing the 2.4 Mb entire human dystrophin gene. Mol. Ther. J. Am. Soc. Gene Ther. 2009, 17, 309–317. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Hoshiya, H.; D’Antona, G.; Gerli, M.F.; Messina, G.; Antonini, S.; Tonlorenzi, R.; Benedetti, S.; Berghella, L.; Torrente, Y.; et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011, 3, 96ra78. [Google Scholar] [CrossRef]
- Tedesco, F.S.; Gerli, M.F.; Perani, L.; Benedetti, S.; Ungaro, F.; Cassano, M.; Antonini, S.; Tagliafico, E.; Artusi, V.; Longa, E.; et al. Transplantation of genetically corrected human iPSC-derived progenitors in mice with limb-girdle muscular dystrophy. Sci. Transl. Med. 2012, 4, 140ra189. [Google Scholar] [CrossRef]
- Kazuki, Y.; Hiratsuka, M.; Takiguchi, M.; Osaki, M.; Kajitani, N.; Hoshiya, H.; Hiramatsu, K.; Yoshino, T.; Kazuki, K.; Ishihara, C.; et al. Complete genetic correction of ips cells from Duchenne muscular dystrophy. Mol. Ther. J. Am.Soc. Gene Ther. 2010, 18, 386–393. [Google Scholar] [CrossRef]
- Benedetti, S.; Uno, N.; Hoshiya, H.; Ragazzi, M.; Ferrari, G.; Kazuki, Y.; Moyle, L.A.; Tonlorenzi, R.; Lombardo, A.; Chaouch, S.; et al. Reversible immortalisation enables genetic correction of human muscle progenitors and engineering of next-generation human artificial chromosomes for Duchenne muscular dystrophy. EMBO Mol. Med. 2018, 10, 254–275. [Google Scholar] [CrossRef]
- Kurosaki, H.; Hiratsuka, M.; Imaoka, N.; Iida, Y.; Uno, N.; Kazuki, Y.; Ishihara, C.; Yakura, Y.; Mimuro, J.; Sakata, Y.; et al. Integration-free and stable expression of FVIII using a human artificial chromosome. J. Hum. Genet. 2011, 56, 727–733. [Google Scholar] [CrossRef]
- Yakura, Y.; Ishihara, C.; Kurosaki, H.; Kazuki, Y.; Komatsu, N.; Okada, Y.; Doi, T.; Takeya, H.; Oshimura, M. An induced pluripotent stem cell-mediated and integration-free factor VIII expression system. Biochem. Biophys. Res. Commun. 2013, 431, 336–341. [Google Scholar] [CrossRef]
- Kazuki, Y.; Kobayashi, K.; Aueviriyavit, S.; Oshima, T.; Kuroiwa, Y.; Tsukazaki, Y.; Senda, N.; Kawakami, H.; Ohtsuki, S.; Abe, S.; et al. Trans-chromosomic mice containing a human CYP3A cluster for prediction of xenobiotic metabolism in humans. Hum. Mol. Genet. 2013, 22, 578–592. [Google Scholar] [CrossRef] [PubMed]
- Co, D.O.; Borowski, A.H.; Leung, J.D.; van der Kaa, J.; Hengst, S.; Platenburg, G.J.; Pieper, F.R.; Perez, C.F.; Jirik, F.R.; Drayer, J.I. Generation of transgenic mice and germline transmission of a mammalian artificial chromosome introduced into embryos by pronuclear microinjection. Chromosome Res. Int. J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2000, 8, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Kuroiwa, Y.; Kasinathan, P.; Choi, Y.J.; Naeem, R.; Tomizuka, K.; Sullivan, E.J.; Knott, J.G.; Duteau, A.; Goldsby, R.A.; Osborne, B.A.; et al. Cloned transchromosomic calves producing human immunoglobulin. Nat. Biotechnol. 2002, 20, 889–894. [Google Scholar] [CrossRef] [PubMed]
- Ebersole, T.; Okamoto, Y.; Noskov, V.N.; Kouprina, N.; Kim, J.H.; Leem, S.H.; Barrett, J.C.; Masumoto, H.; Larionov, V. Rapid generation of long synthetic tandem repeats and its application for analysis in human artificial chromosome formation. Nucleic Acids Res. 2005, 33, e130. [Google Scholar] [CrossRef]
- Kim, J.H.; Kononenko, A.; Erliandri, I.; Kim, T.A.; Nakano, M.; Iida, Y.; Barrett, J.C.; Oshimura, M.; Masumoto, H.; Earnshaw, W.C.; et al. Human artificial chromosome (HAC) vector with a conditional centromere for correction of genetic deficiencies in human cells. Proc. Natl. Acad. Sci. USA 2011, 108, 20048–20053. [Google Scholar] [CrossRef]
- Nakano, M.; Cardinale, S.; Noskov, V.N.; Gassmann, R.; Vagnarelli, P.; Kandels-Lewis, S.; Larionov, V.; Earnshaw, W.C.; Masumoto, H. Inactivation of a human kinetochore by specific targeting of chromatin modifiers. Dev. Cell 2008, 14, 507–522. [Google Scholar] [CrossRef]
- Kouprina, N.; Samoshkin, A.; Erliandri, I.; Nakano, M.; Lee, H.S.; Fu, H.; Iida, Y.; Aladjem, M.; Oshimura, M.; Masumoto, H.; et al. Organization of synthetic alphoid DNA array in human artificial chromosome (HAC) with a conditional centromere. ACS Synth. Biol. 2012, 1, 590–601. [Google Scholar] [CrossRef]
- Kouprina, N.; Earnshaw, W.C.; Masumoto, H.; Larionov, V. A new generation of human artificial chromosomes for functional genomics and gene therapy. Cell. Mol. Life Sci. CMLS 2013, 70, 1135–1148. [Google Scholar] [CrossRef]
- Lee, N.C.; Kononenko, A.V.; Lee, H.S.; Tolkunova, E.N.; Liskovykh, M.A.; Masumoto, H.; Earnshaw, W.C.; Tomilin, A.N.; Larionov, V.; Kouprina, N. Protecting a transgene expression from the HAC-based vector by different chromatin insulators. Cell. Mol. Life Sci. CMLS 2013, 70, 3723–3737. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Skvortsova, E.V.; Liskovykh, M.A.; Ponomartsev, S.V.; Kuzmin, A.A.; Khudiakov, A.A.; Malashicheva, A.B.; Alenina, N.; Larionov, V.; Kouprina, N.; et al. Transfer of Synthetic Human Chromosome into Human Induced Pluripotent Stem Cells for Biomedical Applications. Cells 2018, 7, 261. [Google Scholar] [CrossRef]
- Liskovykh, M.; Lee, N.C.; Larionov, V.; Kouprina, N. Moving toward a higher efficiency of microcell-mediated chromosome transfer. Mol. Ther. Methods Clin. Dev. 2016, 3, 16043. [Google Scholar] [CrossRef] [PubMed]
- Iida, Y.; Kim, J.H.; Kazuki, Y.; Hoshiya, H.; Takiguchi, M.; Hayashi, M.; Erliandri, I.; Lee, H.S.; Samoshkin, A.; Masumoto, H.; et al. Human artificial chromosome with a conditional centromere for gene delivery and gene expression. DNA Res. Int. J. Rapid Publ. Rep. Genes Genomes 2010, 17, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Petrov, N.; Molina, O.; Liskovykh, M.; Pesenti, E.; Ohzeki, J.I.; Masumoto, H.; Earnshaw, W.C.; Larionov, V. Human Artificial Chromosome with Regulated Centromere: A Tool for Genome and Cancer Studies. ACS Synth. Biol. 2018, 7, 1974–1989. [Google Scholar] [CrossRef] [PubMed]
- Ohzeki, J.; Bergmann, J.H.; Kouprina, N.; Noskov, V.N.; Nakano, M.; Kimura, H.; Earnshaw, W.C.; Larionov, V.; Masumoto, H. Breaking the HAC Barrier: Histone H3K9 acetyl/methyl balance regulates CENP-A assembly. EMBO J. 2012, 31, 2391–2402. [Google Scholar] [CrossRef]
- Liskovykh, M.; Ponomartsev, S.; Popova, E.; Bader, M.; Kouprina, N.; Larionov, V.; Alenina, N.; Tomilin, A. Stable maintenance of de novo assembled human artificial chromosomes in embryonic stem cells and their differentiated progeny in mice. Cell Cycle 2015, 14, 1268–1273. [Google Scholar] [CrossRef]
- Kouprina, N.; Tomilin, A.N.; Masumoto, H.; Earnshaw, W.C.; Larionov, V. Human artificial chromosome-based gene delivery vectors for biomedicine and biotechnology. Expert Opin. Drug Deliv. 2014, 11, 517–535. [Google Scholar] [CrossRef]
- Sinenko, S.A.; Ponomartsev, S.V.; Tomilin, A.N. Human artificial chromosomes for pluripotent stem cell-based tissue replacement therapy. Exp. Cell Res. 2020, 389, 111882. [Google Scholar] [CrossRef]
- High, K.A. Update on progress and hurdles in novel genetic therapies for hemophilia. Hematol. Am. Soc. Hematol. Educ. Program. 2007, 1, 466–472. [Google Scholar] [CrossRef]
- Scott, D.W.; Lozier, J.N. Gene therapy for haemophilia: Prospects and challenges to prevent or reverse inhibitor formation. Br. J. Haematol. 2012, 156, 295–302. [Google Scholar] [CrossRef]
- Skvortsova, E.V.; Sinenko, S.A.; Tomilin, A. Immortalized murine fibroblast cell lines are refractory to reprogramming to pluripotent state. Oncotarget 2018, 9, 35241–35250. [Google Scholar] [CrossRef]
- Tang, Y.; Garson, K.; Li, L.; Vanderhyden, B.C. Optimization of lentiviral vector production using polyethylenimine-mediated transfection. Oncol. Lett. 2015, 9, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Malashicheva, A.; Kanzler, B.; Tolkunova, E.; Trono, D.; Tomilin, A. Lentivirus as a tool for lineage-specific gene manipulations. Genesis 2007, 45, 456–459. [Google Scholar] [CrossRef] [PubMed]
- Kuzmin, A.A.; Ermakova, V.V.; Sinenko, S.A.; Ponomartsev, S.V.; Starkova, T.Y.; Skvortsova, E.V.; Cherepanova, O.; Tomilin, A.N. Genetic tool for fate mapping of Oct4 (Pou5f1)-expressing cells and their progeny past the pluripotency stage. Stem Cell Res. Ther. 2019, 10, 391. [Google Scholar] [CrossRef] [PubMed]
- Carey, B.W.; Markoulaki, S.; Hanna, J.; Saha, K.; Gao, Q.; Mitalipova, M.; Jaenisch, R. Reprogramming of murine and human somatic cells using a single polycistronic vector. Proc. Natl. Acad. Sci. USA 2009, 106, 157–162. [Google Scholar] [CrossRef]
- Suzuki, T.; Kazuki, Y.; Oshimura, M.; Hara, T. Highly Efficient Transfer of Chromosomes to a Broad Range of Target Cells Using Chinese Hamster Ovary Cells Expressing Murine Leukemia Virus-Derived Envelope Proteins. PLoS ONE 2016, 11, e0157187. [Google Scholar] [CrossRef]
- Ikeno, M.; Grimes, B.; Okazaki, T.; Nakano, M.; Saitoh, K.; Hoshino, H.; McGill, N.I.; Cooke, H.; Masumoto, H. Construction of YAC-based mammalian artificial chromosomes. Nat. Biotechnol. 1998, 16, 431–439. [Google Scholar] [CrossRef]
- Pesenti, E.; Kouprina, N.; Liskovykh, M.; Aurich-Costa, J.; Larionov, V.; Masumoto, H.; Earnshaw, W.C.; Molina, O. Generation of a Synthetic Human Chromosome with Two Centromeric Domains for Advanced Epigenetic Engineering Studies. ACS Synth. Biol. 2018, 7, 1116–1130. [Google Scholar] [CrossRef]
- Bi, L.; Lawler, A.M.; Antonarakis, S.E.; High, K.A.; Gearhart, J.D.; Kazazian, H.H., Jr. Targeted disruption of the mouse factor VIII gene produces a model of haemophilia A. Nat. Genet. 1995, 10, 119–121. [Google Scholar] [CrossRef]
- Uno, N.; Uno, K.; Zatti, S.; Ueda, K.; Hiratsuka, M.; Katoh, M.; Oshimura, M. The transfer of human artificial chromosomes via cryopreserved microcells. Cytotechnology 2013, 65, 803–809. [Google Scholar] [CrossRef]
- Bakhmet, E.I.; Nazarov, I.B.; Gazizova, A.R.; Vorobyeva, N.E.; Kuzmin, A.A.; Gordeev, M.N.; Sinenko, S.A.; Aksenov, N.D.; Artamonova, T.O.; Khodorkovskii, M.A.; et al. hnRNP-K Targets Open Chromatin in Mouse Embryonic Stem Cells in Concert with Multiple Regulators. Stem Cells 2019, 37, 1018–1029. [Google Scholar] [CrossRef]
- Hasegawa, K.; Nakatsuji, N. Insulators prevent transcriptional interference between two promoters in a double gene construct for transgenesis. FEBS Lett. 2002, 520, 47–52. [Google Scholar] [CrossRef][Green Version]
- Suzuki, N.; Nishii, K.; Okazaki, T.; Ikeno, M. Human artificial chromosomes constructed using the bottom-up strategy are stably maintained in mitosis and efficiently transmissible to progeny mice. J. Biol. Chem. 2006, 281, 26615–26623. [Google Scholar] [CrossRef] [PubMed]
- Kazuki, Y.; Hoshiya, H.; Takiguchi, M.; Abe, S.; Iida, Y.; Osaki, M.; Katoh, M.; Hiratsuka, M.; Shirayoshi, Y.; Hiramatsu, K.; et al. Refined human artificial chromosome vectors for gene therapy and animal transgenesis. Gene Ther. 2011, 18, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Mejia, J.E.; Willmott, A.; Levy, E.; Earnshaw, W.C.; Larin, Z. Functional complementation of a genetic deficiency with human artificial chromosomes. Am. J. Hum. Genet. 2001, 69, 315–326. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Laner, A.; Goussard, S.; Ramalho, A.S.; Schwarz, T.; Amaral, M.D.; Courvalin, P.; Schindelhauer, D.; Grillot-Courvalin, C. Bacterial transfer of large functional genomic DNA into human cells. Gene Ther. 2005, 12, 1559–1572. [Google Scholar] [CrossRef][Green Version]
- Rocchi, L.; Braz, C.; Cattani, S.; Ramalho, A.; Christan, S.; Edlinger, M.; Ascenzioni, F.; Laner, A.; Kraner, S.; Amaral, M.; et al. Escherichia coli-cloned CFTR loci relevant for human artificial chromosome therapy. Hum. Gene Ther. 2010, 21, 1077–1092. [Google Scholar] [CrossRef]
- Breman, A.M.; Steiner, C.M.; Slee, R.B.; Grimes, B.R. Input DNA ratio determines copy number of the 33 kb Factor IX gene on de novo human artificial chromosomes. Mol. Ther. J. Am. Soc. Gene Ther. 2008, 16, 315–323. [Google Scholar] [CrossRef]
- Ikeno, M.; Inagaki, H.; Nagata, K.; Morita, M.; Ichinose, H.; Okazaki, T. Generation of human artificial chromosomes expressing naturally controlled guanosine triphosphate cyclohydrolase I gene. Genes Cells 2002, 7, 1021–1032. [Google Scholar] [CrossRef]
- Voet, T.; Schoenmakers, E.; Carpentier, S.; Labaere, C.; Marynen, P. Controlled transgene dosage and PAC-mediated transgenesis in mice using a chromosomal vector. Genomics 2003, 82, 596–605. [Google Scholar] [CrossRef]
- Ito, M.; Ikeno, M.; Nagata, H.; Yamamoto, T.; Hiroguchi, A.; Fox, I.J.; Miyakawa, S. Treatment of nonalbumin rats by transplantation of immortalized hepatocytes using artificial human chromosome. Transpl. Proc. 2009, 41, 422–424. [Google Scholar] [CrossRef]
- Ito, M.; Ito, R.; Yoshihara, D.; Ikeno, M.; Kamiya, M.; Suzuki, N.; Horiguchi, A.; Nagata, H.; Yamamoto, T.; Kobayashi, N.; et al. Immortalized hepatocytes using human artificial chromosome. Cell Transpl. 2008, 17, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Larionov, V. Transformation-associated recombination (TAR) cloning for genomics studies and synthetic biology. Chromosoma 2016, 125, 621–632. [Google Scholar] [CrossRef] [PubMed]
- Liskovykh, M.; Goncharov, N.V.; Petrov, N.; Aksenova, V.; Pegoraro, G.; Ozbun, L.L.; Reinhold, W.C.; Varma, S.; Dasso, M.; Kumeiko, V.; et al. A novel assay to screen siRNA libraries identifies protein kinases required for chromosome transmission. Genome Res. 2019, 29, 1719–1732. [Google Scholar] [CrossRef] [PubMed]
- Kouprina, N.; Liskovykh, M.; Petrov, N.; Larionov, V. Human artificial chromosome (HAC) for measuring chromosome instability (CIN) and identification of genes required for proper chromosome transmission. Exp. Cell Res. 2019, 387, 111805. [Google Scholar] [CrossRef]
- Suzuki, T.; Kazuki, Y.; Hara, T.; Oshimura, M. Current advances in microcell-mediated chromosome transfer technology and its applications. Exp. Cell Res. 2020, 111915. [Google Scholar] [CrossRef]
- Brown, D.M.; Glass, J.I. Technology used to build and transfer mammalian chromosomes. Exp. Cell Res. 2020, 388, 111851. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Blasticidin Treatment | mESC-HAC-GFP | iPSC-HAC-F8(Cl1) | iPSC-HAC-F8(Cl2) |
|---|---|---|---|
| 20 days with Bsd | 97.24% | 92.66% | 92.52% |
| 15 days without Bsd | 91.50% | 48.61% | 45.78% |
| HAC daily loss | 0.004 | 0.042 | 0.045 |
| Blasticidin Treatment | mES-HAC-GFP | iPSC-HAC-F8(Cl1) | iPS-HAC-F8(Cl2) | iPS-HAC-F8(CMV) |
|---|---|---|---|---|
| 20 days with Bsd | 96.67% | 96.67% | 96.67% | 80.00% |
| At 15 days without Bsd | 85.71% | 36.73% | 59.37% | 73.33% |
| HAC daily loss | 0.008 | 0.062 | 0.032 | 0.006 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ponomartsev, S.V.; Sinenko, S.A.; Skvortsova, E.V.; Liskovykh, M.A.; Voropaev, I.N.; Savina, M.M.; Kuzmin, A.A.; Kuzmina, E.Y.; Kondrashkina, A.M.; Larionov, V.; et al. Human AlphoidtetO Artificial Chromosome as a Gene Therapy Vector for the Developing Hemophilia A Model in Mice. Cells 2020, 9, 879. https://doi.org/10.3390/cells9040879
Ponomartsev SV, Sinenko SA, Skvortsova EV, Liskovykh MA, Voropaev IN, Savina MM, Kuzmin AA, Kuzmina EY, Kondrashkina AM, Larionov V, et al. Human AlphoidtetO Artificial Chromosome as a Gene Therapy Vector for the Developing Hemophilia A Model in Mice. Cells. 2020; 9(4):879. https://doi.org/10.3390/cells9040879
Chicago/Turabian StylePonomartsev, Sergey V., Sergey A. Sinenko, Elena V. Skvortsova, Mikhail A. Liskovykh, Ivan N. Voropaev, Maria M. Savina, Andrey A. Kuzmin, Elena Yu. Kuzmina, Alexandra M. Kondrashkina, Vladimir Larionov, and et al. 2020. "Human AlphoidtetO Artificial Chromosome as a Gene Therapy Vector for the Developing Hemophilia A Model in Mice" Cells 9, no. 4: 879. https://doi.org/10.3390/cells9040879
APA StylePonomartsev, S. V., Sinenko, S. A., Skvortsova, E. V., Liskovykh, M. A., Voropaev, I. N., Savina, M. M., Kuzmin, A. A., Kuzmina, E. Y., Kondrashkina, A. M., Larionov, V., Kouprina, N., & Tomilin, A. N. (2020). Human AlphoidtetO Artificial Chromosome as a Gene Therapy Vector for the Developing Hemophilia A Model in Mice. Cells, 9(4), 879. https://doi.org/10.3390/cells9040879