Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
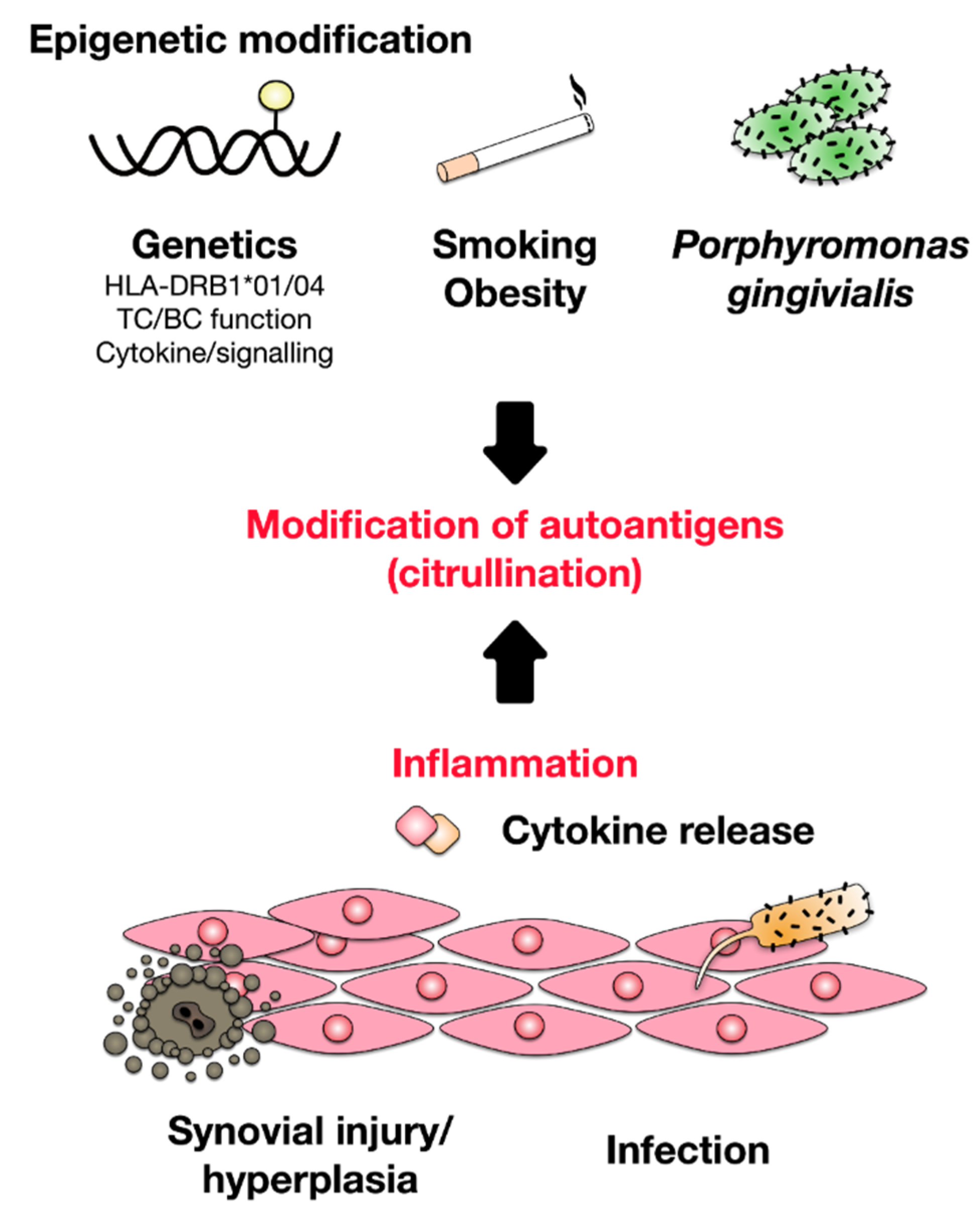
2. Development of Rheumatoid Arthritis
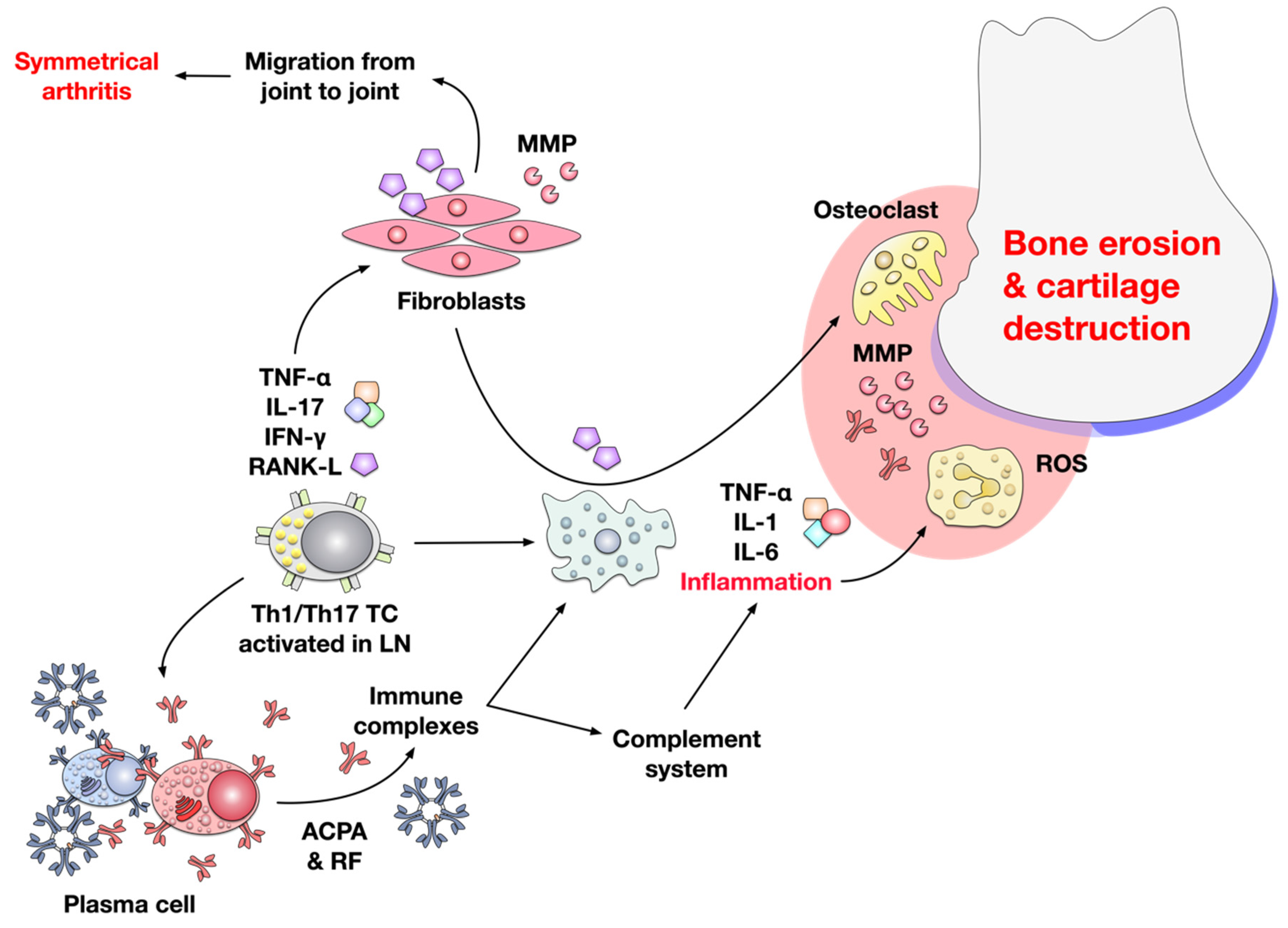
3. Pathomechanism of RA
3.1. Contribution of Dendritic Cells to Establishment and Maintenance of Inflammation in RA
3.2. Joint Inflammation in RA is Mediated by T cells, B cells, Macrophages, and Fibroblasts
3.3. Contribution of Cytokines to Inflammation in RA
3.4. Contribution of B Cells and Autoantibodies to the Pathogenesis of RA
3.5. RA also Results in Neovascularization
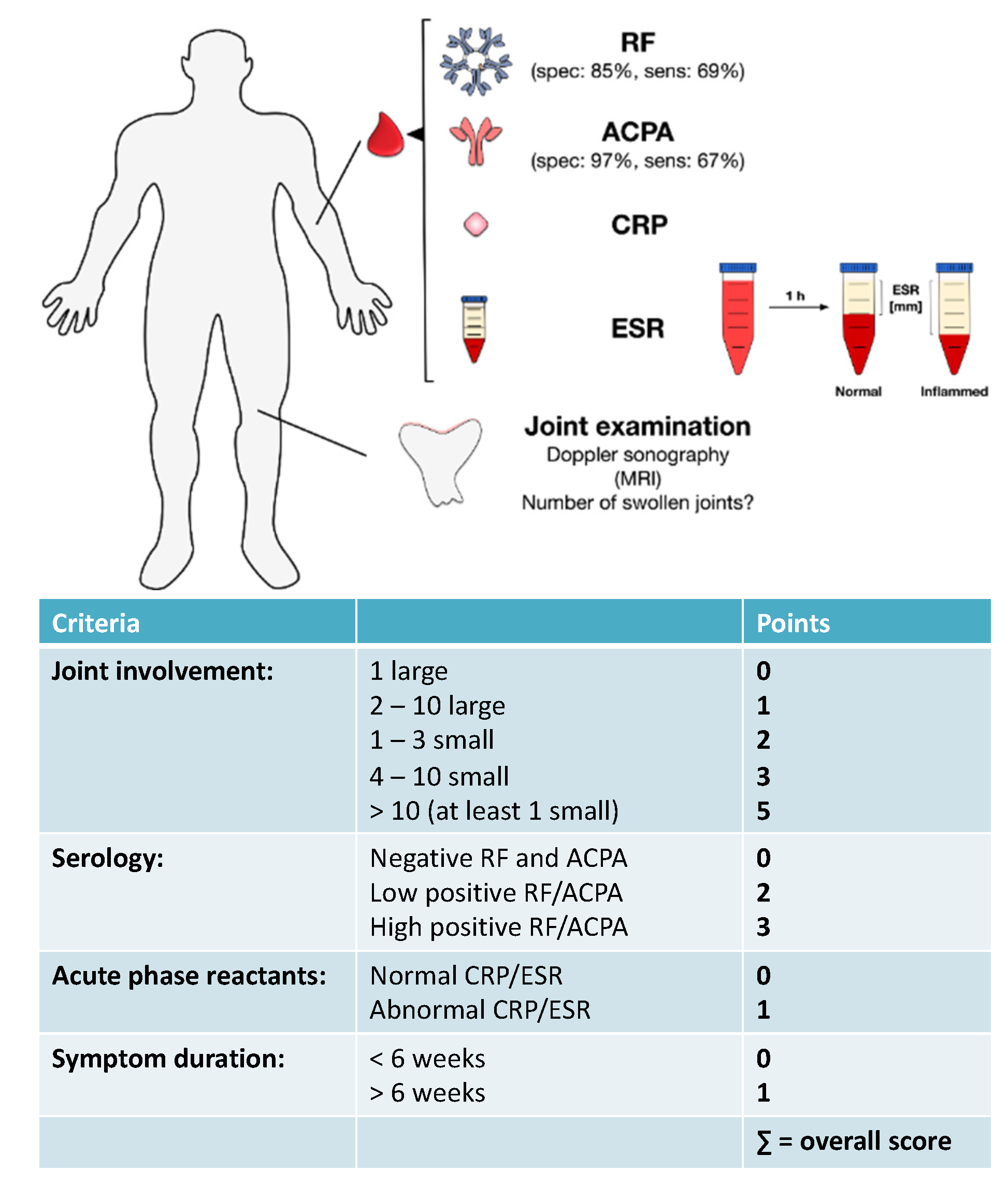
4. Diagnosis of RA
EULAR Criteria for the Diagnosis of RA
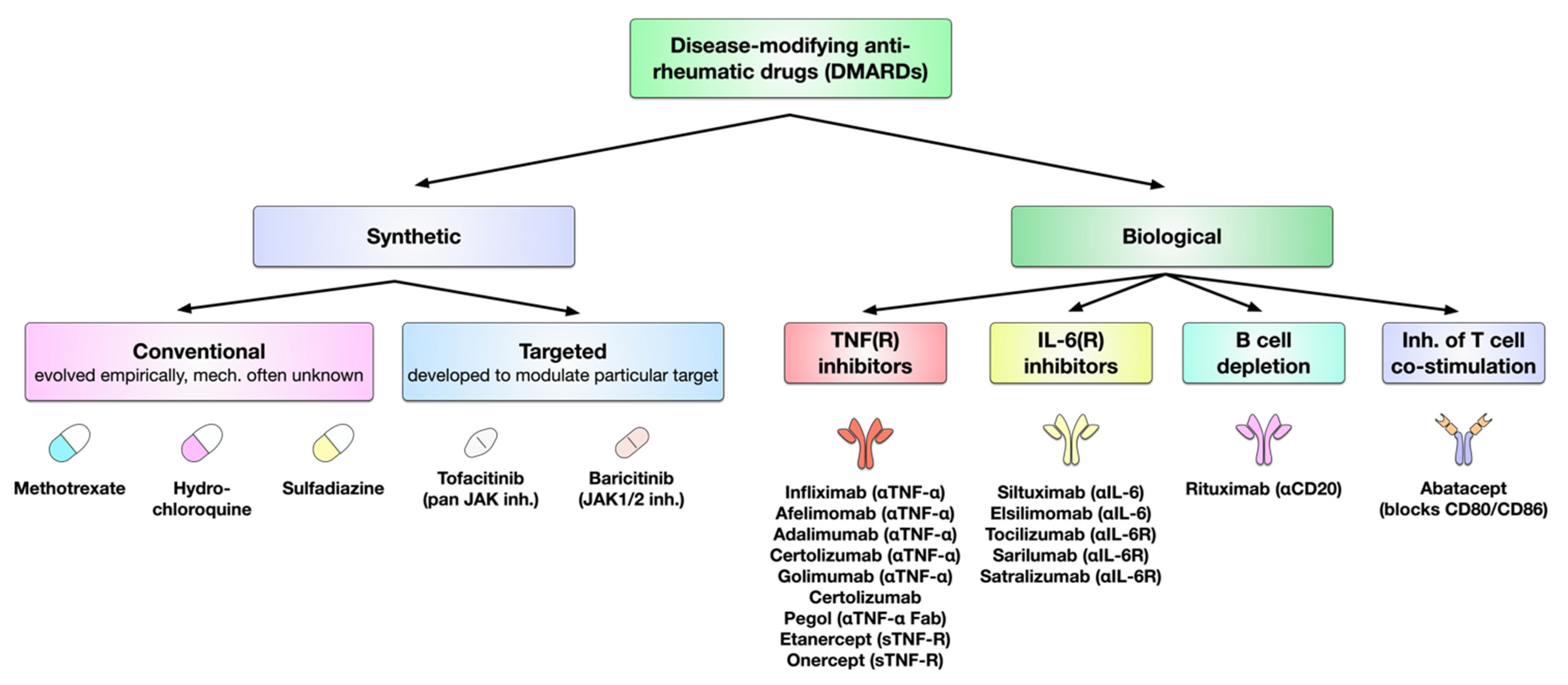
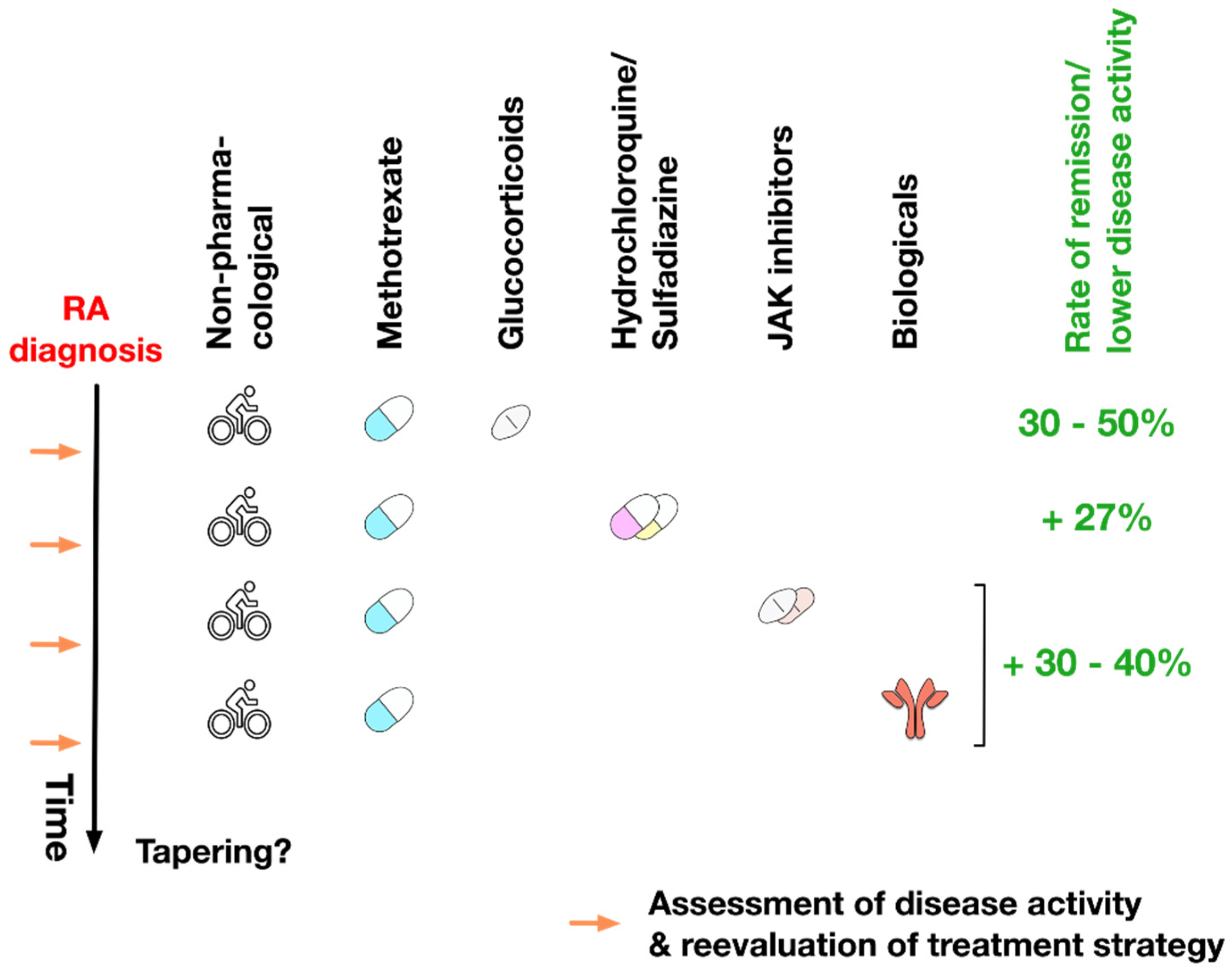
5. Treatment of RA
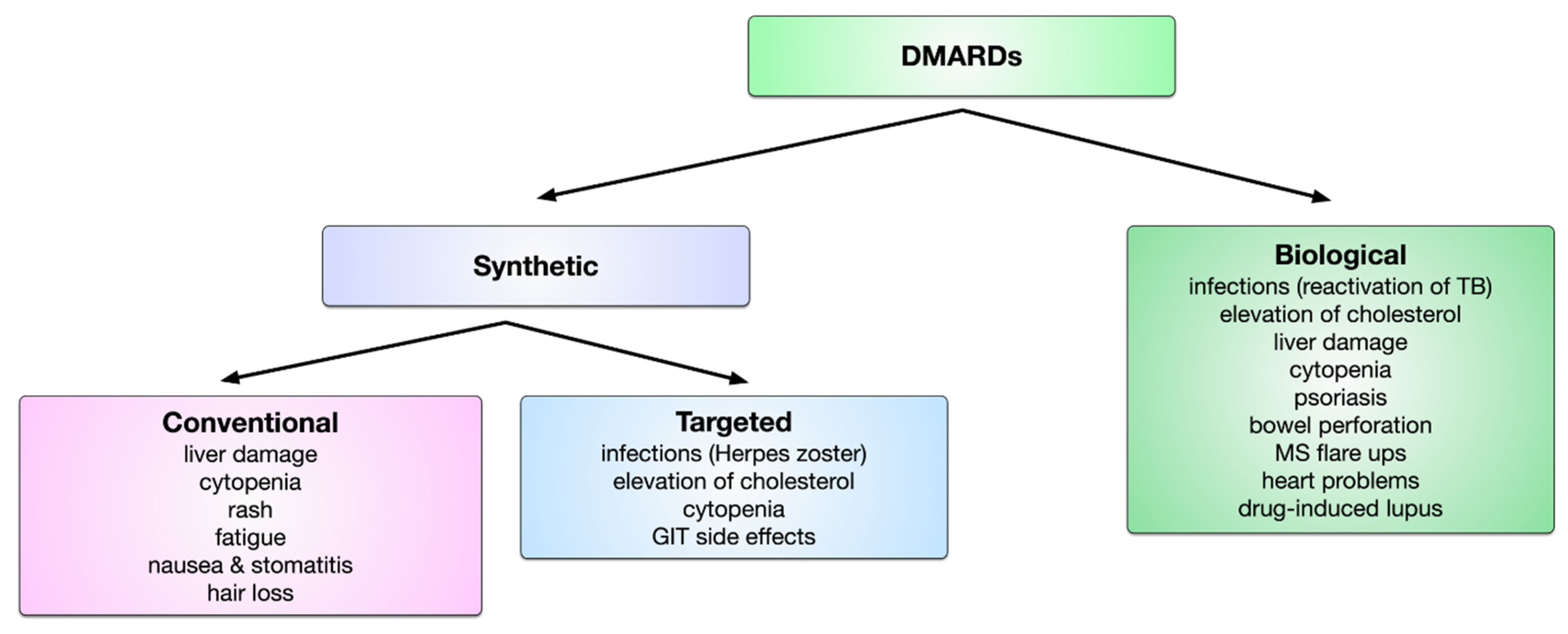
6. DMARDs in the Treatment of RA
6.1. Conventional Synthetic DMARDs
6.1.1. Methotrexate
6.1.2. Sulfasalazin
6.1.3. Hydrochloroquine
6.1.4. Triple Therapy with Synthetic DMARDs
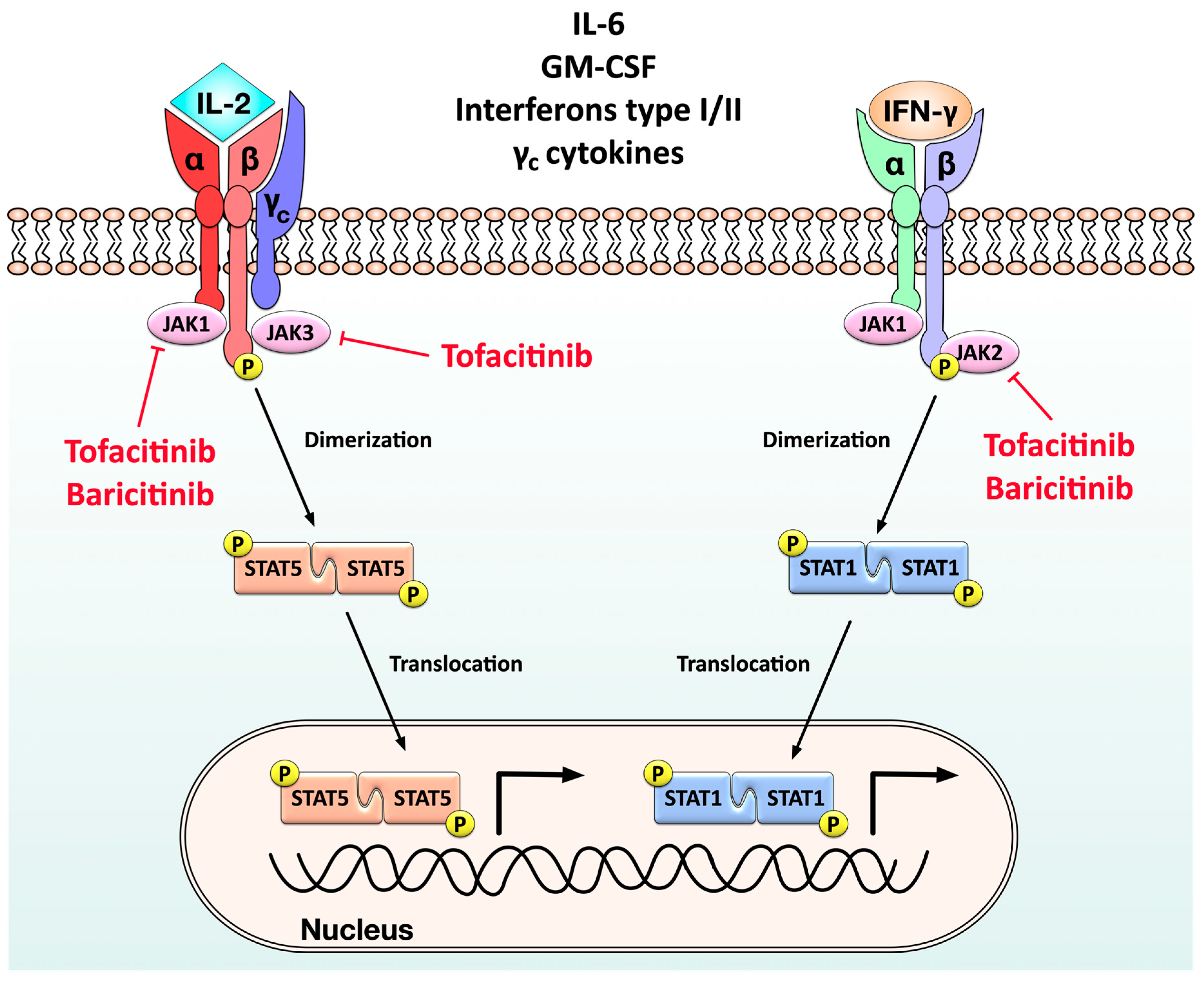
6.2. Targeted Synthetic DMARDs
6.3. Biologic DMARDs
6.3.1. TNF-α Inhibitors
6.3.2. IL-6 Inhibitors, IL-6R Inhibitors
6.3.3. Inhibitors of Co-Stimulation
6.3.4. B Cell Depleting Antibodies
6.4. Limitations of DMARD Therapy
7. Novel Experimental Strategies in the Treatment of RA
7.1. Mesenchymal Stem Cells
7.2. Inhibition of NOD-, LRR-, and Pyrin Domain-Containing Protein 3 (NLRP3)
7.3. Targeting of GM-CSF and GM-CSF Receptor
7.4. Toll-Like Receptor 4 (TLR4) Targeting
8. Treatment Plan of RA
9. Summary and Conclusion
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Smolen, J.S.; Aletaha, D.; McInnes, I.B. Rheumatoid arthritis. Lancet Lond. Engl. 2016, 388, 2023–2038. [Google Scholar] [CrossRef]
- Littlejohn, E.A.; Monrad, S. Early Diagnosis and Treatment of Rheumatoid Arthritis. Prim. Care: Clin. Off. Pr. 2018, 45, 237–255. [Google Scholar] [CrossRef] [PubMed]
- Sacks, J.J.; Luo, Y.-H.; Helmick, C.G. Prevalence of specific types of arthritis and other rheumatic conditions in the ambulatory health care system in the United States, 2001–2005. Arthritis Rheum. 2010, 62, 460–464. [Google Scholar] [CrossRef] [PubMed]
- Sangha, O. Epidemiology of rheumatic diseases. Rheumatology 2000, 39, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Myasoedova, E.; Crowson, C.S.; Kremers, H.M.; Therneau, T.M.; Gabriel, S.E. Is the incidence of rheumatoid arthritis rising? Results from Olmsted County, Minnesota, 1955–2007. Arthritis Rheum. 2010, 62, 1576–1582. [Google Scholar] [CrossRef] [PubMed]
- Brzustewicz, E.; Henc, I.; Daca, A.; Szarecka, M.; Sochocka-Bykowska, M.; Witkowski, J.; Bryl, E. Autoantibodies, C-reactive protein, erythrocyte sedimentation rate and serum cytokine profiling in monitoring of early treatment. Cent. Eur. J. Immunol. 2017, 42, 259–268. [Google Scholar] [CrossRef]
- Aletaha, D.; Ramiro, S. Diagnosis and Management of Rheumatoid Arthritis. JAMA 2018, 320, 1360–1372. [Google Scholar] [CrossRef]
- Deane, K.D.; Demoruelle, M.K.; Kelmenson, L.B.; Kuhn, K.A.; Norris, J.M.; Holers, V.M. Genetic and environmental risk factors for rheumatoid arthritis. Best Pr. Res. Clin. Rheumatol. 2017, 31, 3–18. [Google Scholar] [CrossRef]
- McGraw, W.T.; Potempa, J.; Farley, D.; Travis, J. Purification, Characterization, and Sequence Analysis of a Potential Virulence Factor from Porphyromonas gingivalis, Peptidylarginine Deiminase. Infect. Immun. 1999, 67, 3248–3256. [Google Scholar] [CrossRef]
- Tan, E.M.; Smolen, J.S. Historical observations contributing insights on etiopathogenesis of rheumatoid arthritis and role of rheumatoid factor. J. Exp. Med. 2016, 213, 1937–1950. [Google Scholar] [CrossRef]
- Easlick, K.A. An evaluation of the effect of dental foci of infection on health. J. Am. Dent. Assoc. 1951, 42, 615–697. [Google Scholar] [PubMed]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.-A.; Lundberg, K.; Kinloch, A.J.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef]
- Wilson, C.; Tiwana, H.; Ebringer, A. Molecular mimicry between HLA-DR alleles associated with rheumatoid arthritis and Proteus mirabilis as the aetiological basis for autoimmunity. Microbes Infect. 2000, 2, 1489–1496. [Google Scholar] [CrossRef]
- Tiwana, H.; Wilson, C.; Alvarez, A.; Abuknesha, R.; Bansal, S.; Ebringer, A. Cross-Reactivity between the Rheumatoid Arthritis-Associated Motif EQKRAA and Structurally Related Sequences Found inProteus mirabilis. Infect. Immun. 1999, 67, 2769–2775. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Yu, Y.; Yue, Y.; Zhang, Z.; Su, K. Microbial Infection and Rheumatoid Arthritis. J. Clin. Cell. Immunol. 2013, 4, 174. [Google Scholar] [CrossRef] [PubMed]
- Ospelt, C.; Gay, S.; Klein, K. Epigenetics in the pathogenesis of RA. Semin. Immunopathol. 2017, 39, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.; Bykerk, V.; Cohen, M.D.; et al. Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann. Rheum. Dis. 2010, 69, 1580–1588. [Google Scholar] [CrossRef]
- Orozco, G.; McAllister, K.; Eyre, S. Genetics of rheumatoid arthritis: GWAS and beyond. Open Access Rheumatol. Res. Rev. 2011, 3, 31–46. [Google Scholar] [CrossRef]
- Lenz, T.L.; Deutsch, A.; Han, B.; Hu, X.; Okada, Y.; Eyre, S.; Knapp, M.; Zhernakova, A.; Huizinga, T.W.; Abecasis, G.; et al. Widespread non-additive and interaction effects within HLA loci modulate the risk of autoimmune diseases. Nat. Genet. 2015, 47, 1085–1090. [Google Scholar] [CrossRef]
- Zhang, Q.; Vignali, D.A. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunology 2016, 44, 1034–1051. [Google Scholar] [CrossRef]
- Yap, H.-Y.; Tee, S.Z.-Y.; Wong, M.M.-T.; Chow, S.-K.; Peh, S.-C.; Teow, S.-Y. Pathogenic Role of Immune Cells in Rheumatoid Arthritis: Implications in Clinical Treatment and Biomarker Development. Cells 2018, 7, 161. [Google Scholar] [CrossRef] [PubMed]
- Majorczyk, E.; Jasek, M.; Ploski, R.; Wagner, M.; Kosior, A.; Pawlik, A.; Obojski, A.; Łuszczek, W.; Nowak, I.; Wiśniewski, A.; et al. Association of PTPN22 single nucleotide polymorphism with rheumatoid arthritis but not with allergic asthma. Eur. J. Hum. Genet. 2007, 15, 1043–1048. [Google Scholar] [CrossRef] [PubMed]
- Arleevskaya, M.I.; Kravtsova, O.A.; Lemerle, J.; Renaudineau, Y.; Tsibulkin, A.P. How Rheumatoid Arthritis Can Result from Provocation of the Immune System by Microorganisms and Viruses. Front. Microbiol. 2016, 7, 81. [Google Scholar] [CrossRef] [PubMed]
- Baka, Z.; Buzás, E.; Nagy, G. Rheumatoid arthritis and smoking: Putting the pieces together. Arthritis Res. Ther. 2009, 11, 238. [Google Scholar] [CrossRef] [PubMed]
- Van Drongelen, V.; Holoshitz, J. Human Leukocyte Antigen–Disease Associations in Rheumatoid Arthritis. Rheum. Dis. Clin. North. Am. 2017, 43, 363–376. [Google Scholar] [CrossRef] [PubMed]
- Viatte, S.; Plant, D.; Han, B.; Fu, B.; Yarwood, A.; Thomson, W.; Symmons, D.; Worthington, J.; Young, A.; Hyrich, K.L.; et al. Association of HLA-DRB1 haplotypes with rheumatoid arthritis severity, mortality, and treatment response. JAMA 2015, 313, 1645–1656. [Google Scholar] [CrossRef]
- Chen, J.; Li, J.; Gao, H.; Wang, C.; Luo, J.; Lv, Z.; Li, X. Comprehensive Evaluation of Different T-Helper Cell Subsets Differentiation and Function in Rheumatoid Arthritis. J. Biomed. Biotechnol. 2012, 2012, 1–6. [Google Scholar] [CrossRef]
- Coutant, F.; Miossec, P. Altered dendritic cell functions in autoimmune diseases: Distinct and overlapping profiles. Nat. Rev. Rheumatol. 2016, 12, 703–715. [Google Scholar] [CrossRef]
- Jongbloed, S.L.; Lebre, M.C.; Fraser, A.R.; Gracie, J.A.; Sturrock, R.D.; Tak, P.-P.; McInnes, I. Enumeration and phenotypical analysis of distinct dendritic cell subsets in psoriatic arthritis and rheumatoid arthritis. Arthritis Res. Ther. 2005, 8, R15. [Google Scholar] [CrossRef]
- Page, G.; Miossec, P. Paired synovium and lymph nodes from rheumatoid arthritis patients differ in dendritic cell and chemokine expression. J. Pathol. 2004, 204, 28–38. [Google Scholar] [CrossRef]
- Segura, E.; Touzot, M.; Bohineust, A.; Cappuccio, A.; Chiocchia, G.; Hosmalin, A.; Dalod, M.; Soumelis, V.; Amigorena, S. Human Inflammatory Dendritic Cells Induce Th17 Cell Differentiation. Immunity 2013, 38, 336–348. [Google Scholar] [CrossRef] [PubMed]
- Lebre, M.C.; Jongbloed, S.L.; Tas, S.W.; Smeets, T.J.; McInnes, I.; Tak, P.P. Rheumatoid Arthritis Synovium Contains Two Subsets of CD83−DC-LAMP− Dendritic Cells with Distinct Cytokine Profiles. Am. J. Pathol. 2008, 172, 940–950. [Google Scholar] [CrossRef] [PubMed]
- Tournadre, A.; Lenief, V.; Miossec, P. Immature muscle precursors are a source of interferon-β in myositis: Role of Toll-like receptor 3 activation and contribution to HLA class I up-regulation. Arthritis Rheum. 2012, 64, 533–541. [Google Scholar] [CrossRef] [PubMed]
- Castañeda-Delgado, J.E.; Bastian, Y.; Macias-Segura, N.; Santiago-Algarra, D.; Castillo-Ortiz, J.D.; Alemán-Navarro, A.L.; Martínez-Tejada, P.; Enciso-Moreno, L.; Lira, Y.G.-D.; Olguín-Calderón, D.; et al. Type I Interferon Gene Response Is Increased in Early and Established Rheumatoid Arthritis and Correlates with Autoantibody Production. Front. Immunol. 2017, 8, 279. [Google Scholar] [CrossRef]
- Cooles, F.A.; Anderson, A.; Lendrem, D.; Norris, J.; Pratt, A.; Hilkens, C.M.U.; Isaacs, J.D. The interferon gene signature is increased in patients with early treatment-naive rheumatoid arthritis and predicts a poorer response to initial therapy. J. Allergy Clin. Immunol. 2018, 141, 445–448. [Google Scholar] [CrossRef] [PubMed]
- Page, G.; Lebecque, S.; Miossec, P. Anatomic Localization of Immature and Mature Dendritic Cells in an Ectopic Lymphoid Organ: Correlation with Selective Chemokine Expression in Rheumatoid Synovium. J. Immunol. 2002, 168, 5333–5341. [Google Scholar] [CrossRef]
- Chemin, K.; Gerstner, C.; Malmström, V. Effector Functions of CD4+ T Cells at the Site of Local Autoimmune Inflammation-Lessons from Rheumatoid Arthritis. Front. Immunol. 2019, 10, 353. [Google Scholar] [CrossRef]
- Hume, D.A. The Many Alternative Faces of Macrophage Activation. Front. Immunol. 2015, 6, 370. [Google Scholar] [CrossRef]
- Romagnani, S. T-cell subsets (Th1 versus Th2). Ann. Allergy Asthma Immunol. 2000, 85, 9–18. [Google Scholar] [CrossRef]
- Schmidt, D.; Goronzy, J.J.; Weyand, C.M. CD4+ CD7- CD28- T cells are expanded in rheumatoid arthritis and are characterized by autoreactivity. J. Clin. Investig. 1996, 97, 2027–2037. [Google Scholar] [CrossRef]
- Fasth, A.E.R.; Cao, D.; Van Vollenhoven, R.; Trollmo, C.; Malmström, V. CD28nullCD4+ T Cells - Characterization of an Effector Memory T-Cell Population in Patients with Rheumatoid Arthritis. Scand. J. Immunol. 2004, 60, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Namekawa, T.; Wagner, U.G.; Goronzy, J.J.; Weyand, C.M. Functional subsets of CD4 T cells in rheumatoid synovitis. Arthritis Rheum. 2020, 41, 2108–2116. [Google Scholar] [CrossRef]
- Griffiths, G.M.; Alpert, S.; Lambert, E.; McGuire, J.; Weissman, I.L. Perforin and granzyme A expression identifying cytolytic lymphocytes in rheumatoid arthritis. Proc. Natl. Acad. Sci. USA 1992, 89, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Chemin, K.; Ramsköld, D.; Diaz-Gallo, L.M.; Herrath, J.; Houtman, M.; Tandre, K.; Rönnblom, L.; Catrina, A.; Malmström, V. EOMES-positive CD4+ T cells are increased in PTPN22 (1858T) risk allele carriers. Eur. J. Immunol. 2018, 48, 655–669. [Google Scholar] [CrossRef]
- Wang, J.; Shan, Y.; Jiang, Z.; Feng, J.; Li, C.; Ma, L.; Jiang, Y. High frequencies of activated B cells and T follicular helper cells are correlated with disease activity in patients with new-onset rheumatoid arthritis. Clin. Exp. Immunol. 2013, 174, 212–220. [Google Scholar] [CrossRef]
- Ma, J.; Zhu, C.; Ma, B.; Tian, J.; Baidoo, S.E.; Mao, C.; Wu, W.; Chen, J.-G.; Tong, J.; Yang, M.; et al. Increased Frequency of Circulating Follicular Helper T Cells in Patients with Rheumatoid Arthritis. Clin. Dev. Immunol. 2012, 2012, 1–7. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, Y.; Lv, T.-T.; Yin, Z.-J.; Wang, X.-B. Elevated circulating Th17 and follicular helper CD4 + T cells in patients with rheumatoid arthritis. APMIS 2015, 123, 659–666. [Google Scholar] [CrossRef]
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23–IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef]
- Cascão, R.; Moura, R.A.; Perpetuo, I.; Vieriea-Sousa, E.; Mourao, A.F.; Rodrugues, A.M.; Polido-Pereira, J.; Queiroz, M.V.; Rosario, H.S.; Souto-Carneiro, M.M.M.; et al. Identification of a cytokine network sustaining neutrophil and Th17 activation in untreated early rheumatoid arthritis. Arthritis Res. Ther. 2020. [Google Scholar] [CrossRef]
- Azizi, G.; Jadidi-Niaragh, F.; Mirshafiey, A. Th17 Cells in Immunopathogenesis and treatment of rheumatoid arthritis. Int. J. Rheum. Dis. 2013, 16, 243–253. [Google Scholar] [CrossRef]
- Kaplan, M.J. Role of neutrophils in systemic autoimmune diseases. Arthritis Res. 2013, 15, 219. [Google Scholar] [CrossRef] [PubMed]
- Koenders, M.; Berg, W.B.V.D. Secukinumab for rheumatology: Development and its potential place in therapy. Drug Des. Dev. Ther. 2016, 10, 2069–2080. [Google Scholar] [CrossRef] [PubMed]
- Baker, K.F.; Isaacs, J.D. Novel therapies for immune-mediated inflammatory diseases: What can we learn from their use in rheumatoid arthritis, spondyloarthritis, systemic lupus erythematosus, psoriasis, Crohn’s disease and ulcerative colitis? Ann. Rheum. Dis. 2018, 77, 175–187. [Google Scholar] [CrossRef] [PubMed]
- Evans, H.G.; Roostalu, U.; Walter, G.J.; Gullick, N.; Frederiksen, K.S.; Roberts, C.; Sumner, J.; Baeten, D.L.; Gerwien, J.G.; Cope, A.P.; et al. TNF-α blockade induces IL-10 expression in human CD4+ T cells. Nat. Commun. 2014, 5, 3199. [Google Scholar] [CrossRef] [PubMed]
- Möttönen, M.; Heikkinen-Eloranta, J.; Mustonen, L.; Isomäki, P.; Luukkainen, R.; Lassila, O. CD4+ CD25+ T cells with the phenotypic and functional characteristics of regulatory T cells are enriched in the synovial fluid of patients with rheumatoid arthritis. Clin. Exp. Immunol. 2005, 140, 360–367. [Google Scholar] [CrossRef]
- Komatsu, N.; Okamoto, K.; Sawa, S.; Nakashima, T.; Oh-Hora, M.; Kodama, T.; Tanaka, S.A.; Bluestone, J.; Takayanagi, H. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat. Med. 2013, 20, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Sun, X.; Zhao, J.; Zhang, J.; Zhu, H.; Li, C.; Gao, N.; Jia, Y.; Xu, D.; Huang, F.-P.; et al. Regulatory T cells in rheumatoid arthritis showed increased plasticity toward Th17 but retained suppressive function in peripheral blood. Ann. Rheum. Dis. 2014, 74, 1293–1301. [Google Scholar] [CrossRef]
- Nie, H.; Zheng, Y.; Li, R.; Guo, T.B.; He, N.; Fang, L.; Liu, X.; Xiao, L.; Chen, X.; Wan, B.; et al. Phosphorylation of FOXP3 controls regulatory T cell function and is inhibited by TNF-α in rheumatoid arthritis. Nat. Med. 2013, 19, 322–328. [Google Scholar] [CrossRef]
- Nadkarni, S.; Mauri, C.; Ehernstein, M.R. Anti-TNF-alpha therapy induces a distinct regulatory T cell population in patients with rheumatoid arthritis via TGF-beta. J. Exp. Med. 2007, 204, 33–39. [Google Scholar] [CrossRef]
- Brennan, F.M.; McInnes, I. Evidence that cytokines play a role in rheumatoid arthritis. J. Clin. Investig. 2008, 118, 3537–3545. [Google Scholar] [CrossRef]
- Ma, H.; Xu, M.; Song, Y.; Zhang, T.; Yin, H.; Yin, S. Interferon-γ facilitated adjuvant-induced arthritis at early stage. Scand. J. Immunol. 2019, 89, e12757. [Google Scholar] [CrossRef] [PubMed]
- Dayer, J.M.; Beutler, B.; Cerami, A. Cachectin/tumor necrosis factor stimulates collagenase and prostaglandin E2 production by human synovial cells and dermal fibroblasts. J. Exp. Med. 1985, 162, 2163–2168. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, D.R.; Nedwin, G.E.; Bringman, T.S.; Smith, D.D.; Mundy, G.R. Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nat. 1986, 319, 516–518. [Google Scholar] [CrossRef] [PubMed]
- Marahleh, A.; Kitaura, H.; Ohori, F.; Kishikawa, A.; Ogawa, S.; Shen, W.-R.; Qi, J.; Noguchi, T.; Nara, Y.; Mizoguchi, I. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Front. Immunol. 2019, 10, 2925. [Google Scholar] [CrossRef] [PubMed]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-α induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef]
- Kobayashi, K.; Takahashi, N.; Jimi, E.; Udagawa, N.; Takami, M.; Kotake, S.; Nakagawa, N.; Kinosaki, M.; Yamaguchi, K.; Shima, N.; et al. Tumor Necrosis Factor α Stimulates Osteoclast Differentiation by a Mechanism Independent of the Odf/Rankl–Rank Interaction. J. Exp. Med. 2000, 191, 275–286. [Google Scholar] [CrossRef]
- Azuma, Y.; Kaji, K.; Katogi, R.; Takeshita, S.; Kudo, A. Tumor Necrosis Factor-α Induces Differentiation of and Bone Resorption by Osteoclasts. J. Boil. Chem. 2000, 275, 4858–4864. [Google Scholar] [CrossRef]
- Fossiez, F.; Djossou, O.; Chomarat, P.; Flores-Romo, L.; Ait-Yahia, S.; Maat, C.; Pin, J.J.; Garrone, P.; Garcia, E.; Saeland, S.; et al. T cell interleukin-17 induces stromal cells to produce proinflammatory and hematopoietic cytokines. J. Exp. Med. 1996, 183, 2593–2603. [Google Scholar] [CrossRef]
- Borregaard, N. Neutrophils, from Marrow to Microbes. Immun. 2010, 33, 657–670. [Google Scholar] [CrossRef]
- Robert, M.; Miossec, P. IL-17 in Rheumatoid Arthritis and Precision Medicine: From Synovitis Expression to Circulating Bioactive Levels. Front. Med. 2019, 5, 364. [Google Scholar] [CrossRef]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, M.; Lubberts, E.; Joosten, L.; Berg, W.V.D.; Miossec, P. IL-17 derived from juxta-articular bone and synovium contributes to joint degradation in rheumatoid arthritis. Arthritis Res. 2001, 3, 168–177. [Google Scholar] [CrossRef] [PubMed]
- Van Bezooijen, R.L.; Papapoulos, S.E.; Lowik, C.W. Effect of interleukin-17 on nitric oxide production and osteoclastic bone resorption: Is there dependency on nuclear factor-kappaB and receptor activator of nuclear factor kappaB (RANK)/RANK ligand signaling? Bone 2001, 28, 378–386. [Google Scholar] [CrossRef]
- Chabaud, M.; Garnero, P.; Dayer, J.-M.; Guerne, P.-A.; Fossiez, F.; Miossec, P. Contribution of interleukin 17 to synovium matrix destruction in rheumatoid arthritis. Cytokine 2000, 12, 1092–1099. [Google Scholar] [CrossRef] [PubMed]
- Pickens, S.R.; Volin, M.V.; Mandelin, A.M.; Kolls, J.K.; Pope, R.M.; Shahrara, S. IL-17 contributes to angiogenesis in rheumatoid arthritis. J. Immunol. 2010, 184, 3233–3241. [Google Scholar] [CrossRef]
- Ryu, S.; Lee, J.H.; Kim, S.I. IL-17 increased the production of vascular endothelial growth factor in rheumatoid arthritis synoviocytes. Clin. Rheumatol. 2005, 25, 16–20. [Google Scholar] [CrossRef]
- Xie, Y.-D.; Jin, L.; Yu, Q.-W. [The role of IFN-gamma, IL-10, IL-12 and TRAIL in sera and synovium fluids from patients with rheumatoid arthritis]. Chin. J. Cell. Mol. Immunol. 2007, 23, 536–537. [Google Scholar]
- Kokkonen, H.; Sãderstrãm, I.; Rocklãv, J.; Hallmans, G.; Lejon, K.; Rantapää-Dahlqvist, S.; Söderström, I.; Rocklov, J. Up-regulation of cytokines and chemokines predates the onset of rheumatoid arthritis. Arthritis Rheum. 2010, 62, 383–391. [Google Scholar] [CrossRef]
- Steiner, G.; Tohidast-Akrad, M.; Witzmann, G.; Vesely, M.; Studnicka-Benke, A.; Gal, A.; Kunaver, M.; Zenz, P.; Smolen, J.S. Cytokine production by synovial T cells in rheumatoid arthritis. Rheumatology 1999, 38, 202–213. [Google Scholar] [CrossRef]
- Morita, Y.; Yamamura, M.; Kawashima, M.; Harada, S.; Tsuji, K.; Shibuya, K.; Maruyama, K.; Makino, H. Flow cytometric single-cell analysis of cytokine production by CD4+ T cells in synovial tissue and peripheral blood from patients with rheumatoid arthritis. Arthritis Rheum. 1998, 41, 1669–1676. [Google Scholar] [CrossRef]
- Thanapati, S.; Ganu, M.; Giri, P.; Kulkarni, S.; Sharma, M.; Babar, P.; Ganu, A.; Tripathy, A. Impaired NK cell functionality and increased TNF-α production as biomarkers of chronic chikungunya arthritis and rheumatoid arthritis. Hum. Immunol. 2017, 78, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Olalekan, S.A.; Cao, Y.; Hamel, K.M.; Finnegan, A. B cells expressing IFN-γ suppress Treg-cell differentiation and promote autoimmune experimental arthritis. Eur. J. Immunol. 2015, 45, 988–998. [Google Scholar] [CrossRef]
- Karonitsch, T.; Von Dalwigk, K.; Steiner, C.W.; Blüml, S.; Steiner, G.; Kiener, H.P.; Ramiro, S.; Aringer, M.; Steiner, G. Interferon signals and monocytic sensitization of the interferon-γ signaling pathway in the peripheral blood of patients with rheumatoid arthritis. Arthritis Rheum. 2012, 64, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Bach, E.A.; Aguet, M.; Schreiber, R.D. THE IFNγ RECEPTOR: A Paradigm for Cytokine Receptor Signaling. Annu. Rev. Immunol. 1997, 15, 563–591. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.A.; Farrar, M.; Farrar, M.; Hershey, G.K.; Fernandez-Luna, J. The structure and function of interferon-gamma receptors. Int. J. Immunopharmacol. 1992, 14, 413–419. [Google Scholar] [CrossRef]
- Tang, M.; Tian, L.; Luo, G.; Yu, X. Interferon-Gamma-Mediated Osteoimmunology. Front. Immunol. 2018, 9, 1508. [Google Scholar] [CrossRef] [PubMed]
- Kwak, H.B.; Ha, H.; Kim, H.N.; Lee, J.H.; Kim, H.S.; Lee, S.; Kim, H.M.; Kim, J.Y.; Kim, H.H.; Song, Y.W.; et al. Reciprocal cross-talk between RANKL and interferon-gamma-inducible protein 10 is responsible for bone-erosive experimental arthritis. Arthritis Rheum. 2008, 58, 1332–1342. [Google Scholar] [CrossRef]
- Luster, A.D.; Ravetch, J.V. Biochemical characterization of a gamma interferon-inducible cytokine (IP-10). J. Exp. Med. 1987, 166, 1084–1097. [Google Scholar] [CrossRef]
- Kim, E.Y.; Moudgil, K.D. Immunomodulation of autoimmune arthritis by pro-inflammatory cytokines. Cytokine 2017, 98, 87–96. [Google Scholar] [CrossRef]
- Fuller, K.; Wong, B.; Fox, S.; Choi, Y.; Chambers, T. TRANCE Is Necessary and Sufficient for Osteoblast-mediated Activation of Bone Resorption in Osteoclasts. J. Exp. Med. 1998, 188, 997–1001. [Google Scholar] [CrossRef]
- Okamoto, K.; Takayanagi, H. Regulation of bone by the adaptive immune system in arthritis. Arthritis Res. 2011, 13, 219. [Google Scholar] [CrossRef] [PubMed]
- Yeo, L.; Schmultz, K.; Toellner, K.; Salmon, M.; Filer, A.D.; Buckley, C.; Raza, K.; Scheel-Toellnew, D. Cytokine mRNA profiling identifies B cells as a major source of RANKL in rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 2022–2028. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.M.; Kim, K.W.; Yang, C.-W.; Park, S.-H.; Ju, J.H. Cytokine-Mediated Bone Destruction in Rheumatoid Arthritis. J. Immunol. Res. 2014, 2014, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Pettit, A.; Ji, H.; Von Stechow, D.; Müller, R.; Goldring, S.R.; Choi, Y.; Benoist, C.; Gravallese, E.M. TRANCE/RANKL Knockout Mice Are Protected from Bone Erosion in a Serum Transfer Model of Arthritis. Am. J. Pathol. 2001, 159, 1689–1699. [Google Scholar] [CrossRef]
- Goh, F.G.; Midwood, K.S. Intrinsic danger: Activation of Toll-like receptors in rheumatoid arthritis. Rheumatol. 2011, 51, 7–23. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Bozec, A.; Ramming, A.; Schett, G. Anti-inflammatory and immune-regulatory cytokines in rheumatoid arthritis. Nat. Rev. Rheumatol. 2018, 15, 9–17. [Google Scholar] [CrossRef]
- Reboul, P.; Pelletier, J.P.; Tardif, G.; Cloutier, J.M.; Martel-Pelletier, J. The new collagenase, collagenase-3, is expressed and synthesized by human chondrocytes but not by synoviocytes. A role in osteoarthritis. J. Clin. Investig. 1996, 97, 2011–2019. [Google Scholar] [CrossRef]
- Borden, P.; Solymar, D.; Sucharczuk, A.; Lindman, B.R.; Cannon, P.; Heller, R.A. Cytokine Control of Interstitial Collagenase and Collagenase-3 Gene Expression in Human Chondrocytes. J. Boil. Chem. 1996, 271, 23577–23581. [Google Scholar] [CrossRef]
- Redlich, K.; Smolen, J.S. Inflammatory bone loss: Pathogenesis and therapeutic intervention. Nat. Rev. Drug Discov. 2012, 11, 234–250. [Google Scholar] [CrossRef]
- Lefèvre, S.; Knedla, A.; Tennie, C.; Kampmann, A.; Wunrau, C.; Dinser, R.; Korb, A.; Schnäker, E.-M.; Tarner, I.H.; Robbins, P.D.; et al. Synovial fibroblasts spread rheumatoid arthritis to unaffected joints. Nat. Med. 2009, 15, 1414–1420. [Google Scholar] [CrossRef]
- Smolen, J.S.; Aletaha, D.; Koeller, M.; Weisman, M.H.; Emery, P. New therapies for treatment of rheumatoid arthritis. Lancet Lond. Engl. 2007, 370, 1861–1874. [Google Scholar] [CrossRef]
- McInnes, I.; Schett, G. The Pathogenesis of Rheumatoid Arthritis. N. Engl. J. Med. 2011, 365, 2205–2219. [Google Scholar] [CrossRef] [PubMed]
- Holers, V.M.; Banda, N.K. Complement in the Initiation and Evolution of Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1057. [Google Scholar] [CrossRef] [PubMed]
- Scher, J.U. B-cell therapies for rheumatoid arthritis. Bull. Nyu Hosp. Jt. Dis. 2012, 70, 200–203. [Google Scholar] [PubMed]
- Nishimura, K. Meta-analysis: Diagnostic Accuracy of Anti–Cyclic Citrullinated Peptide Antibody and Rheumatoid Factor for Rheumatoid Arthritis. Ann. Intern. Med. 2007, 146, 797. [Google Scholar] [CrossRef] [PubMed]
- Ingegnoli, F.; Castelli, R.; Gualtierotti, R. Rheumatoid Factors: Clinical Applications. Dis. Markers 2013, 35, 727–734. [Google Scholar] [CrossRef]
- Steiner, G. Auto-antibodies and autoreactive T-cells in rheumatoid arthritis: Pathogenetic players and diagnostic tools. Clin. Rev. Allergy Immunol. 2007, 32, 23–36. [Google Scholar] [CrossRef]
- Wegner, N.; Lundberg, K.; Kinloch, A.J.; Fisher, B.; Malmström, V.; Feldmann, M.; Venables, P.J. Autoimmunity to specific citrullinated proteins gives the first clues to the etiology of rheumatoid arthritis. Immunol. Rev. 2010, 233, 34–54. [Google Scholar] [CrossRef]
- Aggarwal, R.; Liao, K.; Nair, R.; Ringold, S.; Costenbader, K.H. Anti-citrullinated peptide antibody assays and their role in the diagnosis of rheumatoid arthritis. Arthritis Rheum. 2009, 61, 1472–1483. [Google Scholar] [CrossRef]
- Gerlag, D.M.; Safy, M.; Maijer, K.I.; Tang, M.W.; Tas, S.W.; Starmans-Kool, M.J.F.; van Tubergen, A.; Janssen, M.; de Hair, M.; Hansson, M.; et al. Tak PP7F1000Prime recommendation of Effects of B-cell directed therapy on the preclinical stage of rheumatoid arthritis: The PRAIRI study. Ann. Rheum. Dis. 2019, 78, 179–185. [Google Scholar]
- Forslind, K.; Ahlmen, M.; Eberhardt, K.; Hafström, I.; Svensson, B. Prediction of radiological outcome in early rheumatoid arthritis in clinical practice: Role of antibodies to citrullinated peptides (anti-CCP). Ann. Rheum. Dis. 2004, 63, 1090–1095. [Google Scholar] [CrossRef] [PubMed]
- Rönnelid, J.; Wick, M.C.; Lampa, J.; Lindblad, S.; Nordmark, B.; Klareskog, L.; Van Vollenhoven, R.F. Longitudinal analysis of citrullinated protein/peptide antibodies (anti-CP) during 5 year follow up in early rheumatoid arthritis: Anti-CP status predicts worse disease activity and greater radiological progression. Ann. Rheum. Dis. 2005, 64, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- De Rycke, L.; Peene, I.; Hoffman, I.; Kruithof, E.; Union, A.; Meheus, L.; Lebeer, K.; Wyns, B.; Vincent, C.; Mielants, H.; et al. Rheumatoid factor and anticitrullinated protein antibodies in rheumatoid arthritis: Diagnostic value, associations with radiological progression rate, and extra-articular manifestations. Ann. Rheum. Dis. 2004, 63, 1587–1593. [Google Scholar] [CrossRef] [PubMed]
- Coutant, F. Pathogenic effects of anti-citrullinated protein antibodies in rheumatoid arthritis – role for glycosylation. Jt. Bone Spine 2019, 86, 562–567. [Google Scholar] [CrossRef] [PubMed]
- Krishnamurthy, A.; Joshua, V.; Hensvold, A.H.; Jin, T.; Sun, M.; Vivar, N.; Ytterberg, A.; Engström, M.; Fernandes-Cerqueira, C.; Amara, K.; et al. Identification of a novel chemokine-dependent molecular mechanism underlying rheumatoid arthritis-associated autoantibody-mediated bone loss. Ann. Rheum. Dis. 2015, 75, 721–729. [Google Scholar] [CrossRef]
- Scherer, H.U.; Van Der Woude, D.; Ioan-Facsinay, A.; El Bannoudi, H.; Trouw, L.A.; Wang, J.; Häupl, T.; Burmester, G.-R.; Deelder, A.M.; Huizinga, T.W.J.; et al. Glycan profiling of anti-citrullinated protein antibodies isolated from human serum and synovial fluid. Arthritis Rheum. 2010, 62, 1620–1629. [Google Scholar] [CrossRef]
- Nandakumar, K.S.; Collin, M.; Olsén, A.; Nimmerjahn, F.; Blom, A.M.; Ravetch, J.V.; Holmdahl, R. Endoglycosidase treatment abrogates IgG arthritogenicity: Importance of IgG glycosylation in arthritis. Eur. J. Immunol. 2007, 37, 2973–2982. [Google Scholar] [CrossRef]
- Rombouts, Y.; Ewing, E.A.; Van De Stadt, L.; Selman, M.H.J.; Trouw, L.A.; Deelder, A.M.; Huizinga, T.W.J.; Wuhrer, M.; Van Schaardenburg, D.; Toes, R.; et al. Anti-citrullinated protein antibodies acquire a pro-inflammatory Fc glycosylation phenotype prior to the onset of rheumatoid arthritis. Ann. Rheum. Dis. 2013, 74, 234–241. [Google Scholar]
- Ercan, A.; Cui, J.; Chatterton, D.E.W.; Deane, K.D.; Hazen, M.M.; Brintnell, W.; O’Donnell, C.I.; Derber, L.A.; Weinblatt, M.E.; Shadick, N.A.; et al. IgG galactosylation aberrancy precedes disease onset, correlates with disease activity and is prevalent in autoantibodies in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2239–2248. [Google Scholar] [CrossRef]
- Pfeifle, R.; Rothe, T.; Ipseiz, N.; Scherer, H.U.; Culemann, S.; Harre, U.A.; Ackermann, J.; Seefried, M.; Kleyer, A.; Uderhardt, S.; et al. Regulation of autoantibody activity by the IL-23–TH17 axis determines the onset of autoimmune disease. Nat. Immunol. 2016, 18, 104–113. [Google Scholar] [CrossRef]
- Elshabrawy, H.A.; Chen, Z.; Volin, M.V.; Ravella, S.; Virupannavar, S.; Shahrara, S. The pathogenic role of angiogenesis in rheumatoid arthritis. Angiogenesis 2015, 18, 433–448. [Google Scholar] [CrossRef]
- Bartók, B.; Firestein, G.S. Fibroblast-like synoviocytes: Key effector cells in rheumatoid arthritis. Immunol. Rev. 2010, 233, 233–255. [Google Scholar] [CrossRef] [PubMed]
- Fassbender, H.G.; Simmling-Annefeld, M. The potential aggressiveness of synovial tissue in rheumatoid arthritis. J. Pathol. 1983, 139, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Baier, A.; Meineckel, I.; Gay, S.; Pap, T. Apoptosis in rheumatoid arthritis. Curr. Opin. Rheumatol. 2003, 15, 274–279. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Green, D.R.; Keystone, E.C.; Connor, A.; Zollman, S.; Firestein, G.S. p53 tumor suppressor gene mutations in fibroblast-like synoviocytes from erosion synovium and non-erosion synovium in rheumatoid arthritis. Arthritis Res. Ther. 2004, 7, R12–R18. [Google Scholar] [CrossRef]
- Yamanishi, Y.; Boyle, D.L.; Rosengren, S.; Green, D.R.; Zvaifler, N.J.; Firestein, G.S. Regional analysis of p53 mutations in rheumatoid arthritis synovium. Proc. Natl. Acad. Sci. USA 2002, 99, 10025–10030. [Google Scholar] [CrossRef] [PubMed]
- Cha, H.-S.; Rosengren, S.; Boyle, D.L.; Firestein, G.S. PUMA regulation and proapoptotic effects in fibroblast-like synoviocytes. Arthritis Rheum. 2006, 54, 587–592. [Google Scholar] [CrossRef]
- Coutant, F.; Miossec, P. Evolving concepts of the pathogenesis of rheumatoid arthritis with focus on the early and late stages. Curr. Opin. Rheumatol. 2020, 32, 57–63. [Google Scholar] [CrossRef]
- Burmester, G.; Pope, J.E. Novel treatment strategies in rheumatoid arthritis. Lancet 2017, 389, 2338–2348. [Google Scholar] [CrossRef]
- D’Agostino, M.A.; Terslev, L.; Wakefield, R.; Østergaard, M.; Balint, P.; Naredo, E.; Iagnocco, A.; Backhaus, M.; Grassi, W.; Emery, P. Novel algorithms for the pragmatic use of ultrasound in the management of patients with rheumatoid arthritis: From diagnosis to remission. Ann. Rheum. Dis. 2016, 75, 1902–1908. [Google Scholar] [CrossRef]
- Prado, A.D.D.; Staub, H.L.; Bisi, M.C.; Da Silveira, I.G.; Mendonça, J.A.; Pereira, J.P.; Fonseca, J.E. Ultrasound and its clinical use in rheumatoid arthritis: Where do we stand? Adv. Rheumatol. 2018, 58, 19. [Google Scholar] [CrossRef] [PubMed]
- Zayat, A.S.; Ellegaard, K.; Conaghan, P.G.; Terslev, L.; Hensor, E.M.A.; Freeston, J.; Emery, P.; Wakefield, R.J. The specificity of ultrasound-detected bone erosions for rheumatoid arthritis. Ann. Rheum. Dis. 2014, 74, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, R.; Hama, M.; Takase, K.; Ihata, A.; Kishimoto, D.; Terauchi, K.; Watanabe, R.; Uehara, T.; Samukawa, S.; Ueda, A.; et al. Ultrasonography is a potent tool for the prediction of progressive joint destruction during clinical remission of rheumatoid arthritis. Mod. Rheumatol. 2013, 23, 456–465. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, T.; Ikeda, K.; Hosokawa, J.; Yamagata, M.; Tanaka, S.; Norimoto, A.; Sanayama, Y.; Nakagomi, D.; Takahashi, K.; Hirose, K.; et al. Prediction of Relapse after Discontinuation of Biologic Agents by Ultrasonographic Assessment in Patients With Rheumatoid Arthritis in Clinical Remission: High Predictive Values of Total Gray-Scale and Power Doppler Scores That Represent Residual Synovial. Arthritis Rheum. 2014, 66, 1576–1581. [Google Scholar] [CrossRef]
- Takase-Minegishi, K.; Horita, N.; Kobayashi, K.; Yoshimi, R.; Kirino, Y.; Ohno, S.; Kaneko, T.; Nakajima, H.; Wakefield, R.J.; Emery, P. Diagnostic test accuracy of ultrasound for synovitis in rheumatoid arthritis: Systematic review and meta-analysis. Rheumatology 2017, 57, 49–58. [Google Scholar] [CrossRef]
- Cohen, S.; Potter, H.; Deodhar, A.; Emery, P.; Conaghan, P.G.; Østergaard, M. Extremity magnetic resonance imaging in rheumatoid arthritis: Updated literature review. Arthritis Rheum. 2011, 63, 660–665. [Google Scholar] [CrossRef]
- Shrive, A.K.; Holden, D.; Myles, D.A.; Greenhough, T.J. Structure Solution of C-Reactive Proteins: Molecular Replacement With a Twist. Acta Crystallogr. Sect. D Boil. Crystallogr. 1996, 52, 1049–1057. [Google Scholar] [CrossRef]
- Baumann, H.; Gauldie, J. The acute phase response. Immunol. Today 1994, 15, 74–80. [Google Scholar]
- Kuta, A.E.; Baum, L. C-reactive protein is produced by a small number of normal human peripheral blood lymphocytes. J. Exp. Med. 1986, 164, 321–326. [Google Scholar] [CrossRef]
- Calabrò, P.; Chang, D.W.; Willerson, J.T.; Yeh, E.T. Release of C-Reactive Protein in Response to Inflammatory Cytokines by Human Adipocytes: Linking Obesity to Vascular Inflammation. J. Am. Coll. Cardiol. 2005, 46, 1112–1113. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, M.; Samols, D.; Kushner, I. STAT3 Participates in Transcriptional Activation of the C-reactive Protein Gene by Interleukin-6. J. Boil. Chem. 1996, 271, 9503–9509. [Google Scholar] [CrossRef] [PubMed]
- Calabrò, P.; Willerson, J.T.; Yeh, E.T. Inflammatory Cytokines Stimulated C-Reactive Protein Production by Human Coronary Artery Smooth Muscle Cells. Circ. 2003, 108, 1930–1932. [Google Scholar] [CrossRef] [PubMed]
- Siegel, J.; Osmand, A.P.; Wilson, M.F.; Gewurz, H. Interactions of C-reactive protein with the complement system. II. C-reactive protein-mediated consumption of complement by poly-L-lysine polymers and other polycations. J. Exp. Med. 1975, 142, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Mold, C.; Gewurz, H.; Du Clos, T.W. Regulation of complement activation by C-reactive protein. Immunopharmacology 1999, 42, 23–30. [Google Scholar] [CrossRef]
- Bharadwaj, D.; Bharadwaj, D.-P.; Volzer, M.; Mold, C.; Du Clos, T.W. The major receptor for C-reactive protein on leukocytes is fcgamma receptor II. J. Exp. Med. 1999, 190, 585–590. [Google Scholar]
- Lu, J.; Marnell, L.L.; Marjon, K.D.; Mold, C.; Du Clos, T.W.; Sun, P.D. Structural recognition and functional activation of FcγR by innate pentraxins. Nature 2008, 456, 989–992. [Google Scholar] [CrossRef]
- Williams, T.N.; Zhang, C.X.; Game, B.A.; He, L.; Huang, Y. C-reactive protein stimulates MMP-1 expression in U937 histiocytes through Fc[gamma]RII and extracellular signal-regulated kinase pathway: An implication of CRP involvement in plaque destabilization. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 61–66. [Google Scholar] [CrossRef]
- Nabata, A.; Kuroki, M.; Ueba, H.; Hashimoto, S.; Umemoto, T.; Wada, H.; Yasu, T.; Saito, M.; Momomura, S.-I.; Kawakami, M. C-reactive protein induces endothelial cell apoptosis and matrix metalloproteinase-9 production in human mononuclear cells: Implications for the destabilization of atherosclerotic plaque. Atherosclerosis 2008, 196, 129–135. [Google Scholar] [CrossRef]
- Devaraj, S.; Yun, J.-M.; Duncan-Staley, C.; Jialal, I. C-reactive protein induces M-CSF release and macrophage proliferation. J. Leukoc. Boil. 2008, 85, 262–267. [Google Scholar] [CrossRef]
- Han, K.H.; Hong, K.-H.; Park, J.-H.; Ko, J.-S.; Kang, D.-H.; Choi, K.-J.; Hong, M.-K.; Park, S.-W.; Park, S.-J. C-Reactive Protein Promotes Monocyte Chemoattractant Protein-1—Mediated Chemotaxis Through Upregulating CC Chemokine Receptor 2 Expression in Human Monocytes. Circulation 2004, 109, 2566–2571. [Google Scholar] [CrossRef]
- Kim, K.-W.; Kim, B.M.; Moon, H.W.; See, S.H.; Kim, H.R. Role of C-reactive protein in osteoclastogenesis in rheumatoid arthritis. Arthritis Res. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- Mallya, R.K.; De Beer, F.C.; Berry, H.; Hamilton, E.D.; Mace, B.; Pepys, M.B. Correlation of clinical parameters of disease activity in rheumatoid arthritis with serum concentration of C-reactive protein and erythrocyte sedimentation rate. J. Rheumatol. 1982, 9, 224–228. [Google Scholar] [PubMed]
- Matsuno, H.; Yudoh, K.; Nakazawa, F.; Koizumi, F. Relationship between histological findings and clinical findings in rheumatoid arthritis. Pathol. Int. 2002, 52, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F. Comparative usefulness of C-reactive protein and erythrocyte sedimentation rate in patients with rheumatoid arthritis. J. Rheumatol. 1997, 24, 1477–1485. [Google Scholar]
- Van Leeuwen, M.; Van Der Heijde, D.M.; Van Rijswijk, M.H.; Houtman, P.M.; Van Riel, P.L.; Van De Putte, L.B.; Limburg, P.C. Interrelationship of outcome measures and process variables in early rheumatoid arthritis. A comparison of radiologic damage, physical disability, joint counts, and acute phase reactants. J. Rheumatol. 1994, 21, 425–429. [Google Scholar]
- Rhodes, B.; Fürnrohr, B.G.; Vyse, T. C-reactive protein in rheumatology: Biology and genetics. Nat. Rev. Rheumatol. 2011, 7, 282–289. [Google Scholar] [CrossRef]
- Jansen, L.E.; Van Der Horst-Bru, I.; Van Schaardenburg, D.; Bezemer, P.D.; Dijkmans, B.A.C. Predictors of radiographic joint damage in patients with early rheumatoid arthritis. Ann. Rheum. Dis. 2001, 60, 924–927. [Google Scholar] [CrossRef]
- Devlin, J.; Gough, A.; Huissoon, A.; Perkins, P.; Holder, R.; Reece, R.; Arthur, V.; Emery, P. The acute phase and function in early rheumatoid arthritis. C-reactive protein levels correlate with functional outcome. J. Rheumatol. 1997, 24, 9–13. [Google Scholar]
- Isiksacan, Z.; Elbuken, C.; Erel, O. A portable microfluidic system for rapid measurement of the erythrocyte sedimentation rate. Lab. A Chip 2016, 16, 4682–4690. [Google Scholar] [CrossRef]
- Ramsay, E.S.; Lerman, M.A. How to use the erythrocyte sedimentation rate in paediatrics. Arch. Dis. Child. Educ. Pr. Ed. 2014, 100, 30–36. [Google Scholar] [CrossRef]
- Radner, H.; Neogi, T.; Smolen, J.S.; Aletaha, D. Performance of the 2010 ACR/EULAR classification criteria for rheumatoid arthritis: A systematic literature review. Ann. Rheum. Dis. 2013, 73, 114–123. [Google Scholar] [CrossRef]
- Sokka, T.; Kautiainen, H.; Möttönen, T.; Hannonen, P. Work disability in rheumatoid arthritis 10 years after the diagnosis. J. Rheumatol. 1999, 26, 1681–1685. [Google Scholar]
- Wolfe, F. The natural history of rheumatoid arthritis. J. Rheumatol. Suppl. 1996, 44, 13–22. [Google Scholar]
- Fries, J. Current treatment paradigms in rheumatoid arthritis. Rheumatology 2000, 39, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Brune, K.; Patrignani, P. New insights into the use of currently available non-steroidal anti-inflammatory drugs. J. Pain Res. 2015, 8, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Crofford, L.J. Use of NSAIDs in treating patients with arthritis. Arthritis Res. Ther. 2013, 15, S2. [Google Scholar] [CrossRef] [PubMed]
- Van Everdingen, A.A.; Jacobs, J.W.; Van Reesema, D.R.S.; Bijlsma, J.W. Low-dose prednisone therapy for patients with early active rheumatoid arthritis: Clinical efficacy, disease-modifying properties, and side effects: A randomized, double-blind, placebo-controlled clinical trial. Ann. Intern. Med. 2002, 136, 1–12. [Google Scholar] [CrossRef]
- Silverstein, F.E.; Faich, G.; Goldstein, J.L.; Simon, L.S.; Pincus, T.; Whelton, A.; Makuch, R.; Eisen, G.; Agrawal, N.M.; Stenson, W.F.; et al. Gastrointestinal Toxicity with Celecoxib vs. Nonsteroidal Anti-inflammatory Drugs for Osteoarthritis and Rheumatoid Arthritis. JAMA 2000, 284, 1247. [Google Scholar] [CrossRef]
- Cronstein, B.N. Low-Dose Methotrexate: A Mainstay in the Treatment of Rheumatoid Arthritis. Pharmacol. Rev. 2005, 57, 163–172. [Google Scholar] [CrossRef]
- Abbasi, M.; Mousavi, M.J.; Jamalzehi, S.; Alimohammadi, R.; Bezvan, M.H.; Mohammadi, H.; Aslani, S. Strategies toward rheumatoid arthritis therapy; the old and the new. J. Cell. Physiol. 2018, 234, 10018–10031. [Google Scholar] [CrossRef]
- Emery, P.O.; Bingham, C.; Burmester, G.R.; Bykerk, V.P.E.; Furst, D.; Mariette, X.; Van Vollenhoven, R.; Arendt, C.; Mountian, I. Certolizumab pegol in combination with dose-optimised methotrexate in DMARD-naïve patients with early, active rheumatoid arthritis with poor prognostic factors: 1-year results from C-EARLY, a randomised, double-blind, placebo-controlled phase III study. Ann. Rheum. Dis. 2016, 76, 96–104. [Google Scholar] [PubMed]
- Nam, J.L.; Villeneuve, E.; Hensor, E.M.A.; Conaghan, P.G.I.; Keen, H.; Buch, M.H.; Gough, A.K.; Green, M.J.; Helliwell, P.S.; Keenan, A.M.; et al. Remission induction comparing infliximab and high-dose intravenous steroid, followed by treat-to-target: A double-blind, randomised, controlled trial in new-onset, treatment-naive, rheumatoid arthritis (the IDEA study). Ann. Rheum. Dis. 2013, 73, 75–85. [Google Scholar] [PubMed]
- Sethi, M.K.; O’Dell, J.R. Combination conventional DMARDs compared to biologicals. Curr. Opin. Rheumatol. 2015, 27, 183–188. [Google Scholar] [CrossRef] [PubMed]
- Weinblatt, M.E. Methotrexate in Rheumatoid Arthritis: A Quarter Century of Development. Trans. Am. Clin. Clim. Assoc. 2013, 124, 16–25. [Google Scholar]
- Nam, J.L.; Takase-Minegishi, K.; Ramiro, S.; Chatzidionysiou, K.; Smolen, J.S.; Van Der Heijde, D.; Bijlsma, J.W.; Burmester, G.R.; Dougados, M.; Scholte-Voshaar, M.; et al. Efficacy of biological disease-modifying antirheumatic drugs: A systematic literature review informing the 2016 update of the EULAR recommendations for the management of rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1113–1136. [Google Scholar] [CrossRef]
- Cronstein, B.N.; Naime, D.; Ostad, E. The antiinflammatory mechanism of methotrexate. Increased adenosine release at inflamed sites diminishes leukocyte accumulation in an in vivo model of inflammation. J. Clin. Investig. 1993, 92, 2675–2682. [Google Scholar] [CrossRef]
- Rajagopalan, P.T.R.; Zhang, Z.; McCourt, L.; Dwyer, M.; Benkovic, S.J.; Hammes, G.G. Interaction of dihydrofolate reductase with methotrexate: Ensemble and single-molecule kinetics. Proc. Natl. Acad. Sci. USA 2002, 99, 13481–13486. [Google Scholar] [CrossRef]
- Borchers, A.T.; Keen, C.L.; Cheema, G.S.; Gershwin, M.E. The use of methotrexate in rheumatoid arthritis. Semin. Arthritis Rheum. 2004, 34, 465–483. [Google Scholar] [CrossRef]
- Van Ede, A.E.; Laan, R.F.J.M.; Rood, M.J.; Huizinga, T.W.J.; Van De Laar, M.A.F.J.; Van Denderen, C.J.; Westgeest, T.A.A.; Romme, T.C.; De Rooij, D.-J.R.A.M.; Jacobs, M.J.M.; et al. Effect of folic or folinic acid supplementation on the toxicity and efficacy of methotrexate in rheumatoid arthritis: A forty-eight-week, multicenter, randomized, double-blind, placebo-controlled study. Arthritis Rheum. 2001, 44, 1515–1524. [Google Scholar] [CrossRef]
- Hawkes, J.S.; Cleland, L.G.; Proudman, S.M.; James, M.J. The effect of methotrexate on ex vivo lipoxygenase metabolism in neutrophils from patients with rheumatoid arthritis. J. Rheumatol. 1994, 21, 55–58. [Google Scholar]
- Phillips, D.C.; Woollard, K.; Griffiths, H.R. The anti-inflammatory actions of methotrexate are critically dependent upon the production of reactive oxygen species. Br. J. Pharmacol. 2003, 138, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Brody, M.; Böhm, I.; Bauer, R. Mechanism of action of methotrexate: Experimental evidence that methotrexate blocks the binding of interleukin 1 beta to the interleukin 1 receptor on target cells. Eur. J. Clin. Chem. Clin. Biochem. J. Forum Eur. Clin. Chem. Soc. 1993, 31, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Wennerstrand, P.; Mårtensson, L.-G.; Söderhäll, S.; Zimdahl, A.; Appell, M.L. Methotrexate binds to recombinant thiopurine S-methyltransferase and inhibits enzyme activity after high-dose infusions in childhood leukaemia. Eur. J. Clin. Pharmacol. 2013, 69, 1641–1649. [Google Scholar] [CrossRef][Green Version]
- Plosker, G.L.; Croom, K.F. Sulfasalazine: A review of its use in the management of rheumatoid arthritis. Drugs 2005, 65, 1825–1849. [Google Scholar] [CrossRef] [PubMed]
- Sousa, T.; Yadav, V.; Zann, V.; Borde, A.; Abrahamsson, B.; Basit, A.W. On the Colonic Bacterial Metabolism of Azo-Bonded Prodrugsof 5-Aminosalicylic Acid. J. Pharm. Sci. 2014, 103, 3171–3175. [Google Scholar] [CrossRef] [PubMed]
- Situnayake, R.D.; McConkey, B. Which component of sulphasalazine is active in rheumatoid arthritis? Br. Med. J. Clin. Res. Ed. 1985, 291, 138. [Google Scholar] [CrossRef]
- Kumar, P.; Banik, S. Pharmacotherapy Options in Rheumatoid Arthritis. Clin. Med. Insights: Arthritis Musculoskelet. Disord. 2013, 6, 35–43. [Google Scholar] [CrossRef]
- Felson, D.; Anderson, J.J.; Meenan, R. Use of short-term efficacy/toxicity tradeoffs to select second-line drugs in rheumatoid arthritis. A metaanalysis of published clinical trials. Arthritis Rheum. 1992, 35, 1117–1125. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Reda, D.; Henderson, W.; Giobbie-Hurder, A.; Williams, D.; Diani, A.; Docsa, S. Sulfasalazine treatment for rheumatoid arthritis: A metaanalysis of 15 randomized trials. J. Rheumatol. 1999, 26, 2123–2130. [Google Scholar]
- Öğrendik, M. Antibiotics for the treatment of rheumatoid arthritis. Int. J. Gen. Med. 2013, 7, 43–47. [Google Scholar] [CrossRef]
- Rogler, G. Gastrointestinal and liver adverse effects of drugs used for treating IBD. Best Pr. Res. Clin. Gastroenterol. 2010, 24, 157–165. [Google Scholar] [CrossRef] [PubMed]
- Farr, M.; Scott, D.G.I.; Bacon, P.A. Side Effect Profile of 200 Patients with Inflammatory Arthritides Treated with Sulphasalazine. Drugs 1986, 32, 49–53. [Google Scholar] [CrossRef] [PubMed]
- Farr, M.; Tunn, E.; Crockson, A.P.; Bacon, P.A. The long term effects of sulphasalazine in the treatment of rheumatoid arthritis and a comparative study with penicillamine. Clin. Rheumatol. 1984, 3, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Cildag, S.; Senturk, T. Sulfasalazine-Related Hypersensitivity Reactions in Patients with Rheumatic Diseases. Jcr: J. Clin. Rheumatol. 2017, 23, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Alamanos, Y.; Voulgari, P.V.; Drosos, A.A. Incidence and Prevalence of Rheumatoid Arthritis, Based on the 1987 American College of Rheumatology Criteria: A Systematic Review. Semin. Arthritis Rheum. 2006, 36, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Fox, R.I. Mechanism of action of hydroxychloroquine as an antirheumatic drug. Semin. Arthritis Rheum. 1993, 23, 82–91. [Google Scholar] [CrossRef]
- Kyburz, D.; Brentano, F.; Gay, S. Mode of action of hydroxychloroquine in RA—evidence of an inhibitory effect on toll-like receptor signaling. Nat. Clin. Pr. Rheumatol. 2006, 2, 458–459. [Google Scholar] [CrossRef]
- Suarez-Almazor, M.E.; Belseck, E.; Shea, B.; Homik, J.A.; Wells, G.; Tugwell, P. Antimalarials for treating rheumatoid arthritis. Cochrane Database Syst. Rev. 2000; CD000959. [Google Scholar] [CrossRef]
- Shinjo, S.K.; Júnior, O.O.M.; Tizziani, V.A.P.; Morita, C.; Kochen, J.A.L.; Takahashi, W.Y.; Laurindo, I.M.M. Chloroquine-induced bull’s eye maculopathy in rheumatoid arthritis: Related to disease duration? Clin. Rheumatol. 2007, 26, 1248–1253. [Google Scholar] [CrossRef]
- Finbloom, D.S.; Silver, K.; Newsome, D.A.; Gunkel, R. Comparison of hydroxychloroquine and chloroquine use and the development of retinal toxicity. J. Rheumatol. 1985, 12, 692–694. [Google Scholar]
- Marmor, M.F.; Carr, R.E.; Easterbrook, M.; Farjo, A.A.; Mieler, W.F. Recommendations on screening for chloroquine and hydroxychloroquine retinopathy: A report by the American Academy of Ophthalmology. Ophthalmology 2002, 109, 1377–1382. [Google Scholar] [CrossRef]
- Peper, S.M.; Lew, R.; Mikuls, T.; Brophy, M.; Rybin, D.; Wu, H.; O’Dell, J. Rheumatoid Arthritis Treatment After Methotrexate: The Durability of Triple Therapy Versus Etanercept. Arthritis Rheum. 2017, 69, 1467–1472. [Google Scholar] [CrossRef] [PubMed]
- Graudal, N.; Hubeck-Graudal, T.; Tarp, S.; Christensen, R.; Jurgens, G. Effect of Combination Therapy on Joint Destruction in Rheumatoid Arthritis: A Network Meta-Analysis of Randomized Controlled Trials. PLoS ONE 2014, 9, e106408. [Google Scholar] [CrossRef] [PubMed]
- Goekoop-Ruiterman, Y.P.M.; De Vries-Bouwstra, J.K.; Allaart, C.F.; Van Zeben, D.; Kerstens, P.J.S.M.; Hazes, J.M.W.; Zwinderman, A.H.; Ronday, H.K.; Han, K.H.; Westedt, M.L.; et al. Clinical and radiographic outcomes of four different treatment strategies in patients with early rheumatoid arthritis (the BeSt study): A randomized, controlled trial. Arthritis Rheum. 2008, 58, S126–S135. [Google Scholar] [CrossRef] [PubMed]
- Van Vollenhoven, R.F.; Geborek, P.; Forslind, K.; Albertsson, K.; Ernestam, S.; Petersson, I.; Chatzidionysiou, K.; Bratt, J. Conventional combination treatment versus biological treatment in methotrexate-refractory early rheumatoid arthritis: 2 year follow-up of the randomised, non-blinded, parallel-group Swefot trial. Lancet 2012, 379, 1712–1720. [Google Scholar] [CrossRef]
- Moreland, L.W.; O’Dell, J.R.; Paulus, H.E.; Curtis, J.R.; Bathon, J.M.; Clair, E.W.S.; Bridges, S.L.; Zhang, J.; McVie, T.; Howard, G.; et al. A randomized comparative effectiveness study of oral triple therapy versus etanercept plus methotrexate in early aggressive rheumatoid arthritis: The treatment of Early Aggressive Rheumatoid Arthritis Trial. Arthritis Rheum. 2012, 64, 2824–2835. [Google Scholar] [CrossRef] [PubMed]
- O’Dell, J.R.; Taylor, T.H.; Brophy, M.; Warren, S.R.; Cannella, A.C.; Kunkel, G.; Leatherman, S.; Mikuls, T.R.; Ahluwalia, V.; Lew, R.A.; et al. Therapies for Active Rheumatoid Arthritis after Methotrexate Failure. N. Engl. J. Med. 2013, 369, 307–318. [Google Scholar] [CrossRef]
- Sotoudehmanesh, R.; Anvari, B.; Akhlaghi, M.; Shahraeeni, S.; Kolahdoozan, S. Methotrexate Hepatotoxicity in Patients with Rheumatoid Arthritis. Middle East. J. Dig. Dis. 2010, 2, 104–109. [Google Scholar]
- Taylor, W.J.; Korendowych, E.; Nash, P.; Helliwell, P.S.; Choy, E.; Krueger, G.G.; Soriano, E.; McHugh, N.J.; Rosen, C.F. Drug use and toxicity in psoriatic disease: Focus on methotrexate. J. Rheumatol. 2008, 35, 1454–1457. [Google Scholar]
- Aithal, G.P. Hepatotoxicity related to antirheumatic drugs. Nat. Rev. Rheumatol. 2011, 7, 139–150. [Google Scholar] [CrossRef]
- Cummins, L.; Katikireddi, V.S.; Shankaranarayana, S.; Su, K.Y.C.; Duggan, E.; Videm, V.; Pahau, H.; Thomas, R. Safety and retention of combination triple disease-modifying anti-rheumatic drugs in new-onset rheumatoid arthritis. Intern. Med. J. 2015, 45, 1266–1273. [Google Scholar] [CrossRef]
- Kotyla, P.J. Are Janus Kinase Inhibitors Superior over Classic Biologic Agents in RA Patients? BioMed Res. Int. 2018, 2018. [Google Scholar] [CrossRef] [PubMed]
- Damsky, W.; King, B. JAK inhibitors in dermatology: The promise of a new drug class. J. Am. Acad. Dermatol. 2017, 76, 736–744. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C. Clinical efficacy of launched JAK inhibitors in rheumatoid arthritis. Rheumatology 2019, 58, i17–i26. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; Keystone, E.; Van Der Heijde, D.; Weinblatt, M.E.; Morales, L.D.C.; Gonzaga, J.R.; Yakushin, S.; Ishii, T.; Emoto, K.; Beattie, S.; et al. Baricitinib versus Placebo or Adalimumab in Rheumatoid Arthritis. N. Engl. J. Med. 2017, 376, 652–662. [Google Scholar] [CrossRef]
- Winthrop, K.L. The emerging safety profile of JAK inhibitors in rheumatic disease. Nat. Rev. Rheumatol. 2017, 13, 234–243. [Google Scholar] [CrossRef]
- Rein, P.; Mueller, R.B. Treatment with Biologicals in Rheumatoid Arthritis: An Overview. Rheumatol. Ther. 2017, 4, 247–261. [Google Scholar] [CrossRef]
- Charles, P.; Elliott, M.J.; Davis, D.; Potter, A.; Kalden, J.; Antoni, C.; Breedveld, F.C.; Smolen, J.S.; Eberl, G.; DeWoody, K.; et al. Regulation of cytokines, cytokine inhibitors, and acute-phase proteins following anti-TNF-alpha therapy in rheumatoid arthritis. J. Immunol. 1999, 163, 1521–1528. [Google Scholar]
- Curtis, J.R.; Xie, F.; Chen, L.; Muntner, P.; Grijalva, C.G.; Spettell, C.; Fernandes, J.; Mcmahan, R.M.; Baddley, J.W.; Saag, K.G.; et al. Use of a disease risk score to compare serious infections associated with anti-tumor necrosis factor therapy among high- versus lower-risk rheumatoid arthritis patients. Arthritis Rheum. 2012, 64, 1480–1489. [Google Scholar] [CrossRef]
- Leombruno, J.; Einarson, T.R.; Keystone, E.C. The safety of anti-tumour necrosis factor treatments in rheumatoid arthritis: Meta and exposure-adjusted pooled analyses of serious adverse events. Ann. Rheum. Dis. 2008, 68, 1136–1145. [Google Scholar] [CrossRef]
- Dreyer, L.; Mellemkjaer, L.; Andersen, A.R.; Bennett, P.; Poulsen, U.E.; Ellingsen, T.; Hansen, T.H.; Jensen, D.V.; Linde, L.; Lindegaard, H.M.; et al. Incidences of overall and site specific cancers in TNF inhibitor treated patients with rheumatoid arthritis and other arthritides—A follow-up study from the DANBIO Registry. Ann. Rheum. Dis. 2012, 72, 79–82. [Google Scholar] [CrossRef]
- McKenna, M.R.; Stobaugh, D.J.; Deepak, P. Melanoma and non-melanoma skin cancer in inflammatory bowel disease patients following tumor necrosis factor-? Inhibitor monotherapy and in combination with thiopurines: Analysis of the Food and Drug Administration Adverse Event Reporting System. J. Gastrointest. Liver Dis. 2014, 23, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Askling, J.; Fahrbach, K.; Nordstrom, B.; Ross, S.; Schmid, C.H.; Symmons, D. Cancer risk with tumor necrosis factor alpha (TNF) inhibitors: Meta-analysis of randomized controlled trials of adalimumab, etanercept, and infliximab using patient level data. Pharmacoepidemiol. Drug Saf. 2010, 20, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Amari, W.; Zeringue, A.; McDonald, J.R.; Caplan, L.; Eisen, S.A.; Ranganathan, P. Risk of non-melanoma skin cancer in a national cohort of veterans with rheumatoid arthritis. Rheumatology 2011, 50, 1431–1439. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Michaud, K. Biologic treatment of rheumatoid arthritis and the risk of malignancy: Analyses from a large US observational study. Arthritis Rheum. 2007, 56, 2886–2895. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.C.; Pawar, A.; Desai, R.J.; Solomon, D.H.; Gale, S.; Bao, M.; Sarsour, K.; Schneeweiss, S. Risk of malignancy associated with use of tocilizumab versus other biologics in patients with rheumatoid arthritis: A multi-database cohort study. Semin. Arthritis Rheum. 2019, 49, 222–228. [Google Scholar] [CrossRef]
- Mercer, L.K.; Green, A.C.; Galloway, J.; Davies, R.; Lunt, M.; Dixon, W.G.; Watson, K.D.; Symmons, D.; Hyrich, K.L. The influence of anti-TNF therapy upon incidence of keratinocyte skin cancer in patients with rheumatoid arthritis: Longitudinal results from the British Society for Rheumatology Biologics Register. Ann. Rheum. Dis. 2012, 71, 869–874. [Google Scholar] [CrossRef]
- Mariette, X.; Matucci-Cerinic, M.; Pavelka, K.; Taylor, P.; Van Vollenhoven, R.; Heatley, R.; Walsh, C.; Lawson, R.; Reynolds, A.; Emery, P. Malignancies associated with tumour necrosis factor inhibitors in registries and prospective observational studies: A systematic review and meta-analysis. Ann. Rheum. Dis. 2011, 70, 1895–1904. [Google Scholar] [CrossRef]
- Wolfe, F.; Michaud, K. The effect of methotrexate and anti–tumor necrosis factor therapy on the risk of lymphoma in rheumatoid arthritis in 19,562 patients during 89,710 PERSON-YEARS of observation. Arthritis Rheum. 2007, 56, 1433–1439. [Google Scholar] [CrossRef]
- Furst, D.E.; Schiff, M.; Fleischmann, R.; Strand, V.A.; Birbara, C.; Compagnone, D.A.; Fischkoff, S.; Chartash, E.K. Adalimumab, a fully human anti tumor necrosis factor-alpha monoclonal antibody, and concomitant standard antirheumatic therapy for the treatment of rheumatoid arthritis: Results of STAR (Safety Trial of Adalimumab in Rheumatoid Arthritis). J. Rheumatol. 2003, 30, 2563–2571. [Google Scholar]
- Van De Putte, L.B.A.; Atkins, C.; Malaise, M.; Sany, J.; Russell, A.S.; Van Riel, P.L.C.M.; Settas, L.; Bijlsma, J.W.; Todesco, S.; Dougados, M.; et al. Efficacy and safety of adalimumab as monotherapy in patients with rheumatoid arthritis for whom previous disease modifying antirheumatic drug treatment has failed. Ann. Rheum. Dis. 2004, 63, 508–516. [Google Scholar] [CrossRef]
- Weinblatt, M.E.; Keystone, E.; Furst, D.E.; Moreland, L.W.; Weisman, M.H.; Birbara, C.A.; Teoh, L.A.; Fischkoff, S.A.; Chartash, E.K. Adalimumab, a fully human anti–tumor necrosis factor α monoclonal antibody, for the treatment of rheumatoid arthritis in patients taking concomitant methotrexate: The ARMADA trial. Arthritis Rheum. 2003, 48, 35–45. [Google Scholar]
- Keystone, E.; Kavanaugh, A.; Sharp, J.T.; Tannenbaum, H.; Hua, Y.; Teoh, L.S.; Fischkoff, S.A.; Chartash, E.K. Radiographic, clinical, and functional outcomes of treatment with adalimumab (a human anti-tumor necrosis factor monoclonal antibody) in patients with active rheumatoid arthritis receiving concomitant methotrexate therapy: A randomized, placebo-controlled, 52-week trial. Arthritis Rheum. 2004, 50, 1400–1411. [Google Scholar] [PubMed]
- Furst, D.E.; Kavanaugh, A.; Florentinus, S.; Kupper, H.; Karunaratne, M.; Birbara, C.A. Final 10-year effectiveness and safety results from study DE020: Adalimumab treatment in patients with rheumatoid arthritis and an inadequate response to standard therapy. Rheumatology 2015, 54, 2188–2197. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Burmester, G.; Panaccione, R.; Gordon, K.; McIlraith, M.; Lacerda, A. SAT0130 Long-term safety of adalimumab in patients from global clinical trials in rheumatoid arthritis, juvenile idiopathic arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis, and crohn’s disease. Ann. Rheum. Dis. 2013, 71, 514–515. [Google Scholar] [CrossRef]
- Torrente-Segarra, V.; Arana, A.U.; Sanchez-Andrade, A.; Tovar, J.; Muñoz, Á.; Martinez, A.; Gonzalez, J.; Fernández, M.; Vazquez, N.; Corominas, H. RENACER Study: Assessment of 12-month efficacy and safety of 168 certolizumab-PEGol rheumatoid arthritis treated patients from a Spanish multicenter National database. Mod. Rheumatol. 2015, 26, 1–18. [Google Scholar]
- Moreland, L.W.; Tindall, E.A.; Weaver, A.L.; Ettlinger, R.E.; Mohler, K.; Widmer, M.B.; Blosch, C.M.; Cohen, S.; Baumgartner, S.W.; Schiff, M.; et al. Treatment of Rheumatoid Arthritis with a Recombinant Human Tumor Necrosis Factor Receptor (p75)–Fc Fusion Protein. N. Engl. J. Med. 1997, 337, 141–147. [Google Scholar] [CrossRef]
- Alldred, A. Etanercept in rheumatoid arthritis. Expert Opin. Pharmacother. 2001, 2, 1137–1148. [Google Scholar] [CrossRef]
- Maid, P.J.; Xavier, R.; Real, R.M.; Pedersen, R.; Shen, Q.; Marshall, L.; Solano, G.; Borlenghi, C.E.; Hidalgo, R.P. Incidence of Antidrug Antibodies in Rheumatoid Arthritis Patients From Argentina Treated With Adalimumab, Etanercept, or Infliximab in a Real-World Setting. JCR: J. Clin. Rheumatol. 2018, 24, 177–182. [Google Scholar] [CrossRef]
- Moots, R.J.; Xavier, R.M.; Mok, C.C.; Rahman, M.U.; Tsai, W.C.; Al-Maini, M.H. The impact of anti-drug antibodies on drug concentrations and clinical outcomes in rheumatoid arthritis patients treated with adalimumab, etanercept, or infliximab: Results from a multinational, real-world clinical practice, non-interventional study. PLoS ONE 2017, 12, e0175207. [Google Scholar]
- Quistrebert, J.; Hässler, S.; Bachelet, D.; Mbogning, C.; Musters, A.; Tak, P.P.; Wijbrandts, C.A.; Herenius, M.; Bergstra, S.A.; Akdemir, G.; et al. Incidence and risk factors for adalimumab and infliximab anti-drug antibodies in rheumatoid arthritis: A European retrospective multicohort analysis. Semin. Arthritis Rheum. 2019, 48, 967–975. [Google Scholar] [CrossRef]
- Verstappen, S.; McCoy, M.J.; Roberts, C.; Dale, N.E.; Hassell, A.B.; Symmons, D. Beneficial effects of a 3-week course of intramuscular glucocorticoid injections in patients with very early inflammatory polyarthritis: Results of the STIVEA trial. Ann. Rheum. Dis. 2009, 69, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Van Schouwenburg, P.; Rispens, T.; Wolbink, G.J. Immunogenicity of anti-TNF biologic therapies for rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 164–172. [Google Scholar] [CrossRef]
- Krishna, M.; Nadler, S.G. Immunogenicity to Biotherapeutics – The Role of Anti-drug Immune Complexes. Front. Immunol. 2016, 7, 164. [Google Scholar] [CrossRef] [PubMed]
- Korswagen, L.A.; Bartelds, G.M.; Krieckaert, C.L.M.; Turkstra, F.; Nurmohamed, M.T.; Van Schaardenburg, D.; Wijbrandts, C.; Tak, P.P.; Lems, W.F.; Dijkmans, B.A.C.; et al. Venous and arterial thromboembolic events in adalimumab-treated patients with antiadalimumab antibodies: A case series and cohort study. Arthritis Rheum. 2011, 63, 877–883. [Google Scholar] [CrossRef]
- Mihara, M.; Ohsugi, Y.; Kishimoto, T. Tocilizumab, a humanized anti-interleukin-6 receptor antibody, for treatment of rheumatoid arthritis. Open Access Rheumatol. Res. Rev. 2011, 3, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Avci, A.B.; Feist, E.; Burmester, G.R. Targeting IL-6 or IL-6 Receptor in Rheumatoid Arthritis: What’s the Difference? BioDrugs Clin. Immunother. Biopharm. Gene Ther. 2018, 32, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Scott, L.J. Tocilizumab: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1865–1879. [Google Scholar] [CrossRef]
- Dhillon, S. Intravenous Tocilizumab: A Review of Its Use in Adults with Rheumatoid Arthritis. BioDrugs 2013, 28, 75–106. [Google Scholar] [CrossRef]
- Nishimoto, N.; Terao, K.; Mima, T.; Nakahara, H.; Takagi, N.; Kakehi, T. Mechanisms and pathologic significances in increase in serum interleukin-6 (IL-6) and soluble IL-6 receptor after administration of an anti–IL-6 receptor antibody, tocilizumab, in patients with rheumatoid arthritis and Castleman disease. Blood 2008, 112, 3959–3964. [Google Scholar] [CrossRef]
- Ogata, A.; Hirano, T.; Hishitani, Y.; Tanaka, T. Safety and Efficacy of Tocilizumab for the Treatment of Rheumatoid Arthritis. Clin. Med. Insights: Arthritis Musculoskelet. Disord. 2012, 5, 27–42. [Google Scholar] [CrossRef]
- Ogata, A.; Kato, Y.; Higa, S.; Yoshizaki, K. IL-6 inhibitor for the treatment of rheumatoid arthritis: A comprehensive review. Mod. Rheumatol. 2019, 29, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Rafique, A.; Martin, J.; Blome, M.; Huang, T.; Ouyang, A.; Papadopoulos, N. AB0037 Evaluation of the binding kinetics and functional bioassay activity of sarilumab and tocilizumab to the human il-6 receptor (il-6r) alpha. Ann. Rheum. Dis. 2013, 72, 797. [Google Scholar] [CrossRef]
- Raimondo, M.G.; Biggioggero, M.; Crotti, C.; Becciolini, A.; Favalli, E.G. Profile of sarilumab and its potential in the treatment of rheumatoid arthritis. Drug Des. Dev. Ther. 2017, 11, 1593–1603. [Google Scholar] [CrossRef] [PubMed]
- Emery, P.; Rondon, J.; Parrino, J.; Lin, Y.; Pena-Rossi, C.; Van Hoogstraten, H.; Graham, N.M.H.; Liu, N.; Paccaly, A.; Wu, R.; et al. Safety and tolerability of subcutaneous sarilumab and intravenous tocilizumab in patients with rheumatoid arthritis. Rheumatology 2019, 58, 849–858. [Google Scholar] [CrossRef]
- Liang, B.; Song, Z.; Wu, B.; Gardner, D.; Shealy, D.J.; Song, X.-Y.; Wooley, P.H. Evaluation of anti-IL-6 monoclonal antibody therapy using murine type II collagen-induced arthritis. J. Inflamm. 2009, 6, 10. [Google Scholar] [CrossRef]
- Huizinga, T.; Fleischmann, R.M.; Jasson, M.; Radin, A.R.; Van Adelsberg, J.; Fiore, S.; Huang, X.; Yancopoulos, G.D.; Stahl, N.; Genovese, M.C. Sarilumab, a fully human monoclonal antibody against IL-6Rα in patients with rheumatoid arthritis and an inadequate response to methotrexate: Efficacy and safety results from the randomised SARIL-RA-MOBILITY Part A trial. Ann. Rheum. Dis. 2013, 73, 1626–1634. [Google Scholar] [CrossRef]
- Genovese, M.C.; Fleischmann, R.; Kivitz, A.J.; Rell-Bakalarska, M.; Martincova, R.; Fiore, S.; Rohane, P.; Van Hoogstraten, H.; Garg, A.; Fan, C.; et al. Sarilumab plus Methotrexate in Patients With Active Rheumatoid Arthritis and Inadequate Response to Methotrexate: Results of a Phase III Study. Arthritis Rheumatol. 2015, 67, 1424–1437. [Google Scholar] [CrossRef]
- Smolen, J.S.; Weinblatt, M.E.; Sheng, S.; Zhuang, Y.; Hsu, B. Sirukumab, a human anti-interleukin-6 monoclonal antibody: A randomised, 2-part (proof-of-concept and dose-finding), phase II study in patients with active rheumatoid arthritis despite methotrexate therapy. Ann. Rheum. Dis. 2014, 73, 1616–1625. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, D.; Salvadore, G.; Hsu, B.; Curran, M.; Casper, C.; Vermeulen, J.; Kent, J.M.; Singh, J.; Drevets, W.C.; et al. The effects of interleukin-6 neutralizing antibodies on symptoms of depressed mood and anhedonia in patients with rheumatoid arthritis and multicentric Castleman’s disease. Brain behav. Immun. 2017, 66, 156–164. [Google Scholar] [CrossRef]
- Bozec, A.; Luo, Y.; Engdahl, C.; Figueiredo, C.P.; Bang, H.; Schett, G. Abatacept blocks anti-citrullinated protein antibody and rheumatoid factor mediated cytokine production in human macrophages in IDO-dependent manner. Arthritis Res. 2018, 20, 24. [Google Scholar] [CrossRef]
- Maxwell, L.J.; Singh, J. Abatacept for Rheumatoid Arthritis: A Cochrane Systematic Review. J. Rheumatol. 2010, 37, 234–245. [Google Scholar] [CrossRef] [PubMed]
- Peichl, P.; Alten, R.; Galeazzi, M.; Lorenz, H.-M.; Nüßlein, H.; Navarro, F.; Elbez, Y.; Chartier, M.; Hackl, R.; Rauch, C.; et al. Abatacept retention and clinical outcomes in Austrian patients with rheumatoid arthritis: Real-world data from the 2-year ACTION study. Wien. Med. Wochenschr. 2019, 170, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Okada, H.; Kajiya, H.; Omata, Y.; Matsumoto, T.; Sato, Y.; Kobayashi, T.; Nakamura, S.; Kaneko, Y.; Nakamura, S.; Koyama, T.; et al. CTLA4-Ig Directly Inhibits Osteoclastogenesis by Interfering With Intracellular Calcium Oscillations in Bone Marrow Macrophages. J. Bone Miner. Res. 2019, 34, 1744–1752. [Google Scholar] [CrossRef]
- Zou, Q.-F.; Li, L.; Han, Q.-R.; Wang, Y.-J.; Wang, X.-B. Abatacept alleviates rheumatoid arthritis development by inhibiting migration of fibroblast-like synoviocytes via MAPK pathway. Eur. Rev. Med. Pharmacol. Sci. 2019, 23, 3105–3111. [Google Scholar]
- Lorenzetti, R.; Janowska, I.; Smulski, C.R.; Frede, N.; Henneberger, N.; Walter, L.; Schleyer, M.-T.; Hüppe, J.M.; Staniek, J.; Salzer, U.; et al. Abatacept modulates CD80 and CD86 expression and memory formation in human B-cells. J. Autoimmun. 2019, 101, 145–152. [Google Scholar] [CrossRef] [PubMed]
- Scarsi, M.; Paolini, L.; Ricotta, D.; Pedrini, A.; Piantoni, S.; Caimi, L.; Tincani, A.; Airò, P. Abatacept Reduces Levels of Switched Memory B Cells, Autoantibodies, and Immunoglobulins in Patients with Rheumatoid Arthritis. J. Rheumatol. 2014, 41, 666–672. [Google Scholar] [CrossRef] [PubMed]
- Sokolove, J.; Schiff, M.; Fleischmann, R.; Weinblatt, M.E.; Connolly, S.E.; Johnsen, A.; Zhu, J.; Maldonado, M.A.; Patel, S.; Robinson, W.H. Robinson, Impact of baseline anti-cyclic citrullinated peptide-2 antibody concentration on efficacy outcomes following treatment with subcutaneous abatacept or adalimumab: 2-year results from the AMPLE trial. Ann. Rheum. Dis. 2015, 75, 709–714. [Google Scholar]
- Alten, R.; Nüßlein, H.G.; Mariette, X.; Galeazzi, M.; Lorenz, H.-M.; Cantagrel, A.; Chartier, M.; Poncet, C.; Rauch, C.; Le Bars, M. Baseline autoantibodies preferentially impact abatacept efficacy in patients with rheumatoid arthritis who are biologic naïve: 6-month results from a real-world, international, prospective study. Rmd Open 2017, 3, e000345. [Google Scholar] [CrossRef]
- Genovese, M.C.; Tena, C.P.; Covarrubias, A.; Leon, G.; Mysler, E.; Keiserman, M.; Valente, R.; Nash, P.; Simon-Campos, J.A.; Box, J.; et al. Subcutaneous Abatacept for the Treatment of Rheumatoid Arthritis: Longterm Data from the ACQUIRE Trial. J. Rheumatol. 2014, 41, 629–639. [Google Scholar] [CrossRef]
- Westhovens, R.; Robles, M.; Ximenes, A.C.; Nayiager, S.; Wollenhaupt, J.; Durez, P.; Gomez-Reino, J.; Grassi, W.; Haraoui, B.; Shergy, W.; et al. Clinical efficacy and safety of abatacept in methotrexate-naive patients with early rheumatoid arthritis and poor prognostic factors. Ann. Rheum. Dis. 2009, 68, 1870–1877. [Google Scholar] [CrossRef]
- Emery, P.; Burmester, G.R.; Bykerk, V.P.; Combe, B.G.; Furst, D.E.; Barré, E.; Wong, D.A.; Huizinga, T.W.J. Evaluating drug-free remission with abatacept in early rheumatoid arthritis: Results from the phase 3b, multicentre, randomised, active-controlled AVERT study of 24 months, with a 12-month, double-blind treatment period. Ann. Rheum. Dis. 2014, 74, 19–26. [Google Scholar] [PubMed]
- Fleischmann, R.; Weinblatt, M.E.; Schiff, M.; Khanna, D.; Maldonado, M.A.; Nadkarni, A.; Furst, D.E. Patient-Reported Outcomes From a Two-Year Head-to-Head Comparison of Subcutaneous Abatacept and Adalimumab for Rheumatoid Arthritis. Arthritis Rheum. 2016, 68, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Schiff, M.; Weinblatt, M.E.; Valente, R.; Van Der Heijde, D.; Citera, G.; Elegbe, A.; Maldonado, M.; Fleischmann, R. Head-to-head comparison of subcutaneous abatacept versus adalimumab for rheumatoid arthritis: Two-year efficacy and safety findings from AMPLE trial. Ann. Rheum. Dis. 2013, 73, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Alemao, E.; Johal, S.; Al, M.J.; Molken, M.R.-V. Cost-Effectiveness Analysis of Abatacept Compared with Adalimumab on Background Methotrexate in Biologic-Naive Adult Patients with Rheumatoid Arthritis and Poor Prognosis. Value Heal. 2018, 21, 193–202. [Google Scholar] [CrossRef]
- Blair, H.A.; Deeks, E.D. Abatacept: A Review in Rheumatoid Arthritis. Drugs 2017, 77, 1221–1233. [Google Scholar] [CrossRef]
- Ogawa, N.; Ohashi, H.; Ota, Y.; Kobori, K.; Suzuki, M.; Tsuboi, S.; Hayakawa, M.; Goto, Y.; Karahashi, T.; Kimoto, O.; et al. Multicenter, observational clinical study of abatacept in Japanese patients with rheumatoid arthritis. Immunol. Med. 2019, 42, 29–38. [Google Scholar] [CrossRef]
- Ozen, G.; Pedro, S.; Schumacher, R.; Simon, T.A.; Michaud, K. Safety of abatacept compared with other biologic and conventional synthetic disease-modifying antirheumatic drugs in patients with rheumatoid arthritis: Data from an observational study. Arthritis Res. 2019, 21, 141. [Google Scholar] [CrossRef]
- Ramwadhdoebe, T.H.; Van Baarsen, L.; Boumans, M.J.H.; Bruijnen, S.; Safy, M.; Berger, F.H.; Semmelink, J.F.; Van Der Laken, C.J.; Gerlag, D.M.; Thurlings, R.M.; et al. Effect of rituximab treatment on T and B cell subsets in lymph node biopsies of patients with rheumatoid arthritis. Rheumatology 2019, 58, 1075–1085. [Google Scholar] [CrossRef]
- Finckh, A.; Simard, J.F.; Duryea, J.; Liang, M.H.; Huang, J.; Daneel, S.; Forster, A.; Gabay, C.; Guerne, P.-A. For the Swiss Clinical Quality Management in Rheumatoid Arthritis Project The effectiveness of anti–tumor necrosis factor therapy in preventing progressive radiographic joint damage in rheumatoid arthritis: A population-based study. Arthritis Rheum. 2005, 54, 54–59. [Google Scholar] [CrossRef]
- Edwards, J.C.W.; Szczepanski, L.; Szchechinski, J.; Filipowiz-Sosonowska, A.; Emery, P.; Close, D.R.; Stevens, R.M.; Shaw, T. Efficacy of B-cell-targeted therapy with rituximab in patients with rheumatoid arthritis. N. Engl. J. Med. 2004, 350, 2572–2581. [Google Scholar] [CrossRef]
- Tavakolpour, S.; AleSaeidi, S.; Darvishi, M.; Ghasemiadl, M.; Darabi-Monadi, S.; Akhlaghdoust, M.; Behjati, S.E.; Jafarieh, A. A comprehensive review of rituximab therapy in rheumatoid arthritis patients. Clin. Rheumatol. 2019, 38, 2977–2994. [Google Scholar] [CrossRef] [PubMed]
- Pollastro, S.; Klarenbeek, P.L.; Doorenspleet, B.D.C.; Esveldt, R.E.E.; Thurlings, R.M.; Boumans, M.J.H.; Gerlag, D.M.; Tak, P.P.; Vos, K. Non-response to rituximab therapy in rheumatoid arthritis is associated with incomplete disruption of the B cell receptor repertoire. Ann. Rheum. Dis. 2019, 78, 1339–1345. [Google Scholar]
- Plosker, G.L.; Figgitt, D.P. Rituximab: A review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs 2003, 63, 803–843. [Google Scholar] [CrossRef] [PubMed]
- Pescovitz, M.D. Rituximab, an Anti-CD20 Monoclonal Antibody: History and Mechanism of Action. Arab. Archaeol. Epigr. 2006, 6, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Ramiro, S.; Gaujoux-Viala, C.; Nam, J.L.; Smolen, J.S.; Buch, M.; Gossec, L.; Van Der Heijde, D.; Winthrop, K.; Landewé, R. Safety of synthetic and biological DMARDs: A systematic literature review informing the 2013 update of the EULAR recommendations for management of rheumatoid arthritis. Ann. Rheum. Dis. 2014, 73, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Bongartz, T.; Sutton, A.J.; Sweeting, M.; Buchan, I.; Matteson, E.L.; Montori, V.M. Anti-TNF Antibody Therapy in Rheumatoid Arthritis and the Risk of Serious Infections and Malignancies. JAMA 2006, 295, 2275. [Google Scholar] [CrossRef]
- Mohan, N.; Edwards, E.T.; Cupps, T.R.; Oliverio, P.J.; Sandberg, G.; Crayton, H.; Richert, J.R.; Siegel, J.N. Demyelination occurring during anti-tumor necrosis factor α therapy for inflammatory arthritides. Arthritis Rheum. 2001, 44, 2862–2869. [Google Scholar] [CrossRef]
- Cohen, S.B.; Tanaka, Y.; Mariette, X.; Curtis, J.R.; Lee, E.B.; Nash, P.; Winthrop, K.L.; Charles-Schoeman, C.; Thirunavukkarasu, K.; Demasi, R.; et al. Long-term safety of tofacitinib for the treatment of rheumatoid arthritis up to 8.5 years: Integrated analysis of data from the global clinical trials. Ann. Rheum. Dis. 2017, 76, 1253–1262. [Google Scholar] [CrossRef]
- Tanaka, Y.; Hirata, S.; Kubo, S.; Fukuyo, S.; Hanami, K.; Sawamukai, N.; Nakano, K.; Nakayamada, S.; Yamaoka, K.; Sawamura, F.; et al. Discontinuation of adalimumab after achieving remission in patients with established rheumatoid arthritis: 1-year outcome of the HONOR study. Ann. Rheum. Dis. 2013, 74, 389–395. [Google Scholar] [CrossRef]
- Wassenberg, S.; Rau, R.; Steinfeld, P.; Zeidler, H. Low-Dose Prednisolone Therapy Study Group Very low-dose prednisolone in early rheumatoid arthritis retards radiographic progression over two years: A multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2005, 52, 3371–3380. [Google Scholar] [CrossRef]
- Julsgaard, M.; Christensen, L.A.; Gibson, P.R.; Gearry, R.B.; Fallingborg, J.; Hvas, C.L.; Bibby, B.M.; Uldbjerg, N.; Connell, W.; Rosella, O.; et al. Concentrations of Adalimumab and Infliximab in Mothers and Newborns, and Effects on Infection. Gastroenterology 2016, 151, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Förger, F.; Zbinden, A.; Villiger, P.M. Certolizumab treatment during late pregnancy in patients with rheumatic diseases: Low drug levels in cord blood but possible risk for maternal infections. A case series of 13 patients. Jt. Bone Spine 2016, 83, 341–343. [Google Scholar] [CrossRef] [PubMed]
- Mariette, X.; Förger, F.; Abraham, B.; Flynn, A.D.; Molto, A.; Flipo, R.-M.; Van Tubergen, A.; Shaughnessy, L.; Simpson, J.; Teil, M.; et al. Lack of placental transfer of certolizumab pegol during pregnancy: Results from CRIB, a prospective, postmarketing, pharmacokinetic study. Ann. Rheum. Dis. 2017, 77, 228–233. [Google Scholar] [CrossRef] [PubMed]
- Clowse, M.E.; Wolf, U.C.; Förger, F.; Cush, J.J.; Golembesky, A.; Shaughnessy, L.; De Cuyper, D.; Mahadevan, U. Pregnancy Outcomes in Subjects Exposed to Certolizumab Pegol. J. Rheumatol. 2015, 42, 2270–2278. [Google Scholar] [CrossRef] [PubMed]
- Shimada, H.; Kameda, T.; Kanenishi, K.; Miyatake, N.; Nakashima, S.; Wakiya, R.; Kato, M.; Miyagi, T.; Mansour, M.M.F.; Hata, T.; et al. Effect of biologic disease-modifying anti-rheumatic drugs for patients with rheumatoid arthritis who hope to become mothers. Clin. Rheumatol. 2019, 38, 1453–1458. [Google Scholar] [CrossRef] [PubMed]
- Komaki, F.; Komaki, Y.; Micic, D.; Ido, A.; Sakuraba, A. Outcome of pregnancy and neonatal complications with anti-tumor necrosis factor-α use in females with immune mediated diseases; a systematic review and meta-analysis. J. Autoimmun. 2017, 76, 38–52. [Google Scholar] [CrossRef]
- Manova, M.; Savova, A.; Vasileva, M.; Terezova, S.; Kamusheva, M.; Grekova, D.; Petkova, V.; Petrova, G. Comparative Price Analysis of Biological Products for Treatment of Rheumatoid Arthritis. Front. Pharmacol. 2018, 9, 1070. [Google Scholar] [CrossRef]
- Klontzas, M.E.; Kenanidis, E.I.; Heliotis, M.; Tsiridis, E.; Mantalaris, A. Bone and cartilage regeneration with the use of umbilical cord mesenchymal stem cells. Expert Opin. Boil. Ther. 2015, 15, 1541–1552. [Google Scholar] [CrossRef]
- Di Nicola, M.; Carlo-Stella, C.; Magni, M.; Milanesi, M.; Longoni, P.D.; Matteucci, P.; Grisanti, S.; Gianni, A.M. Human bone marrow stromal cells suppress T-lymphocyte proliferation induced by cellular or nonspecific mitogenic stimuli. Blood 2002, 99, 3838–3843. [Google Scholar] [CrossRef]
- Krampera, M.; Glennie, S.; Dyson, J.; Scott, D.; Laylor, R.; Simpson, E.; Dazzi, F. Bone marrow mesenchymal stem cells inhibit the response of naive and memory antigen-specific T cells to their cognate peptide. Blood 2003, 101, 3722–3729. [Google Scholar] [CrossRef]
- Aggarwal, S.; Pittenger, M.F. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood 2005, 105, 1815–1822. [Google Scholar] [CrossRef]
- Zheng, Z.H.; Li, X.Y.; Ding, J.; Jia, J.F.; Zhu, P. Allogeneic mesenchymal stem cell and mesenchymal stem cell-differentiated chondrocyte suppress the responses of type II collagen-reactive T cells in rheumatoid arthritis. Rheumatology 2008, 47, 22–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Li, Q.; Zhu, J.; Guo, H.; Zhai, Q.; Li, B.; Jin, Y.; He, X.; Jin, F. Comparison of therapeutic effects of different mesenchymal stem cells on rheumatoid arthritis in mice. PeerJ 2019, 7, e7023. [Google Scholar] [CrossRef] [PubMed]
- Park, E.H.; Lim, H.-S.; Lee, S.; Roh, K.; Seo, K.-W.; Kang, K.-S.; Shin, K. Intravenous Infusion of Umbilical Cord Blood-Derived Mesenchymal Stem Cells in Rheumatoid Arthritis: A Phase Ia Clinical Trial. Stem Cells Transl. Med. 2018, 7, 636–642. [Google Scholar] [CrossRef] [PubMed]
- Ghoryani, M.; Shariati-Sarabi, Z.; Afshari, J.T.; Ghasemi, A.; Poursamimi, J.; Mohammadi, M. Amelioration of clinical symptoms of patients with refractory rheumatoid arthritis following treatment with autologous bone marrow-derived mesenchymal stem cells: A successful clinical trial in Iran. Biomed. Pharmacother. 2018, 109, 1834–1840. [Google Scholar] [CrossRef]
- Swanson, K.V.; Deng, M.; Ting, J.P.-Y. The NLRP3 inflammasome: Molecular activation and regulation to therapeutics. Nat. Rev. Immunol. 2019, 19, 477–489. [Google Scholar] [CrossRef]
- Kolly, L.; Busso, N.; Palmer, G.; Talabot-Ayer, D.; Chobaz, V.; So, A. Expression and function of the NALP3 inflammasome in rheumatoid synovium. Immunology 2010, 129, 178–185. [Google Scholar] [CrossRef]
- Guo, C.; Fu, R.; Wang, S.; Huang, Y.; Li, X.; Zhou, M.; Zhao, J.; Yang, N. NLRP3 inflammasome activation contributes to the pathogenesis of rheumatoid arthritis. Clin. Exp. Immunol. 2018, 194, 231–243. [Google Scholar] [CrossRef]
- Williamson, D.J.; Begley, C.G.; Vadas, M.A.; Metcalf, D. The detection and initial characterization of colony-stimulating factors in synovial fluid. Clin. Exp. Immunol. 1988, 72, 67–73. [Google Scholar]
- Xu, W.D.; Firestein, G.S.; Taetle, R.; Kaushansky, K.; Zvaifler, N.J. Cytokines in chronic inflammatory arthritis. II. Granulocyte-macrophage colony-stimulating factor in rheumatoid synovial effusions. J. Clin. Investig. 1989, 83, 876–882. [Google Scholar] [CrossRef]
- Berenbaum, F.; Rajzbaum, G.; Amor, B.; Toubert, A. Evidence for GM-CSF receptor expression in synovial tissue. An analysis by semi-quantitative polymerase chain reaction on rheumatoid arthritis and osteoarthritis synovial biopsies. Eur. Cytokine Netw. 1994, 5, 43–46. [Google Scholar] [PubMed]
- Cook, A.D.; Braine, E.L.; Campbell, I.K.; Rich, M.J.; Hamilton, J.A. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): Requirement for GM-CSF in the effector phase of disease. Arthritis Res. 2001, 3, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Plater-Zyberk, C.; Joosten, L.A.B.; Helsen, M.M.A.; Hepp, J.; Baeuerle, P.A.; Berg, W.B.V.D. GM-CSF neutralisation suppresses inflammation and protects cartilage in acute streptococcal cell wall arthritis of mice. Ann. Rheum. Dis. 2006, 66, 452–457. [Google Scholar] [CrossRef]
- Burmester, G.; Weinblatt, M.E.; McInnes, I.; Porter, D.; Barbarash, O.; Vatutin, M.; Szombati, I.; Esfandiari, E.; Sleeman, M.A.; Kane, C.; et al. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Ann. Rheum. Dis. 2012, 72, 1445–1452. [Google Scholar] [CrossRef]
- Burmester, G.R.; McInnes, I.; Kremer, J.; Miranda, P.; Korkosz, M.; Vencovský, J.; Rubbert-Roth, A.; Mysler, E.; Sleeman, M.A.; Godwood, A.; et al. A randomised phase IIb study of mavrilimumab, a novel GM–CSF receptor alpha monoclonal antibody, in the treatment of rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1020–1030. [Google Scholar] [CrossRef]
- Šenolt, L. Emerging therapies in rheumatoid arthritis: Focus on monoclonal antibodies. F1000Research 2019, 8, 1549. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.C.; for the NEXUS Study Group; Saurigny, D.; Vencovský, J.; Takeuchi, T.; Nakamura, T.; Matsievskaia, G.; Hunt, B.; Wagner, T.; Souberbielle, B.; et al. Efficacy and safety of namilumab, a human monoclonal antibody against granulocyte-macrophage colony-stimulating factor (GM-CSF) ligand in patients with rheumatoid arthritis (RA) with either an inadequate response to background methotrexate therapy or an inadequate response or intolerance to an anti-TNF (tumour necrosis factor) biologic therapy: A randomized, controlled trial. Arthritis Res. 2019, 21, 101. [Google Scholar]
- Roelofs, M.F.; Boelens, W.C.; Joosten, L.; Abdollahi-Roodsaz, S.; Geurts, J.; Wunderink, L.U.; Schreurs, B.W.; Berg, W.B.V.D.; Radstake, T.R.D.J. Identification of small heat shock protein B8 (HSP22) as a novel TLR4 ligand and potential involvement in the pathogenesis of rheumatoid arthritis. J. Immunol. 2006, 176, 7021–7027. [Google Scholar] [CrossRef]
- Midwood, K.S.; Sacre, S.; Piccinini, A.M.; Inglis, J.; Trebaul, A.; Chan, E.; Drexler, S.; Sofat, N.; Kashiwagi, M.; Orend, G.; et al. Tenascin-C is an endogenous activator of Toll-like receptor 4 that is essential for maintaining inflammation in arthritic joint disease. Nat. Med. 2009, 15, 774–780. [Google Scholar] [CrossRef]
- Pierer, M.; Wagner, U.; Rossol, M.; Ibrahim, S. Toll-Like Receptor 4 Is Involved in Inflammatory and Joint Destructive Pathways in Collagen-Induced Arthritis in DBA1J Mice. PLoS ONE 2011, 6, e23539. [Google Scholar] [CrossRef]
- Monnet, E.; Choy, E.H.; McInnes, I.; Kobakhidze, T.; De Graaf, K.; Jacqmin, P.; Lapeyre, G.; De Min, C. Efficacy and safety of NI-0101, an anti-toll-like receptor 4 monoclonal antibody, in patients with rheumatoid arthritis after inadequate response to methotrexate: A phase II study. Ann. Rheum. Dis. 2019, 79, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Erickson, A.R.; O’Dell, J.R. Lessons Learned From the RACAT Trial: A Comparison of Rheumatoid Arthritis Therapies. Fed. Pr. 2016, 33, 17–21. [Google Scholar]
- Ducreux, J.; Durez, P.; Galant, C.; Toukap, A.N.; Eynde, B.V.D.; Houssiau, F.A.; Lauwerys, B.R. Global Molecular Effects of Tocilizumab Therapy in Rheumatoid Arthritis Synovium. Arthritis Rheumatol. 2013, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Szumski, A.; Koening, A.S.; Jones, T.V.; Marchall, L. Predictors of remission with etanercept-methotrexate induction therapy and loss of remission with etanercept maintenance, reduction, or withdrawal in moderately active rheumatoid arthritis: Results of the PRESERVE trial. Arthritis Res. Ther. 2018, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Smolen, J.S.; Nash, P.; Durez, P.; Hall, S.; Ilivanoca, E.; Irazoque-Palazuelos, F.; Miranda, P.; Park, M.C.; Pavelka, K.; Pedersen, R.; et al. Maintenance, reduction, or withdrawal of etanercept after treatment with etanercept and methotrexate in patients with moderate rheumatoid arthritis (PRESERVE): A randomised controlled trial. Lancet Lond. Engl. 2013, 381, 918–929. [Google Scholar] [CrossRef]
- Bos, W.H.; Dijkmans, B.A.C.; Boers, M.; Van De Stadt, R.J.; Van Schaardenburg, D. Effect of dexamethasone on autoantibody levels and arthritis development in patients with arthralgia: A randomised trial. Ann. Rheum. Dis. 2009, 69, 571–574. [Google Scholar] [CrossRef]
- Machold, K.; Landewé, R.; Ramiro, S.; Stamm, T.A.; Van Der Heijde, D.; Verpoort, K.N.; Brickmann, K.; Vázquez-Mellado, J.; Karateev, D.; Breedveld, F.C.; et al. The Stop Arthritis Very Early (SAVE) trial, an international multicentre, randomised, double-blind, placebo-controlled trial on glucocorticoids in very early arthritis. Ann. Rheum. Dis. 2010, 69, 495–502. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, Y.-J.; Anzaghe, M.; Schülke, S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells 2020, 9, 880. https://doi.org/10.3390/cells9040880
Lin Y-J, Anzaghe M, Schülke S. Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells. 2020; 9(4):880. https://doi.org/10.3390/cells9040880
Chicago/Turabian StyleLin, Yen-Ju, Martina Anzaghe, and Stefan Schülke. 2020. "Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis" Cells 9, no. 4: 880. https://doi.org/10.3390/cells9040880
APA StyleLin, Y.-J., Anzaghe, M., & Schülke, S. (2020). Update on the Pathomechanism, Diagnosis, and Treatment Options for Rheumatoid Arthritis. Cells, 9(4), 880. https://doi.org/10.3390/cells9040880