Human Embryonic Stem Cell-Derived Wilson’s Disease Model for Screening Drug Efficacy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Gene Editing
2.3. Embryonic Body Generation
2.4. Differentiation into Hepatocyte-Like Cells (HLCs)
2.5. Immunostaining
2.6. Flow Cytometry
2.7. Real-Time PCR
2.8. Periodic Acid-Schiff (PAS) Staining
2.9. Cell Viability Assay
2.10. Chemical Treatment
2.11. Measuring Copper Concentration
2.12. RNA Sequencing
2.13. Statistical Analysis
3. Results
3.1. Generation of Wilson’s Disease Model Using the CRISPR/Cas9 System
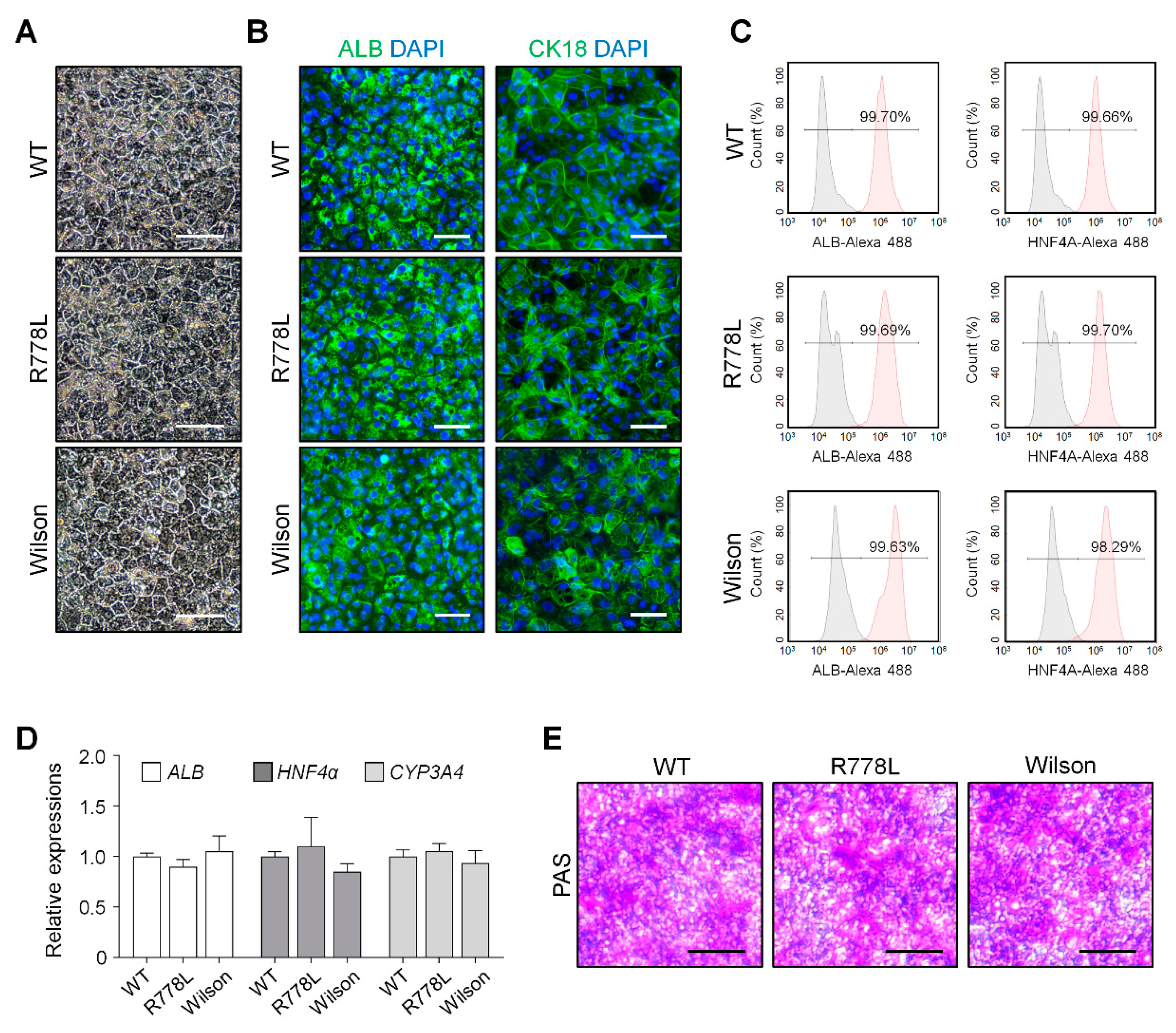
3.2. Differentiation of WT, R778L-Introduced hESCs, and Wilson iPSCs into Hepatocyte-Like Cells (HLCs)
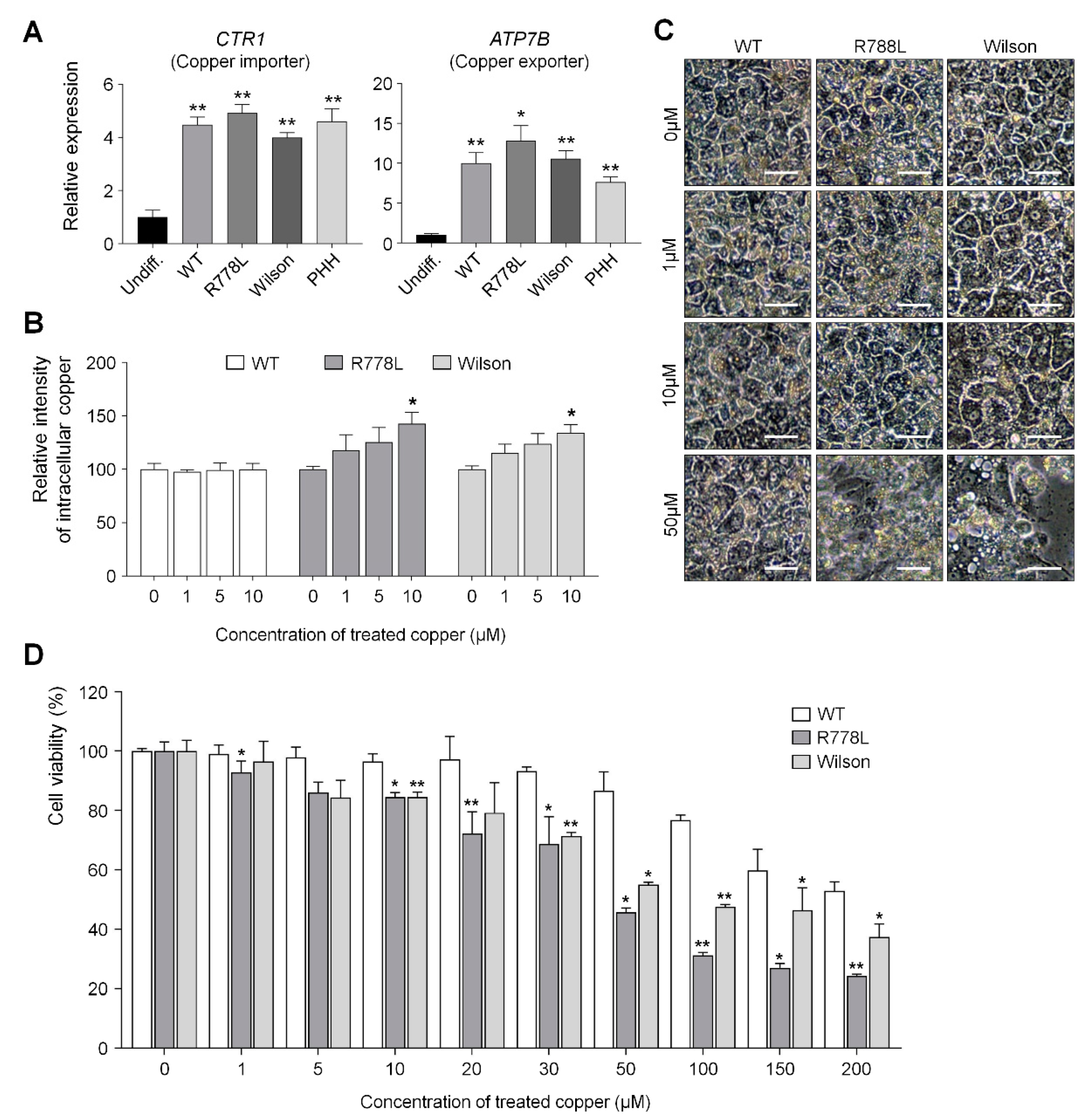
3.3. Comparing Vulnerability Against Copper in WT-HLCs, R778L-Introduced HLCs, and Wilson hiPSC-HLCs
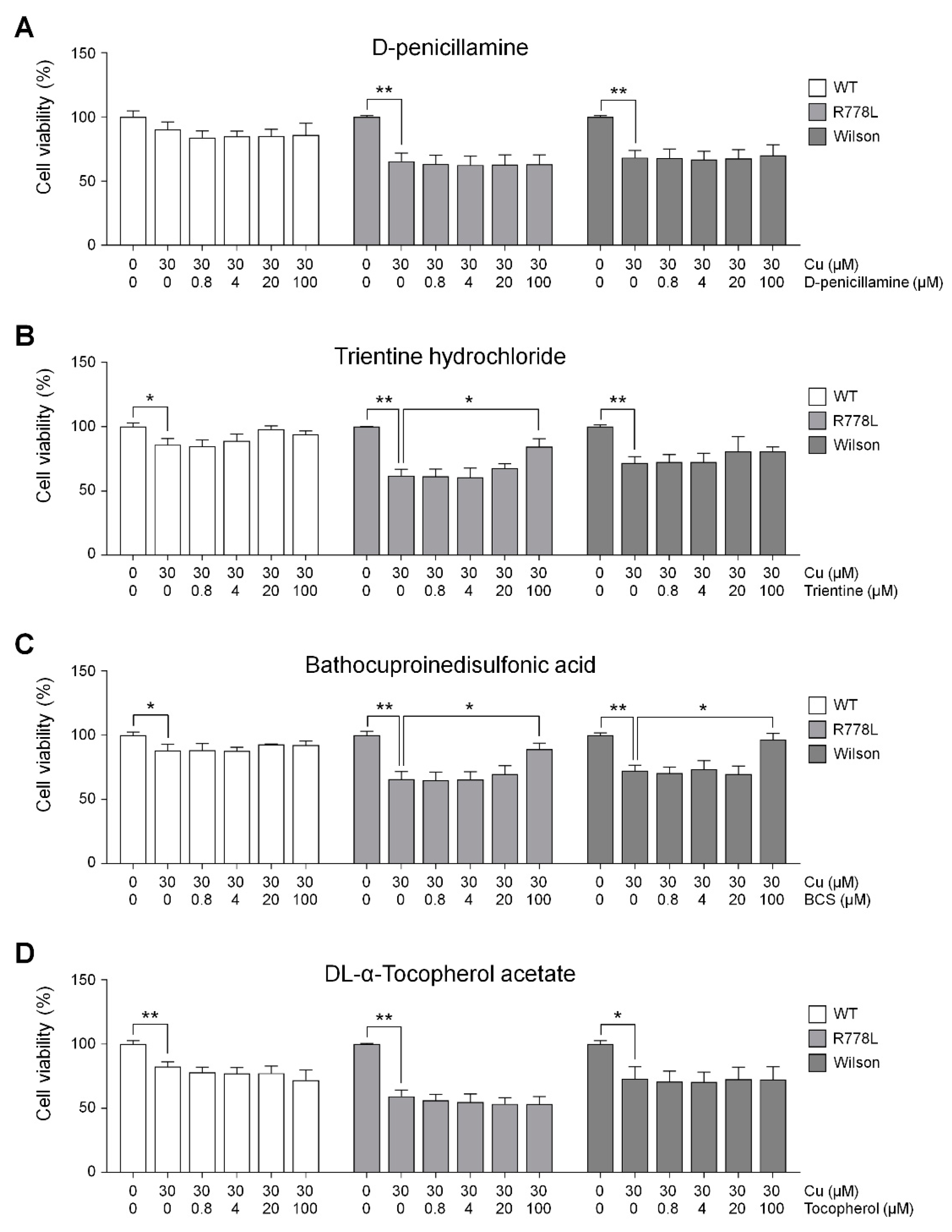
3.4. Evaluation of In Vitro Wilson’s Disease Model for Drug Screening
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wu, F.; Wang, J.; Pu, C.; Qiao, L.; Jiang, C. Wilson’s disease: A comprehensive review of the molecular mechanisms. Int. J. Mol. Sci. 2015, 16, 6419–6431. [Google Scholar] [CrossRef]
- Prohaska, J.R. Genetic diseases of copper metabolism. Clin. Physiol. Biochem. 1986, 4, 87–93. [Google Scholar] [PubMed]
- Forbes, J.R.; Cox, D.W. Copper-dependent trafficking of Wilson disease mutant ATP7B proteins. Hum. Mol. Genet. 2000, 9, 1927–1935. [Google Scholar] [CrossRef] [PubMed]
- Huster, D.; Kuhne, A.; Bhattacharjee, A.; Raines, L.; Jantsch, V.; Noe, J.; Schirrmeister, W.; Sommerer, I.; Sabri, O.; Berr, F.; et al. Diverse functional properties of Wilson disease ATP7B variants. Gastroenterology 2012, 142, 947.e945–956.e945. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, R.F. Wilson’s Disease. Semin. Neurol. 2007, 27, 123–132. [Google Scholar] [CrossRef]
- Dziezyc-Jaworska, K.; Litwin, T.; Czlonkowska, A. Clinical manifestations of Wilson disease in organs other than the liver and brain. Ann. Transl. Med. 2019, 7, S62. [Google Scholar] [CrossRef]
- Hedera, P. Clinical management of Wilson disease. Ann. Transl. Med. 2019, 7, S66. [Google Scholar] [CrossRef]
- Mohr, I.; Weiss, K.H. Current anti-copper therapies in management of Wilson disease. Ann. Transl. Med. 2019, 7, S69. [Google Scholar] [CrossRef]
- Ferenci, P.; Caca, K.; Loudianos, G.; Mieli-Vergani, G.; Tanner, S.; Sternlieb, I.; Schilsky, M.; Cox, D.; Berr, F. Diagnosis and phenotypic classification of Wilson disease. Liver Int. 2003, 23, 139–142. [Google Scholar] [CrossRef]
- Catana, A.M.; Medici, V. Liver transplantation for Wilson disease. World J. Hepatol. 2012, 4, 5–10. [Google Scholar] [CrossRef]
- Roberts, E.A.; Schilsky, M.L.; American Association for Study of Liver Diseases (AASLD). Diagnosis and treatment of Wilson disease: An update. Hepatology 2008, 47, 2089–2111. [Google Scholar] [CrossRef]
- Ebert, A.D.; Liang, P.; Wu, J.C. Induced pluripotent stem cells as a disease modeling and drug screening platform. J. Cardiovasc. Pharmacol. 2012, 60, 408–416. [Google Scholar] [CrossRef]
- Sterneckert, J.L.; Reinhardt, P.; Scholer, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef]
- Avior, Y.; Sagi, I.; Benvenisty, N. Pluripotent stem cells in disease modelling and drug discovery. Nat. Rev. Mol. Cell Biol. 2016, 17, 170–182. [Google Scholar] [CrossRef]
- Hockemeyer, D.; Jaenisch, R. Induced Pluripotent Stem Cells Meet Genome Editing. Cell Stem Cell 2016, 18, 573–586. [Google Scholar] [CrossRef]
- Parisi, S.; Polishchuk, E.V.; Allocca, S.; Ciano, M.; Musto, A.; Gallo, M.; Perone, L.; Ranucci, G.; Iorio, R.; Polishchuk, R.S.; et al. Characterization of the most frequent ATP7B mutation causing Wilson disease in hepatocytes from patient induced pluripotent stem cells. Sci. Rep. 2018, 8, 6247. [Google Scholar] [CrossRef]
- Yi, F.; Qu, J.; Li, M.; Suzuki, K.; Kim, N.Y.; Liu, G.H.; Belmonte, J.C. Establishment of hepatic and neural differentiation platforms of Wilson’s disease specific induced pluripotent stem cells. Protein Cell 2012, 3, 855–863. [Google Scholar] [CrossRef]
- Zhang, S.; Chen, S.; Li, W.; Guo, X.; Zhao, P.; Xu, J.; Chen, Y.; Pan, Q.; Liu, X.; Zychlinski, D.; et al. Rescue of ATP7B function in hepatocyte-like cells from Wilson’s disease induced pluripotent stem cells using gene therapy or the chaperone drug curcumin. Hum. Mol. Genet. 2011, 20, 3176–3187. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef]
- Cong, L.; Zhang, F. Genome engineering using CRISPR-Cas9 system. Methods Mol. Biol. 2015, 1239, 197–217. [Google Scholar] [CrossRef]
- Hsu, P.D.; Lander, E.S.; Zhang, F. Development and applications of CRISPR-Cas9 for genome engineering. Cell 2014, 157, 1262–1278. [Google Scholar] [CrossRef]
- Woo, D.H.; Chen, Q.; Yang, T.L.; Glineburg, M.R.; Hoge, C.; Leu, N.A.; Johnson, F.B.; Lengner, C.J. Enhancing a Wnt-Telomere Feedback Loop Restores Intestinal Stem Cell Function in a Human Organotypic Model of Dyskeratosis Congenita. Cell Stem Cell 2016, 19, 397–405. [Google Scholar] [CrossRef]
- Park, C.Y.; Kim, D.H.; Son, J.S.; Sung, J.J.; Lee, J.; Bae, S.; Kim, J.H.; Kim, D.W.; Kim, J.S. Functional Correction of Large Factor VIII Gene Chromosomal Inversions in Hemophilia A Patient-Derived iPSCs Using CRISPR-Cas9. Cell Stem Cell 2015, 17, 213–220. [Google Scholar] [CrossRef]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef]
- Kim, J.H.; Wang, M.; Lee, J.; Park, H.J.; Han, C.; Hong, H.S.; Kim, J.S.; An, G.H.; Park, K.; Park, H.K.; et al. Prediction of hepatotoxicity for drugs using human pluripotent stem cell-derived hepatocytes. Cell Biol. Toxicol. 2018, 34, 51–64. [Google Scholar] [CrossRef]
- Kathawala, M.; Hirschfield, G.M. Insights into the management of Wilson’s disease. Therap. Adv. Gastroenterol. 2017, 10, 889–905. [Google Scholar] [CrossRef]
- Patil, M.; Sheth, K.A.; Krishnamurthy, A.C.; Devarbhavi, H. A review and current perspective on Wilson disease. J. Clin. Exp. Hepatol. 2013, 3, 321–336. [Google Scholar] [CrossRef]
- Zhang, L.P.; Takahara, T.; Yata, Y.; Furui, K.; Jin, B.; Kawada, N.; Watanabe, A. Increased expression of plasminogen activator and plasminogen activator inhibitor during liver fibrogenesis of rats: Role of stellate cells. J. Hepatol. 1999, 31, 703–711. [Google Scholar] [CrossRef]
- Schumacher, J.D.; Guo, G.L. Regulation of Hepatic Stellate Cells and Fibrogenesis by Fibroblast Growth Factors. Biomed. Res. Int. 2016, 2016, 8323747. [Google Scholar] [CrossRef]
- Patsenker, E.; Stickel, F. Role of integrins in fibrosing liver diseases. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 301, G425–G434. [Google Scholar] [CrossRef]
- Butler, N.S.; Schmidt, N.W. Erythropoietin-producing hepatocellular receptor B2 receptor tyrosine kinase: A novel regulator of infection- and inflammation-induced liver fibrosis. Hepatology 2015, 62, 680–683. [Google Scholar] [CrossRef]
- Xia, Z.; Huang, X.; Chen, K.; Wang, H.; Xiao, J.; He, K.; Huang, R.; Duan, X.; Liu, H.; Zhang, J.; et al. Proapoptotic Role of Potassium Ions in Liver Cells. Biomed. Res. Int. 2016, 2016, 1729135. [Google Scholar] [CrossRef]
- Syn, W.K.; Oo, Y.H.; Pereira, T.A.; Karaca, G.F.; Jung, Y.; Omenetti, A.; Witek, R.P.; Choi, S.S.; Guy, C.D.; Fearing, C.M.; et al. Accumulation of natural killer T cells in progressive nonalcoholic fatty liver disease. Hepatology 2010, 51, 1998–2007. [Google Scholar] [CrossRef]
- Sole, C.; Sola, E.; Morales-Ruiz, M.; Fernandez, G.; Huelin, P.; Graupera, I.; Moreira, R.; de Prada, G.; Ariza, X.; Pose, E.; et al. Characterization of Inflammatory Response in Acute-on-Chronic Liver Failure and Relationship with Prognosis. Sci. Rep. 2016, 6, 32341. [Google Scholar] [CrossRef]
- Czlonkowska, A.; Gajda, J.; Rodo, M. Effects of long-term treatment in Wilson’s disease with D-penicillamine and zinc sulphate. J. Neurol. 1996, 243, 269–273. [Google Scholar] [CrossRef]
- Walshe, J.M. Treatment of Wilson’s disease with trientine (triethylene tetramine) dihydrochloride. Lancet 1982, 1, 643–647. [Google Scholar] [CrossRef]
- Schilsky, M.L. Treatment of Wilson’s disease: What are the relative roles of penicillamine, trientine, and zinc supplementation? Curr. Gastroenterol. Rep. 2001, 3, 54–59. [Google Scholar] [CrossRef]
- Fryer, M.J. Potential of vitamin E as an antioxidant adjunct in Wilson’s disease. Med. Hypotheses 2009, 73, 1029–1030. [Google Scholar] [CrossRef]
- Shen, L.; Ji, H.F. Adjunctive vitamin E treatment in Wilson disease and suggestions for future trials. Hepatology 2010, 51, 1864–1865. [Google Scholar] [CrossRef]
- Chandhok, G.; Horvath, J.; Aggarwal, A.; Bhatt, M.; Zibert, A.; Schmidt, H.H. Functional analysis and drug response to zinc and D-penicillamine in stable ATP7B mutant hepatic cell lines. World J. Gastroenterol. 2016, 22, 4109–4119. [Google Scholar] [CrossRef]
- European Association For The Study Of The Liver. EASL Clinical Practice Guidelines: Wilson’s disease. J. Hepatol. 2012, 56, 671–685. [Google Scholar] [CrossRef]
- Delangle, P.; Mintz, E. Chelation therapy in Wilson’s disease: From D-penicillamine to the design of selective bioinspired intracellular Cu(I) chelators. Dalton Trans. 2012, 41, 6359–6370. [Google Scholar] [CrossRef]
- Prats-Puig, A.; Soriano-Rodriguez, P.; Oliveras, G.; Carreras-Badosa, G.; Espuna, S.; Diaz-Roldan, F.; de Zegher, F.; Ibanez, L.; Bassols, J.; Puig, T.; et al. Soluble CRTC3: A Newly Identified Protein Released by Adipose Tissue That Is Associated with Childhood Obesity. Clin. Chem. 2016, 62, 476–484. [Google Scholar] [CrossRef]
- Song, Y.; Altarejos, J.; Goodarzi, M.O.; Inoue, H.; Guo, X.; Berdeaux, R.; Kim, J.H.; Goode, J.; Igata, M.; Paz, J.C.; et al. CRTC3 links catecholamine signalling to energy balance. Nature 2010, 468, 933–939. [Google Scholar] [CrossRef]
- Grasedyck, K. [D-penicillamine--side effects, pathogenesis and decreasing the risks]. Z. Rheumatol. 1988, 47, 17–19. [Google Scholar]
- Weiss, K.H.; Thurik, F.; Gotthardt, D.N.; Schafer, M.; Teufel, U.; Wiegand, F.; Merle, U.; Ferenci-Foerster, D.; Maieron, A.; Stauber, R.; et al. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin. Gastroenterol. Hepatol. 2013, 11, 1028.e1-2–1035.e1-2. [Google Scholar] [CrossRef]
- Kim, B.; Chung, S.J.; Shin, H.W. Trientine-induced neurological deterioration in a patient with Wilson’s disease. J. Clin. Neurosci. 2013, 20, 606–608. [Google Scholar] [CrossRef]
- Aggarwal, A.; Bhatt, M. Advances in Treatment of Wilson Disease. Tremor Other Hyperkinet Mov. (NY) 2018, 8, 525. [Google Scholar] [CrossRef]
- Ko, H.C.; Gelb, B.D. Concise review: Drug discovery in the age of the induced pluripotent stem cell. Stem Cells Transl. Med. 2014, 3, 500–509. [Google Scholar] [CrossRef]
- Luz, A.L.; Tokar, E.J. Pluripotent Stem Cells in Developmental Toxicity Testing: A Review of Methodological Advances. Toxicol. Sci. 2018, 165, 31–39. [Google Scholar] [CrossRef]
- Nair, M.; Sandhu, S.S.; Sharma, A.K. Induced Pluripotent Stem Cell Technology: A Paradigm Shift in Medical Science for Drug Screening and Disease Modeling. Curr. Med. Chem. 2017, 24, 4368–4398. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, D.; Kim, S.-B.; Ryu, J.L.; Hong, H.; Chang, J.-H.; Yoo, T.-J.; Jin, X.; Park, H.-J.; Han, C.; Lee, B.H.; et al. Human Embryonic Stem Cell-Derived Wilson’s Disease Model for Screening Drug Efficacy. Cells 2020, 9, 872. https://doi.org/10.3390/cells9040872
Kim D, Kim S-B, Ryu JL, Hong H, Chang J-H, Yoo T-J, Jin X, Park H-J, Han C, Lee BH, et al. Human Embryonic Stem Cell-Derived Wilson’s Disease Model for Screening Drug Efficacy. Cells. 2020; 9(4):872. https://doi.org/10.3390/cells9040872
Chicago/Turabian StyleKim, Dongkyu, Su-Bin Kim, Jung Lim Ryu, Heesu Hong, Jin-Hyuk Chang, Tack-Jin Yoo, Xiong Jin, Han-Jin Park, Choongseong Han, Beom Hee Lee, and et al. 2020. "Human Embryonic Stem Cell-Derived Wilson’s Disease Model for Screening Drug Efficacy" Cells 9, no. 4: 872. https://doi.org/10.3390/cells9040872
APA StyleKim, D., Kim, S.-B., Ryu, J. L., Hong, H., Chang, J.-H., Yoo, T.-J., Jin, X., Park, H.-J., Han, C., Lee, B. H., Choi, J.-H., Yoo, H.-W., Kim, J.-H., & Woo, D.-H. (2020). Human Embryonic Stem Cell-Derived Wilson’s Disease Model for Screening Drug Efficacy. Cells, 9(4), 872. https://doi.org/10.3390/cells9040872