High-Capacity Adenoviral Vectors Permit Robust and Versatile Testing of DMD Gene Repair Tools and Strategies in Human Cells


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
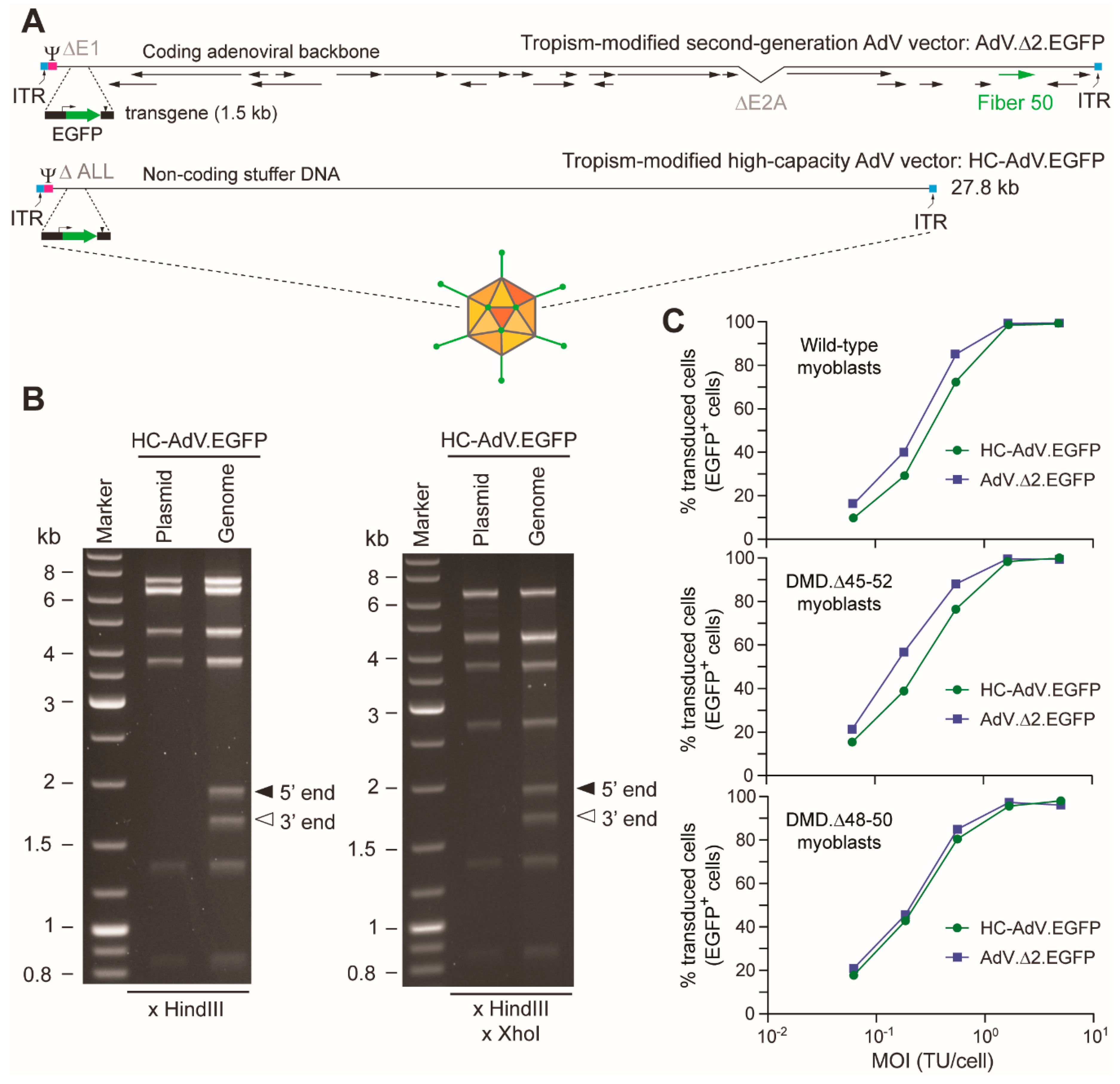
2.2. Production and Characterization of Adenoviral Vectors
2.3. Transduction Experiments
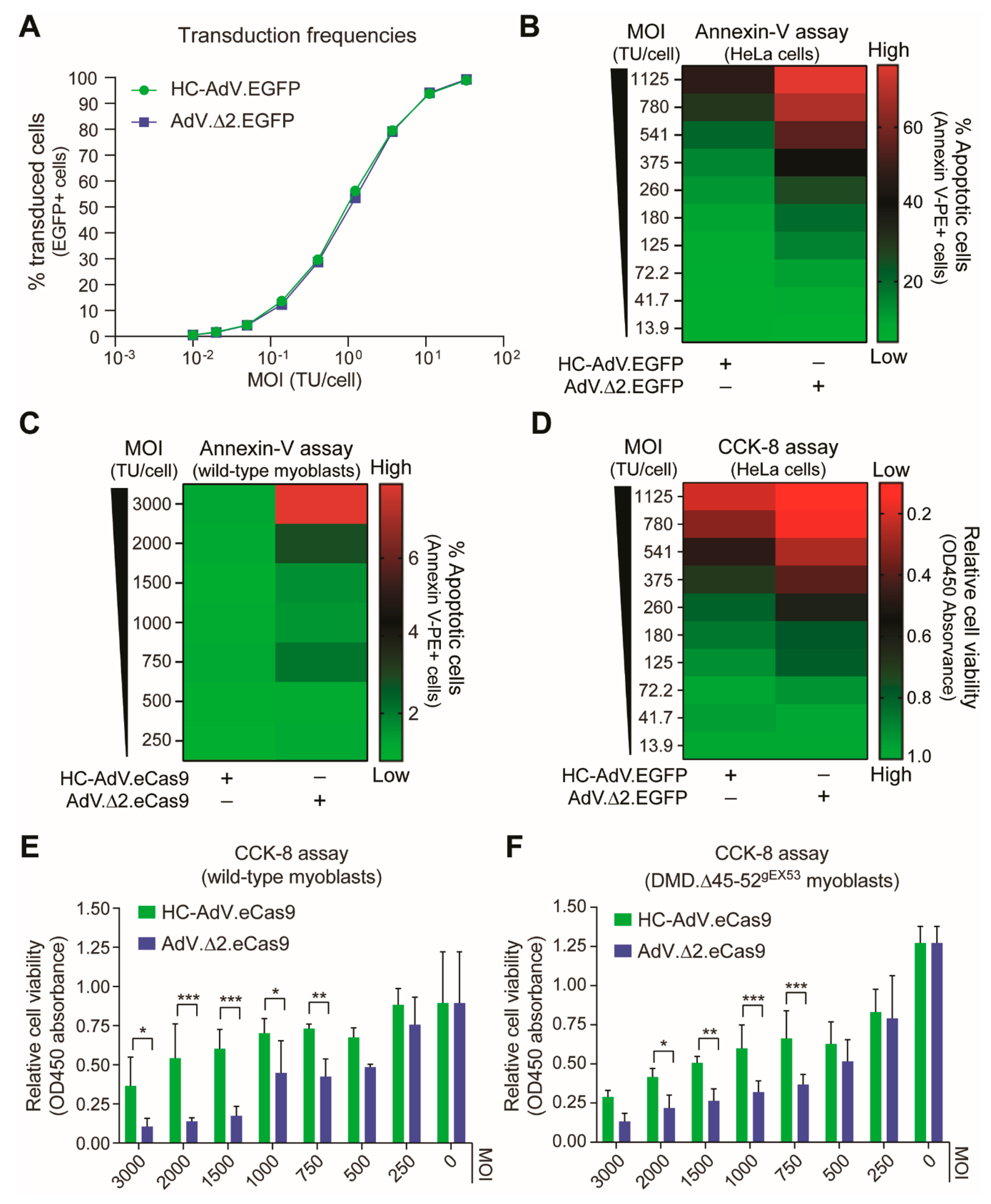
2.4. Cell Viability Assays
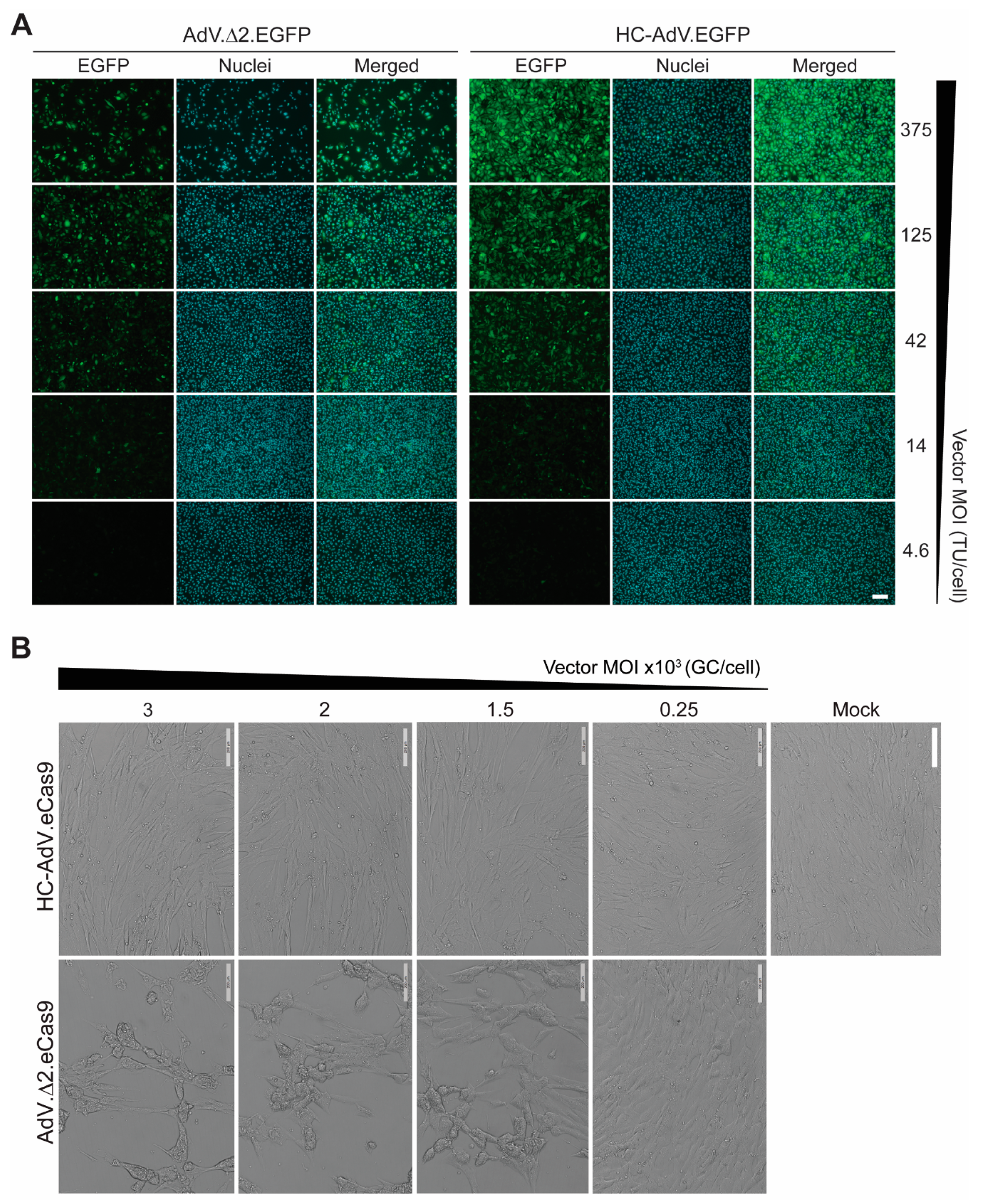
2.5. Live-Cell Light Microscopy
2.6. Flow Cytometry
2.7. qPCR Assays
2.8. Immunofluorescence Microscopy Analyses
2.9. Western Blot Analysis
2.10. Statistical Analyses
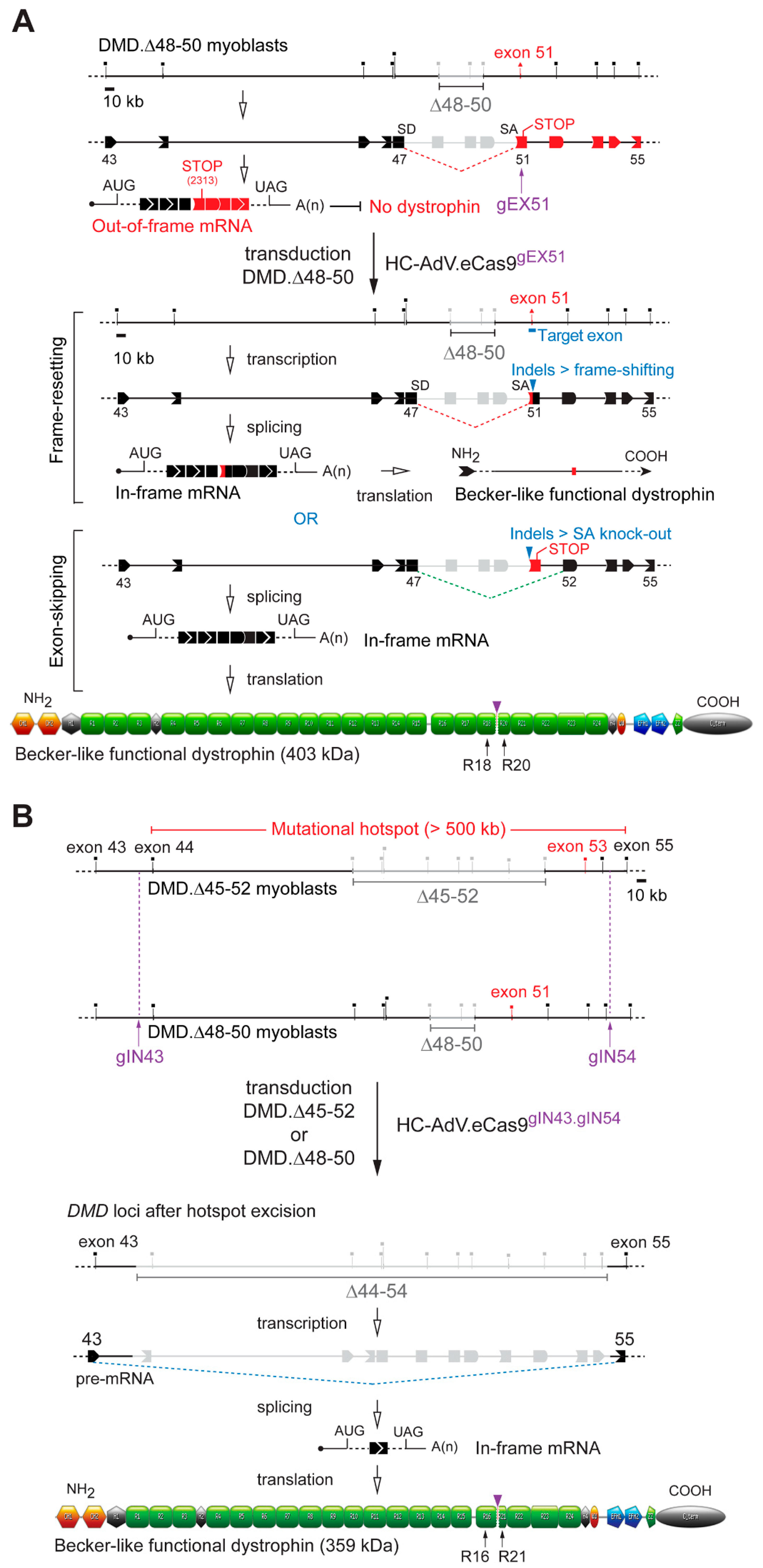
3. Results
3.1. At High Doses HC-AdVs Are Significantly Less Cytotoxic Than Second-Generation AdVs
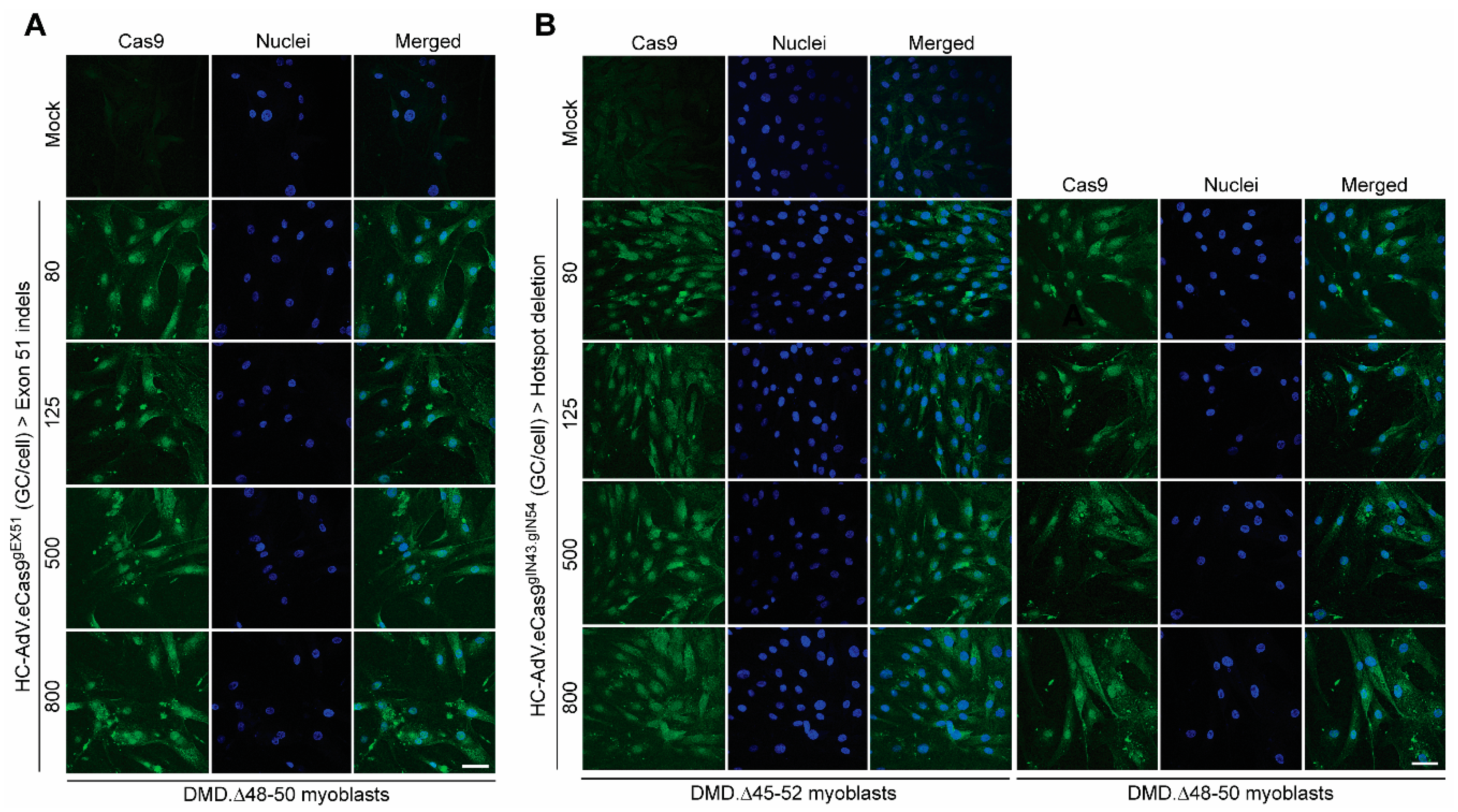
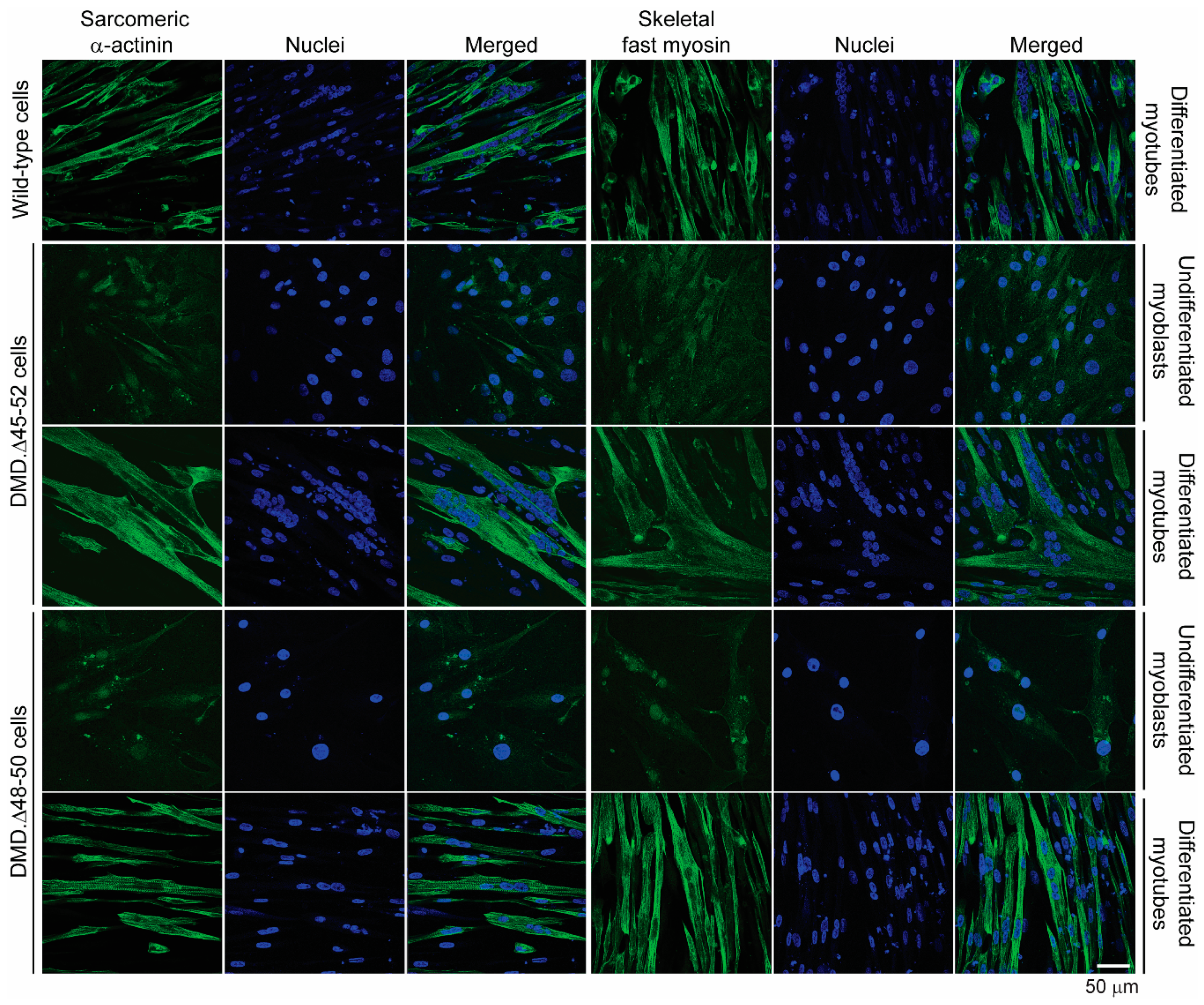
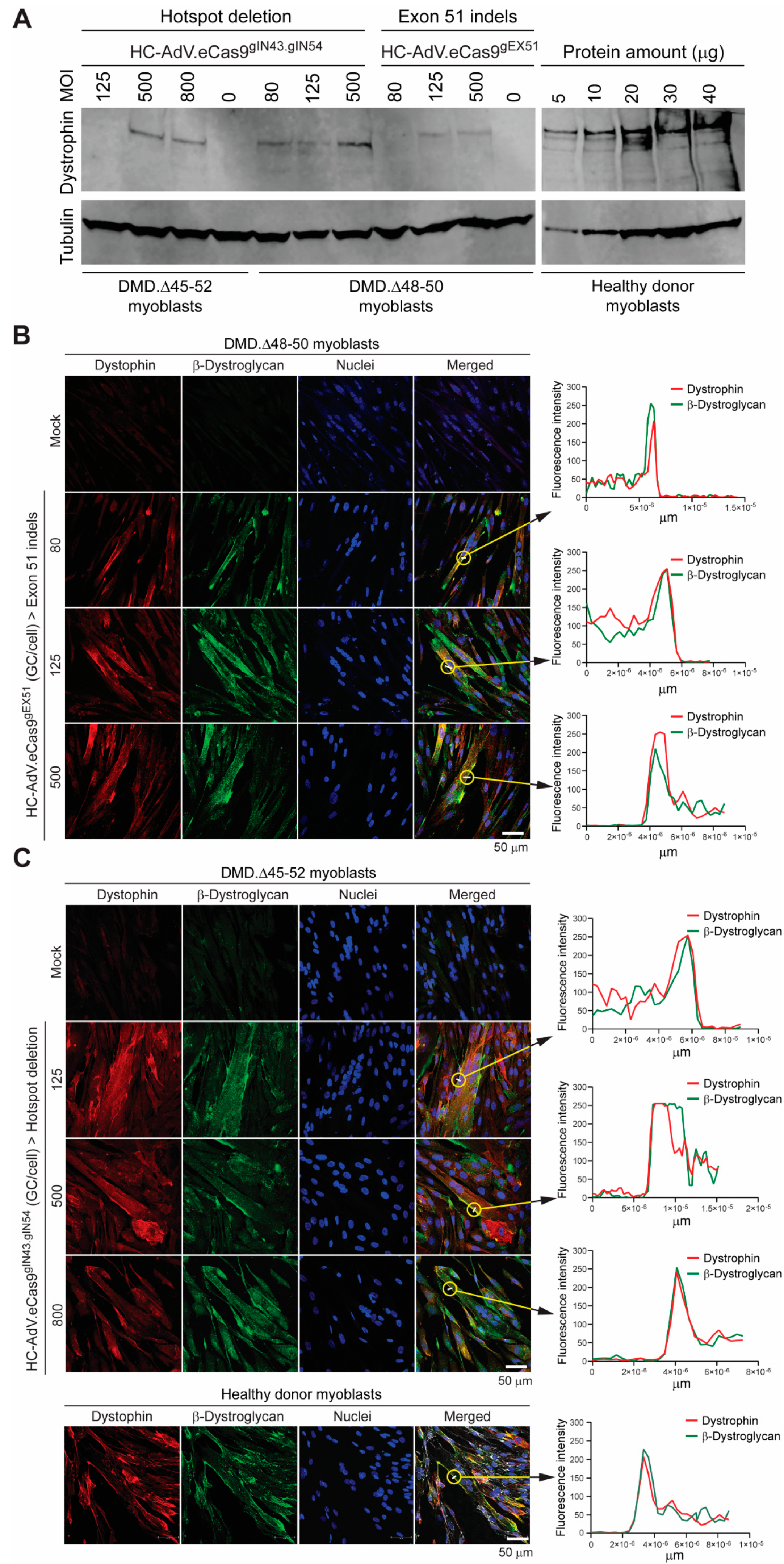
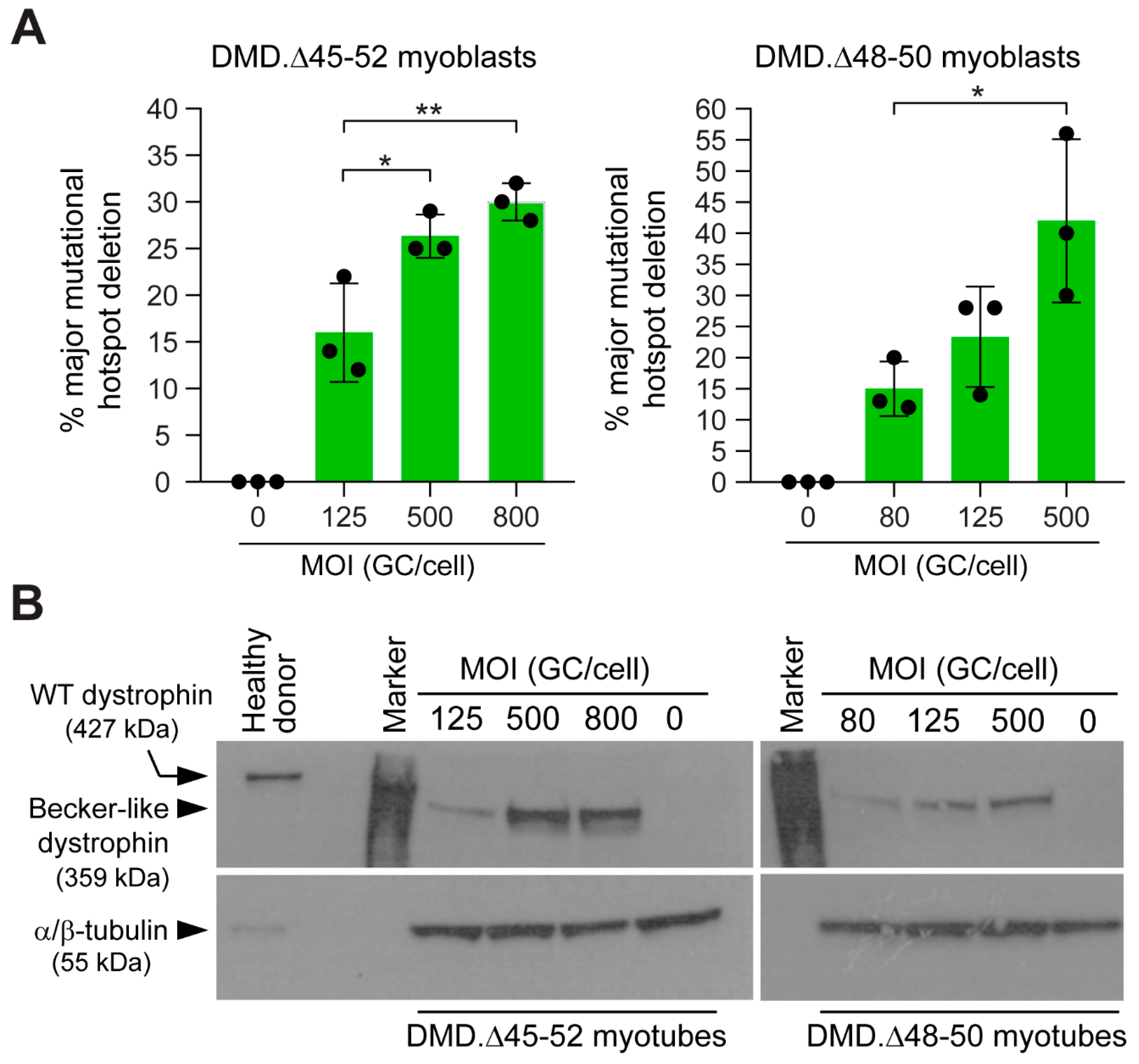
3.2. HC-AdVs Encoding Single or Dual RGNs Induce Robust Dystrophin Synthesis in Unselected DMD Muscle Cell Populations
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mah, J.K.; Korngut, L.; Fiest, K.M.; Dykeman, J.; Day, L.J.; Pringsheim, T.; Jette, N. A systematic review and meta-analysis on the epidemiology of the muscular dystrophies. Can. J. Neurol. Sci. 2016, 43, 163–177. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, E.P.; Brown Jr, R.H.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef]
- Hoffman, E.P.; Fischbeck, K.H.; Brown, R.H.; Johnson, M.; Medori, R.; Loire, J.D.; Harris, J.B.; Waterston, R.; Brooke, M.; Specht, L. Characterization of dystrophin in muscle-biopsy specimens from patients with Duchenne’s or Becker’s muscular dystrophy. N. Engl. J. Med. 1988, 318, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Ahn, A.H.; Kunkel, L.M. The structural and functional diversity of dystrophin. Nat. Gen. 1993, 3, 283–291. [Google Scholar] [CrossRef]
- Guiraud, S.; Aartsma-Rus, A.; Vieira, N.M.; Davies, K.E.; van Ommen, G.-J.B.; Kunkel, L.M. The pathogenesis and therapy of muscular dystrophies. Annu. Rev. Genomics Hum. Gen. 2015, 16, 281–308. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7,000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- Koenig, M.; Beggs, A.H.; Moyer, M.; Scherpf, S.; Heindrich, K.; Bettecken, T.; Meng, G.; Müller, C.R.; Lindlöf, M.; Kaariainen, H.; et al. The molecular basis for Duchenne versus Becker muscular dystrophy: Correlation of severity with type of deletion. Am. J. Hum. Genet. 1989, 45, 498–506. [Google Scholar]
- Verhaart, I.E.; Aartsma-Rus, A. Therapeutic developments for Duchenne muscular dystrophy. Nat. Rev. Neurol. 2019, 15, 373–386. [Google Scholar] [CrossRef]
- Nelson, C.E.; Robinson-Hamm, J.N.; Gersbach, C.A. Genome engineering: A new approach to gene therapy for neuromuscular disorders. Nat. Rev. Neurol. 2017, 13, 647–661. [Google Scholar] [CrossRef]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef]
- Doudna, J.A.; Charpentier, E. The new frontier of genome engineering with CRISPR-Cas9. Science 2014, 346, 1258096. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gonçalves, M.A. DNA, RNA and Protein Tools for Editing the Genetic Information in Human Cells. iScience 2018, 6, 247–263. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Gonçalves, M.A. Genome editing at the crossroads of delivery, specificity, and fidelity. Trends Biotechnol. 2015, 33, 280–291. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef] [PubMed]
- Li, H.L.; Fujimoto, N.; Sasakawa, N.; Shirai, S.; Ohkame, T.; Sakuma, T.; Tanaka, M.; Amano, N.; Watanabe, A.; Sakurai, H. Precise correction of the dystrophin gene in duchenne muscular dystrophy patient induced pluripotent stem cells by TALEN and CRISPR-Cas9. Stem Cell Reports 2015, 4, 143–154. [Google Scholar] [CrossRef]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Multiplex CRISPR/Cas9-based genome editing for correction of dystrophin mutations that cause Duchenne muscular dystrophy. Nat. Commun. 2015, 6, 6244. [Google Scholar] [CrossRef]
- Ousterout, D.G.; Kabadi, A.M.; Thakore, P.I.; Perez-Pinera, P.; Brown, M.T.; Majoros, W.H.; Reddy, T.E.; Gersbach, C.A. Correction of dystrophin expression in cells from Duchenne muscular dystrophy patients through genomic excision of exon 51 by zinc finger nucleases. Mol. Ther. 2015, 23, 523–532. [Google Scholar] [CrossRef]
- Maggio, I.; Chen, X.; Gonçalves, M.A. The emerging role of viral vectors as vehicles for DMD gene editing. Genome Med. 2016, 8, 59. [Google Scholar] [CrossRef]
- Gonçalves, M.A.; de Vries, A.A. Adenovirus: From foe to friend. Rev. Medical Virol. 2006, 16, 167–186. [Google Scholar] [CrossRef]
- Gonçalves, M.A. Adeno-associated virus: From defective virus to effective vector. Virol. J. 2005, 2, 43. [Google Scholar] [CrossRef]
- Maggio, I.; Stefanucci, L.; Janssen, J.M.; Liu, J.; Chen, X.; Mouly, V.; Gonçalves, M.A. Selection-free gene repair after adenoviral vector transduction of designer nucleases: Rescue of dystrophin synthesis in DMD muscle cell populations. Nucleic Acids Res. 2016, 44, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Liu, J.; Janssen, J.M.; Chen, X.; Gonçalves, M.A. Adenoviral vectors encoding CRISPR/Cas9 multiplexes rescue dystrophin synthesis in unselected populations of DMD muscle cells. Sci. Rep. 2016, 6, 37051. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Park, K.H.; Zhao, L.; Xu, J.; El Refaey, M.; Gao, Y.; Zhu, H.; Ma, J.; Han, R. CRISPR-mediated genome editing restores dystrophin expression and function in mdx mice. Mol. Ther. 2016, 24, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Long, C.; McAnally, J.R.; Shelton, J.M.; Mireault, A.A.; Bassel-Duby, R.; Olson, E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science 2014, 345, 1184–1188. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Rivera, R.M.C.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef]
- Miller, D.G.; Rutledge, E.A.; Russell, D.W. Chromosomal effects of adeno-associated virus vector integration. Nature Genet. 2002, 30, 147–148. [Google Scholar] [CrossRef]
- Porteus, M.H.; Cathomen, T.; Weitzman, M.D.; Baltimore, D. Efficient gene targeting mediated by adeno-associated virus and DNA double-strand breaks. Mol. Cell. Biol. 2003, 23, 3558–3565. [Google Scholar] [CrossRef]
- Miller, D.G.; Petek, L.M.; Russell, D.W. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat. Genet. 2004, 36, 767–773. [Google Scholar] [CrossRef]
- Li, H.; Haurigot, V.; Doyon, Y.; Li, T.; Wong, S.Y.; Bhagwat, A.S.; Malani, N.; Anguela, X.M.; Sharma, R.; Ivanciu, L.; et al. In vivo genome editing restores haemostasis in a mouse model of haemophilia. Nature 2011, 475, 217–221. [Google Scholar] [CrossRef]
- Anguela, X.M.; Sharma, R.; Doyon, Y.; Miller, J.C.; Li, H.; Haurigot, V.; Rohde, M.E.; Wong, S.Y.; Davidson, R.J.; Zhou, S.; et al. Robust ZFN-mediated genome editing in adult hemophilic mice. Blood 2013, 122, 3283–3287. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Wu, Y.; Gemberling, M.P.; Oliver, M.L.; Waller, M.A.; Bohning, J.D.; Robinson-Hamm, J.N.; Bulaklak, K.; Rivera, R.M.C.; Collier, J.H.; et al. Long-term evaluation of AAV-CRISPR genome editing for Duchenne muscular dystrophy. Nat. Med. 2019, 25, 427–432. [Google Scholar] [CrossRef] [PubMed]
- Hanlon, K.S.; Kleinstiver, B.P.; Garcia, S.P.; Zaborowski, M.P.; Volak, A.; Spirig, S.E.; Muller, A.; Sousa, A.A.; Tsai, S.Q.; Bengtsson, N.E.; et al. High levels of AAV vector integration into CRISPR-induced DNA breaks. Nat. Commun. 2019, 10, 4439. [Google Scholar] [CrossRef] [PubMed]
- Bergelson, J.M.; Cunningham, J.A.; Droguett, G.; Kurt-Jones, E.A.; Krithivas, A.; Hong, J.S.; Horwitz, M.S.; Crowell, R.L.; Finberg, R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science 1997, 275, 1320–1323. [Google Scholar] [CrossRef]
- Knaän-Shanzer, S.; van de Watering, M.J.M.; van der Velde, I.; Gonçalves, M.A.F.V.; Valerio, D.; de Vries, A.A.F. Endowing human adenovirus serotype 5 vectors with fiber domains of species B greatly enhances gene transfer into human mesenchymal stem cells. Stem Cells 2005, 23, 1598–1607. [Google Scholar] [CrossRef]
- Gonçalves, M.A.F.V.; de Vries, A.A.F.; Holkers, M.; van de Watering, M.J.M.; van der Velde, I.; van Nierop, G.P.; Valerio, D.; Knaän-Shanzer, S. Human mesenchymal stem cells ectopically expressing full-length dystrophin can complement Duchenne muscular dystrophy myotubes by cell fusion. Hum. Mol. Genet. 2005, 15, 213–221. [Google Scholar] [CrossRef]
- Gonçalves, M.A.F.V.; Holkers, M.; Cudré-Mauroux, C.; Van Nierop, G.P.; Knaän-Shanzer, S.; Van Der Velde, I.; Valerio, D.; De Vries, A.A. Transduction of myogenic cells by retargeted dual high-capacity hybrid viral vectors: Robust dystrophin synthesis in duchenne muscular dystrophy muscle cells. Mol. Ther. 2006, 13, 976–986. [Google Scholar] [CrossRef]
- Gaggar, A.; Shayakhmetov, D.M.; Lieber, A. CD46 is a cellular receptor for group B adenoviruses. Nat. Med. 2003, 9, 1408–1412. [Google Scholar] [CrossRef]
- Shayakhmetov, D.M.; Papayannopoulou, T.; Stamatoyannopoulos, G.; Lieber, A. Efficient gene transfer into human CD34+ cells by a retargeted adenovirus vector. J. Virol. 2000, 74, 2567–2583. [Google Scholar] [CrossRef]
- Knaän-Shanzer, S.; Van Der Velde, I.; Havenga, M.J.E.; Lemckert, A.A.C.; De Vries, A.A.F.; Valerio, D. Highly efficient targeted transduction of undifferentiated human hematopoietic cells by adenoviral vectors displaying fiber knobs of subgroup B. Hum. Gene Ther. 2001, 12, 1989–2005. [Google Scholar] [CrossRef]
- Morsy, M.A.; Gu, M.; Motzel, S.; Zhao, J.; Lin, J.; Su, Q.; Allen, H.; Franlin, L.; Parks, R.J.; Graham, F.L. An adenoviral vector deleted for all viral coding sequences results in enhanced safety and extended expression of a leptin transgene. Proc. Natl. Acad. Sci. USA 1998, 95, 7866–7871. [Google Scholar] [CrossRef] [PubMed]
- Schiedner, G.; Morral, N.; Parks, R.J.; Wu, Y.; Koopmans, S.C.; Langston, C.; Graham, F.L.; Beaudet, A.L.; Kochanek, S. Genomic DNA transfer with a high-capacity adenovirus vector results in improved in vivo gene expression and decreased toxicity. Nat. Genet. 1998, 18, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Morral, N.; O’Neal, W.; Rice, K.; Leland, M.; Kaplan, J.; Piedra, P.A.; Zhou, H.; Parks, R.J.; Velji, R.; Aguilar-Córdova, E.; et al. Administration of helper-dependent adenoviral vectors and sequential delivery of different vector serotype for long-term liver-directed gene transfer in baboons. Proc. Natl. Acad. Sci. USA 1999, 96, 12816–12821. [Google Scholar] [CrossRef] [PubMed]
- Ehrke-Schulz, E.; Schiwon, M.; Leitner, T.; Dávid, S.; Bergmann, T.; Liu, J.; Ehrhardt, A. CRISPR/Cas9 delivery with one single adenoviral vector devoid of all viral genes. Sci. Rep. 2017, 7, 17113. [Google Scholar] [CrossRef] [PubMed]
- Schiwon, M.; Ehrke-Schulz, E.; Oswald, A.; Bergmann, T.; Michler, T.; Protzer, U.; Ehrhardt, A. One-vector system for multiplexed CRISPR/Cas9 against hepatitis B virus cccDNA utilizing high-capacity adenoviral vectors. Mol. Ther. Nucleic Acids 2018, 12, 242–253. [Google Scholar] [CrossRef] [PubMed]
- Mamchaoui, K.; Trollet, C.; Bigot, A.; Negroni, E.; Chaouch, S.; Wolff, A.; Kandalla, P.K.; Marie, S.; Di Santo, J.; St Guily, J.L.; et al. Immortalized pathological human myoblasts: Towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle 2011, 1, 34. [Google Scholar] [CrossRef]
- Fallaux, F.J.; Bout, A.; van der Velde, I.; van den Wollenberg, D.J.M.; Hehir, K.M.; Keegan, J.; Auger, C.; Cramer, S.J.; van Ormondt, H.; van der Eb, A.J.; et al. New helper cells and matched early region 1-deleted adenovirus vectors prevent generation of replication-competent adenoviruses. Hum. Gene Ther. 1998, 9, 1909–1917. [Google Scholar] [CrossRef]
- Ensinger, M.J.; Ginsberg, H.S. Selection and preliminary characterization of temperature-sensitive mutants of type 5 adenovirus. J. Virol. 1972, 10, 328–339. [Google Scholar] [CrossRef]
- Van Der Vliet, P.C.; Levine, A.J.; Ensinger, M.J.; Ginsberg, H.S. Thermolabile DNA binding proteins from cells infected with a temperature-sensitive mutant of adenovrius defective in viral DNA synthesis. J. Virol. 1975, 15, 348–354. [Google Scholar] [CrossRef]
- Havenga, M.; Holterman, L.; Melis, I.; Smits, S.; Kaspers, J.; Heemskerk, E.; Vlugt, R.v.d.; Koldijk, M.; Schouten, G.; Hateboer, G. Serum-free transient protein production system based on adenoviral vector and PER. C6 technology: High yield and preserved bioactivity. Biotechnol. Bioeng. 2008, 100, 273–283. [Google Scholar] [CrossRef]
- Holkers, M.; Maggio, I.; Henriques, S.F.; Janssen, J.M.; Cathomen, T.; Gonçalves, M.A. Adenoviral vector DNA for accurate genome editing with engineered nucleases. Nat. Methods 2014, 11, 1051–1057. [Google Scholar] [CrossRef] [PubMed]
- Dang, Y.; Jia, G.; Choi, J.; Ma, H.; Anaya, E.; Ye, C.; Shankar, P.; Wu, H. Optimizing sgRNA structure to improve CRISPR-Cas9 knockout efficiency. Genome Biol. 2015, 16, 280. [Google Scholar] [CrossRef] [PubMed]
- Maggio, I.; Zittersteijn, H.A.; Wang, Q.; Liu, J.; Janssen, J.M.; Ojeda, I.T.; van der Maarel, S.M.; Lankester, A.C.; Hoeben, R.C.; Gonçalves, M.A.F.V. Integrating gene delivery and gene-editing technologies by adenoviral vector transfer of optimized CRISPR-Cas9 components. Gene Ther. 2020. [Google Scholar] [CrossRef] [PubMed]
- Janssen, J.M.; Liu, J.; Skokan, J.; Gonçalves, M.A.; de Vries, A.A. Development of an AdEasy-based system to produce first-and second-generation adenoviral vectors with tropism for CAR-or CD46-positive cells. J. Gene Med. 2013, 15, 1–11. [Google Scholar] [CrossRef]
- Holkers, M.; Cathomen, T.; Gonçalves, M.A.F.V. Construction and characterization of adenoviral vectors for the delivery of TALENs into human cells. Methods 2014, 69, 179–187. [Google Scholar] [CrossRef]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]
- Matsumura, K.; Campbell, K.P. Deficiency of dystrophin-associated proteins: A common mechanism leading to muscle cell necrosis in severe childhood muscular dystrophies. Neuromuscul. Disord. 1993, 3, 109–118. [Google Scholar] [CrossRef]
- Wasala, N.B.; Hakim, C.H.; Chen, S.-J.; Yang, N.N.; Duan, D. Questions Answered and Unanswered by the First CRISPR Editing Study in a Canine Model of Duchenne Muscular Dystrophy. Hum. Gene Ther. 2019, 30, 535–543. [Google Scholar] [CrossRef]
- Sun, C.; Serra, C.; Lee, G.; Wagner, K.R. Stem cell-based therapies for Duchenne muscular dystrophy. Exp. Neurol. 2020, 323, 113086. [Google Scholar] [CrossRef]
- Chen, X.; Rinsma, M.; Janssen, J.M.; Liu, J.; Maggio, I.; Gonçalves, M.A.F.V. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016, 44, 6482–6492. [Google Scholar] [CrossRef]
- Daer, R.M.; Cutts, J.P.; Brafman, D.A.; Haynes, K.A. The impact of chromatin dynamics on Cas9-mediated genome editing in human cells. ACS Synt. Biol. 2016, 6, 428–438. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Lucchetti-Miganeh, C.; Yaou, R.B.; Kaplan, J.-C.; Chelly, J.; Leturcq, F.; Barloy-Hubler, F.; Le Rumeur, E. Assessment of the structural and functional impact of in-frame mutations of the DMD gene, using the tools included in the eDystrophin online database. Orphanet J. Rare Dis. 2012, 7, 45. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, A.; Raguénès-Nicol, C.; Ben Yaou, R.; Ameziane-Le Hir, S.; Chéron, A.; Vié, V.; Claustres, M.; Leturcq, F.; Delalande, O.; Hubert, J.-F.; et al. Becker muscular dystrophy severity is linked to the structure of dystrophin. Hum. Mol. Genet. 2014, 24, 1267–1279. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brescia, M.; Janssen, J.M.; Liu, J.; Gonçalves, M.A.F.V. High-Capacity Adenoviral Vectors Permit Robust and Versatile Testing of DMD Gene Repair Tools and Strategies in Human Cells. Cells 2020, 9, 869. https://doi.org/10.3390/cells9040869
Brescia M, Janssen JM, Liu J, Gonçalves MAFV. High-Capacity Adenoviral Vectors Permit Robust and Versatile Testing of DMD Gene Repair Tools and Strategies in Human Cells. Cells. 2020; 9(4):869. https://doi.org/10.3390/cells9040869
Chicago/Turabian StyleBrescia, Marcella, Josephine M. Janssen, Jin Liu, and Manuel A. F. V. Gonçalves. 2020. "High-Capacity Adenoviral Vectors Permit Robust and Versatile Testing of DMD Gene Repair Tools and Strategies in Human Cells" Cells 9, no. 4: 869. https://doi.org/10.3390/cells9040869
APA StyleBrescia, M., Janssen, J. M., Liu, J., & Gonçalves, M. A. F. V. (2020). High-Capacity Adenoviral Vectors Permit Robust and Versatile Testing of DMD Gene Repair Tools and Strategies in Human Cells. Cells, 9(4), 869. https://doi.org/10.3390/cells9040869