The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
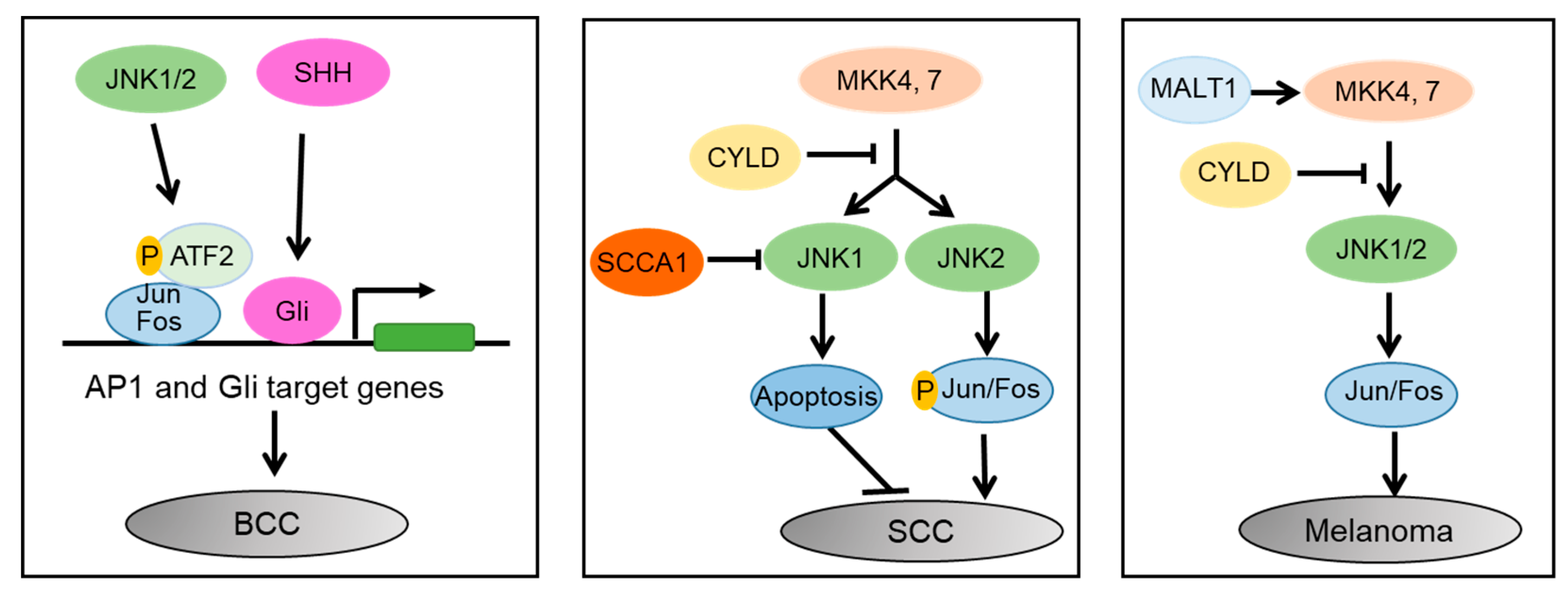{kind=link}
Abstract
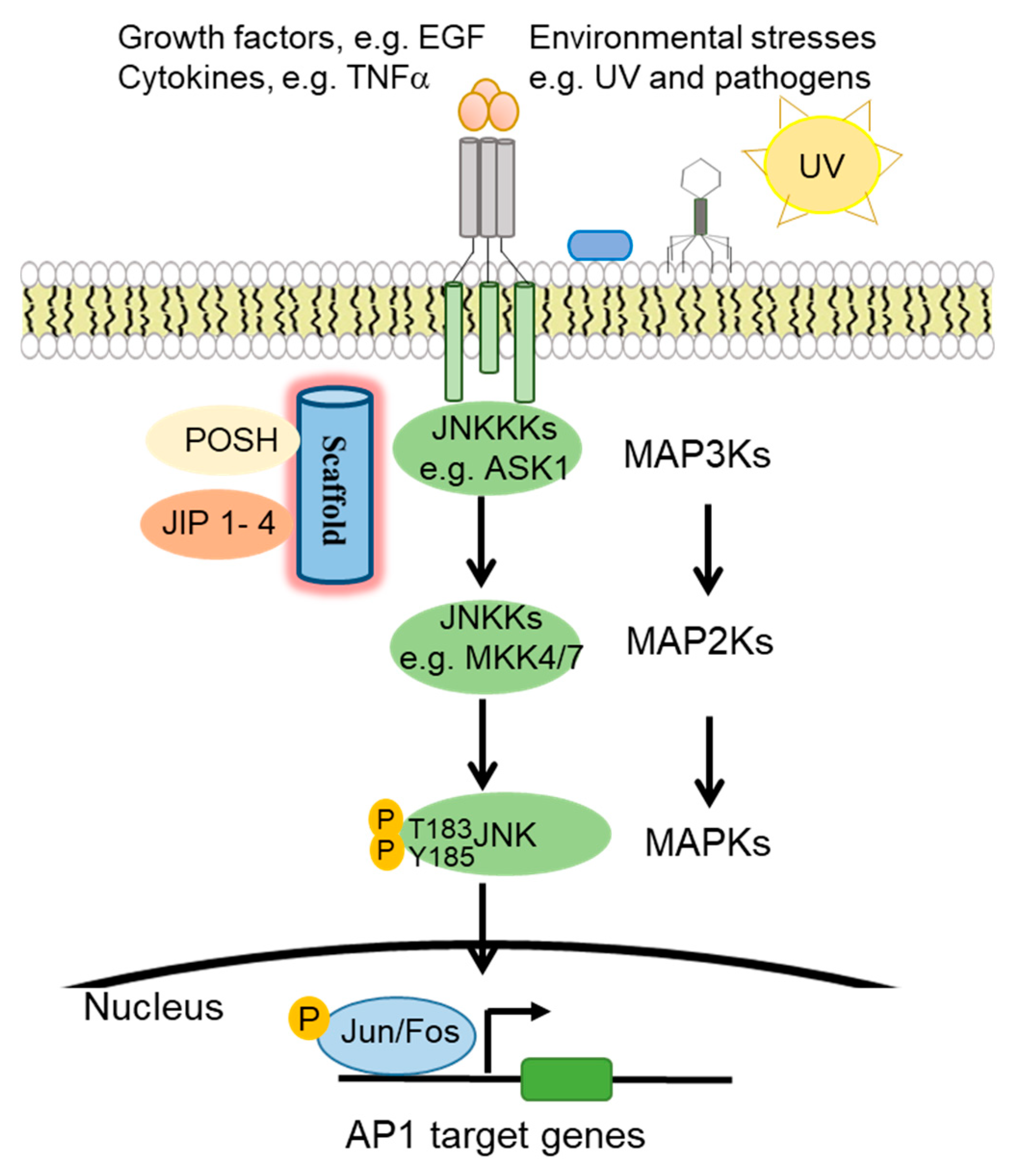
1. The c-Jun N-Terminal Kinase (JNK) Signaling Pathway
1.1. JNK Pathway Components
1.2. JNK Regulation of Cell Cycle Progression
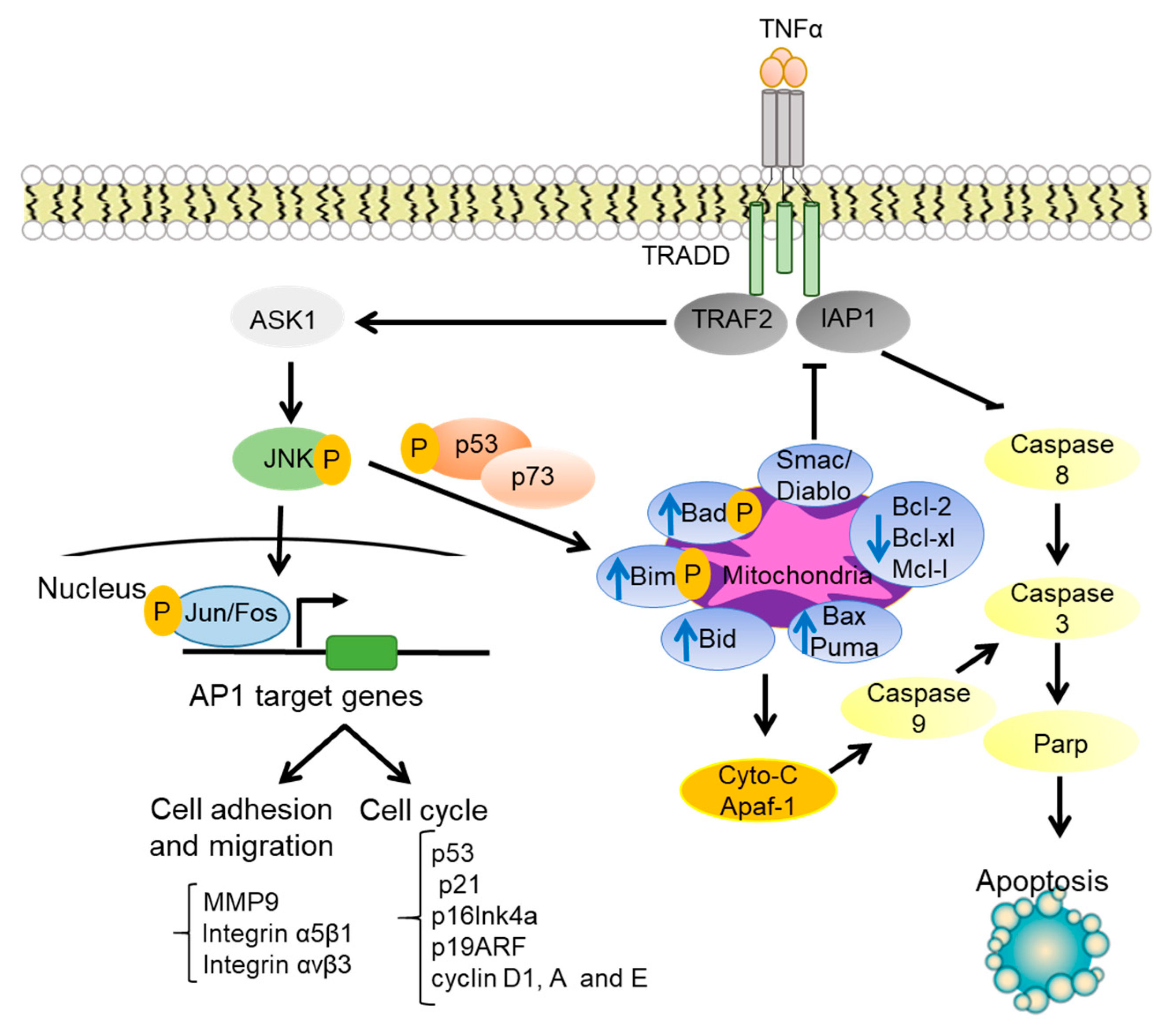
1.3. JNK Regulation of Cell Survival and Apoptosis
2. JNK Signaling in Immunological Skin Disorders
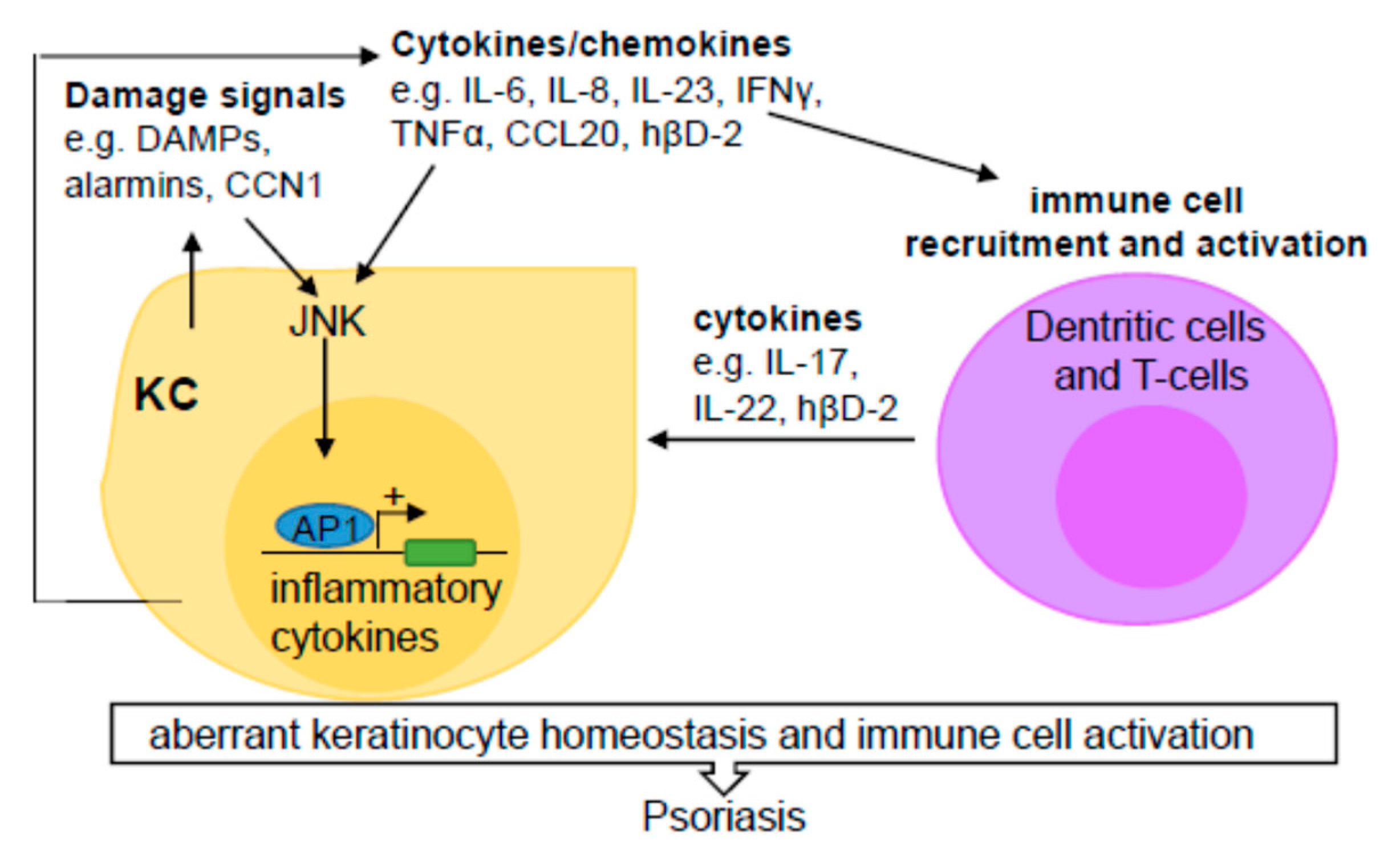
2.1. JNK Regulation of Immune Responses
2.2. JNK Contribution to Psoriasis
2.2.1. Pathogenesis of Psoriasis
2.2.2. JNK and NF-κB Pathway Regulators in Psoriasis
2.2.3. JNK Regulation of Dermal and Epidermal Interactions
2.2.4. JNK as an Effector of Neuropeptide-Induced Inflammation
2.2.5. JNK Links Gap Junction Defects to Psoriasis
2.2.6. JNK Regulation of Barrier Protein Defects
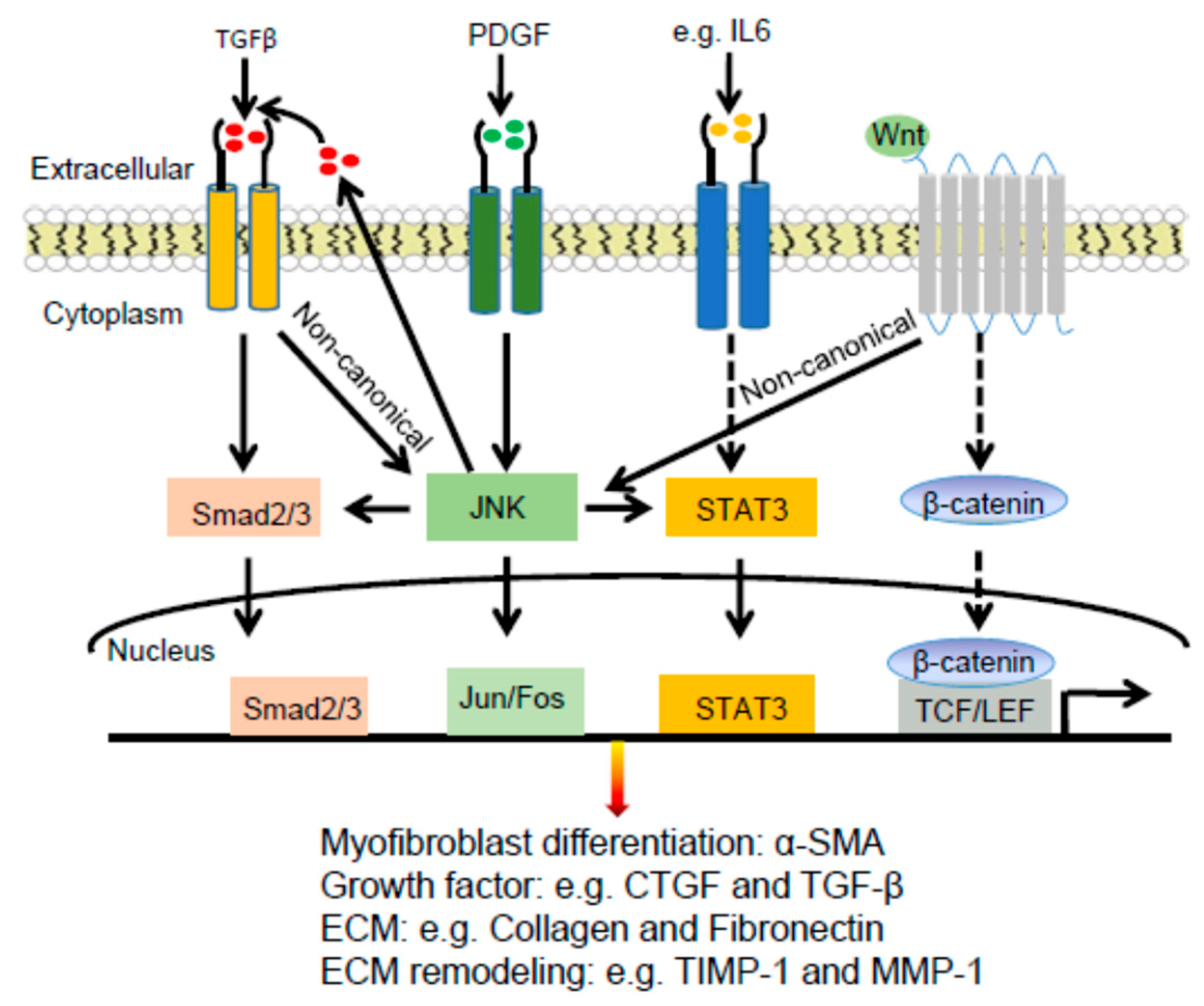
2.3. Dermal Fibrosis
2.3.1. Pathogenesis of Dermal Fibrosis
2.3.2. JNK Connections with TGFβ and PDGF in Dermal Fibrosis
2.3.3. JNK Connections with STAT3 and WNT Signaling Pathways in Dermal Fibrosis
2.3.4. JNK Regulation of Extracellular Matrix Proteins in Dermal Fibrosis
3. JNK Signaling in Skin Cancer
3.1. Differential Roles of JNK1 and JNK2 in SCC
3.2. JNK as a Key Mediator of the SHH, YAP, and WNT Signaling Pathways in BCC
3.3. Melanoma
3.3.1. JNK1 and JNK2 in Melanoma Growth and Progression
3.3.2. Paradoxical Roles of JNK in Melanoma Cell Survival, Apoptosis, and Therapy
4. JNK as a Therapeutic Target
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Kannan, N.; Neuwald, A.F. Evolutionary constraints associated with functional specificity of the CMGC protein kinases MAPK, CDK, GSK, SRPK, DYRK, and CK2alpha. Protein Sci. 2004, 13, 2059–2077. [Google Scholar] [CrossRef] [PubMed]
- Fedorov, O.; Marsden, B.; Pogacic, V.; Rellos, P.; Müller, S.; Bullock, A.N.; Schwaller, J.; Sundström, M.; Knapp, S. A systematic interaction map of validated kinase inhibitors with Ser/Thr kinases. Proc. Natl. Acad. Sci. USA 2007, 104, 20523–20528. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. The functional contrariety of JNK. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef]
- Gupta, S.; Barrett, T.; Whitmarsh, A.J.; Cavanagh, J.; Sluss, H.K.; Dérijard, B.; Davis, R.J. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996, 15, 2760–2770. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Kobe, B. Uses for JNK: The many and varied substrates of the c-Jun N-terminal kinases. Microbiol. Mol. Biol. Rev. 2006, 70, 1061–1095. [Google Scholar] [CrossRef]
- Biteau, B.; Karpac, J.; Hwangbo, D.; Jasper, H. Regulation of Drosophila lifespan by JNK signaling. Exp. Gerontol. 2011, 46, 349–354. [Google Scholar] [CrossRef]
- Seki, E.; Brenner, D.A.; Karin, M. A Liver Full of JNK: Signaling in Regulation of Cell Function and Disease Pathogenesis, and Clinical Approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef]
- Kusumaningrum, N.; Lee, D.H.; Yoon, H.-S.; Kim, Y.K.; Park, C.-H.; Chung, J.H. Gasdermin C is induced by ultraviolet light and contributes to MMP-1 expression via activation of ERK and JNK pathways. J. Dermatol. Sci. 2018, 90, 180–189. [Google Scholar] [CrossRef]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Kyriakis, J.M.; Avruch, J. Mammalian Mitogen-Activated Protein Kinase Signal Transduction Pathways Activated by Stress and Inflammation. Physiol. Rev. 2001, 81, 807–869. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Destrument, A.; Tournier, C. Physiological Roles of MKK4 and MKK7: Insights from Animal Models. Biochim. Biophys. Acta 2007, 1773, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Chadee, D.N.; Kyriakis, J.M. Activation of SAPK/JNKs in vitro. Methods Mol. Biol. 2010, 661, 59–73. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.M.; Yan, Q.; Yang, T.; Cheng, H.; Du, J.; Yoshioka, K.; Kung, S.K.P.; Ding, G.H. Scaffold protein JLP is critical for CD40 signaling in B lymphocytes. J. Biol. Chem. 2015, 290, 5256–5266. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Kukekov, N.V.; Schmit, T.L.; Biagas, K.V.; Sproul, A.A.; Gire, S.; Maes, M.E.; Xu, Z.; Greene, L.A. Sh3rf2/POSHER protein promotes cell survival by ring-mediated proteasomal degradation of the c-Jun N-terminal kinase scaffold POSH (plenty of SH3s) protein. J. Biol. Chem. 2012, 287, 2247–2256. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K.; Davis, R.J. Regulation of MAP Kinase Signaling Modules by Scaffold Proteins in Mammals. Annu. Rev. Cell Dev. Biol. 2003, 19, 91–118. [Google Scholar] [CrossRef]
- Yoshioka, K. Scaffold Proteins in Mammalian MAP Kinase Cascades. J. Biochem. 2004, 135, 657–661. [Google Scholar] [CrossRef][Green Version]
- Kutluk Oktay, E.B.O.O.M.O.; Filippo, G.G. The c-Jun N-terminal kinase JNK functions upstream of Aurora B to promote entry into mitosis. Cell Cyle 2008, 7, 533–541. [Google Scholar] [CrossRef]
- Ramsdale, R.; Jorissen, R.N.; Li, F.Z.; Al-Obaidi, S.; Ward, T.; Sheppard, K.E.; Bukczynska, P.E.; Young, R.J.; Boyle, S.E.; Shackleton, M.; et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci. Signal. 2015, 8, ra82. [Google Scholar] [CrossRef]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2015, 6, 321. [Google Scholar] [CrossRef]
- Wada, T.; Joza, N.; Cheng, H.-Y.M.; Sasaki, T.; Kozieradzki, I.; Bachmaier, K.; Katada, T.; Schreiber, M.; Wagner, E.F.; Nishina, H.; et al. MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nat. Cell Biol. 2004, 6, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Ichijo, H.; Korsmeyer, S.J. BCL-2 is phosphorylated and inactivated by an ASK1/Jun N-terminal protein kinase pathway normally activated at G(2)/M. Mol. Cell. Biol. 1999, 19, 8469–8478. [Google Scholar] [CrossRef] [PubMed]
- MacCorkle-Chosnek, R.A.; VanHooser, A.; Goodrich, D.W.; Brinkley, B.R.; Tan, T.-H. Cell Cycle Regulation of c-Jun N-Terminal Kinase Activity at the Centrosomes. Biochem. Biophys. Res. Commun. 2001, 289, 173–180. [Google Scholar] [CrossRef] [PubMed]
- Bakiri, L.; Lallemand, D.; Bossy-Wetzel, E.; Yaniv, M. Cell cycle-dependent variations in c-Jun and JunB phosphorylation: A role in the control of cyclin D1 expression. EMBO J. 2000, 19, 2056–2068. [Google Scholar] [CrossRef] [PubMed]
- Gazon, H.; Barbeau, B.; Mesnard, J.M.; Peloponese, J.M. Hijacking of the AP-1 Signaling Pathway during Development of ATL; Frontiers Media S.A.: Lausanne, Switzerland, 2018; Volume 8. [Google Scholar]
- Feehan, R.P.; Shantz, L.M. Molecular signaling cascades involved in nonmelanoma skin carcinogenesis. Biochem. J. 2016, 473, 2973–2994. [Google Scholar] [CrossRef]
- Shaulian, E.; Karin, M. AP-1 in Cell Proliferation and Survival. Oncogene 2001, 20, 2390–2400. [Google Scholar] [CrossRef]
- Jin, Y.-J.; Park, I.; Hong, I.-K.; Byun, H.-J.; Choi, J.; Kim, Y.-M.; Lee, H. Fibronectin and vitronectin induce AP-1-mediated matrix metalloproteinase-9 expression through integrin α5β1/αvβ3-dependent Akt, ERK and JNK signaling pathways in human umbilical vein endothelial cells. Cell. Signal. 2011, 23, 125–134. [Google Scholar] [CrossRef]
- Chen, Y.R.; Tan, T.H. The c-Jun N-Terminal Kinase Pathway and Apoptotic Signaling (Review). Int. J. Oncol. 2000, 16, 651–662. [Google Scholar] [CrossRef]
- Lamb, J.A.; Ventura, J.-J.; Hess, P.; Flavell, R.A.; Davis, R.J. JunD mediates survival signaling by the JNK signal transduction pathway. Mol. Cell 2003, 11, 1479–1489. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Angiolilli, C.; Hartkamp, L.M.; Van Baarsen, L.G.M.; Tak, P.P.; Reedquist, K.A. JNK-dependent downregulation of FoxO1 is required to promote the survival of fibroblast-like synoviocytes in rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 1763–1771. [Google Scholar] [CrossRef]
- Wu, Q.; Wu, W.; Fu, B.; Shi, L.; Wang, X.; Kuca, K. JNK Signaling in Cancer Cell Survival; John Wiley and Sons Inc.: Hoboken, NJ, USA, 2019; Volume 39, pp. 2082–2104. [Google Scholar]
- Jones, E.V.; Dickman, M.J.; Whitmarsh, A.J. Regulation of p73-mediated apoptosis by c-Jun N-terminal kinase. Biochem. J. 2007, 405, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Wolf, E.R.; McAtarsney, C.P.; Bredhold, K.E.; Kline, A.M.; Mayo, L.D. Mutant and wild-type p53 form complexes with p73 upon phosphorylation by the kinase JNK. Sci. Signal. 2018, 11, eaao4170. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Zhang, T.; Liu, D.; Guan, G.; Huang, J.; Proksch, P.; Chen, X.; Lin, W. Notoamide-type alkaloid induced apoptosis and autophagy: Via a P38/JNK signaling pathway in hepatocellular carcinoma cells. RSC Adv. 2019, 9, 19855–19868. [Google Scholar] [CrossRef]
- Gong, X.; Wang, M.; Tashiro, S.-I.; Onodera, S.; Ikejima, T. Involvement of JNK-Initiated p53 Accumulation and Phosphorylation of p53 in Pseudolaric Acid B Induced Cell Death. Exp. Mol. Med. 2006, 428–434. [Google Scholar] [CrossRef]
- Annunziato, F.; Romagnani, C.; Romagnani, S. The 3 major types of innate and adaptive cell-mediated effector immunity. J. Allergy Clin. Immunol. 2015, 135, 626–635. [Google Scholar] [CrossRef] [PubMed]
- van Oosterhout, A.J.M.; Motta, A.C. Th1/Th2 paradigm: Not seeing the forest for the trees? Eur. Respir. J. 2005, 25, 591. [Google Scholar] [CrossRef]
- Kaiko, G.E.; Horvat, J.C.; Beagley, K.W.; Hansbro, P.M. Immunological decision-making: How does the immune system decide to mount a helper T-cell response? Immunology 2008, 123, 326–338. [Google Scholar] [CrossRef]
- Kidd, P. Th1/Th2 balance: The hypothesis, its limitations, and implications for health and disease. Altern. Med. Rev. 2003, 8, 223–246. [Google Scholar]
- Gieseck, R.L.; Wilson, M.S.; Wynn, T.A. Type 2 Immunity in Tissue Repair and Fibrosis; Nature Publishing Group: Berlin, Germany, 2018; Volume 18, pp. 62–76. [Google Scholar]
- Davis, R.J. Signal transduction by the JNK group of MAP kinases. Cell 2000, 103, 239–252. [Google Scholar] [CrossRef]
- Dong, C.; Davis, R.J.; Flavell, R.A. MAP K INASES IN THE I MMUNE R ESPONSE. Annu. Rev. Immunol. 2002, 20, 55–72. [Google Scholar] [CrossRef]
- Ghosh, S.; Karin, M. Missing pieces in the NF-kappaB puzzle. Cell 2002, 109, S81–S96. [Google Scholar] [CrossRef]
- Rincón, M.; Davis, R.J. Regulation of the immune response by stress-activated protein kinases. Immunol. Rev. 2009, 228, 212–224. [Google Scholar] [CrossRef] [PubMed]
- Huang, G.; Shi, L.Z.; Chi, H. Regulation of JNK and p38 MAPK in the immune system: Signal integration, propagation and termination. Cytokine 2009, 48, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK Expression by Macrophages Promotes Obesity-Induced Insulin Resistance and Inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C.A.; Cardwell, L.N.; Guan, Y.; Teixeiro, E.; Daniels, M.A. POSH Regulates CD4+ T Cell Differentiation and Survival. J. Immunol. 2016, 196, 4003–4013. [Google Scholar] [CrossRef] [PubMed]
- Shebzukhov, Y.V.; Stanislawiak, S.; Bezhaeva, T.R.; Nedospasov, S.A.; Kuprash, D.V. Low level of Lck kinase in Th2 cells limits expression of CD4 co-receptor and S73 phosphorylation of transcription factor c-Jun. Sci. Rep. 2017, 7, 2339. [Google Scholar] [CrossRef] [PubMed]
- Coquet, J.M.; Middendorp, S.; van der Horst, G.; Kind, J.; Veraar, E.A.M.; Xiao, Y.; Jacobs, H.; Borst, J. The CD27 and CD70 Costimulatory Pathway Inhibits Effector Function of T Helper 17 Cells and Attenuates Associated Autoimmunity. Immunity 2013, 38, 53–65. [Google Scholar] [CrossRef]
- Di Meglio, P.; Villanova, F.; Nestle, F.O. Psoriasis. Cold Spring Harb. Perspect. Med. 2014, 4, a015354. [Google Scholar] [CrossRef]
- Boehncke, W.-H.; Schön, M.P. Psoriasis. Lancet 2015, 386, 983–994. [Google Scholar] [CrossRef]
- Lowes, M.A.; Kikuchi, T.; Fuentes-Duculan, J.; Cardinale, I.; Zaba, L.C.; Haider, A.S.; Bowman, E.P.; Krueger, J.G. Psoriasis vulgaris lesions contain discrete populations of Th1 and Th17 T cells. J. Investig. Dermatol. 2008, 128, 1207–1211. [Google Scholar] [CrossRef]
- Chiricozzi, A.; Romanelli, P.; Volpe, E.; Borsellino, G.; Romanelli, M. Scanning the Immunopathogenesis of Psoriasis. Int. J. Mol. Sci. 2018, 19, 179. [Google Scholar] [CrossRef] [PubMed]
- Lowes, M.A.; Suárez-Fariñas, M.; Krueger, J.G. Immunology of Psoriasis. Annu. Rev. Immunol. 2014, 32, 227–255. [Google Scholar] [CrossRef] [PubMed]
- Kotb, I.S.; Lewis, B.J.; Barker, R.N.; Ormerod, A.D. Differential effects of phototherapy, adalimumab and betamethasone-calcipotriol on effector and regulatory T cells in psoriasis. Br. J. Derm. 2018, 179, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Elder, J.T. PSORS1: Linking Genetics and Immunology. J. Investig. Dermatol. 2006, 126, 1205–1206. [Google Scholar] [CrossRef]
- Jordan, C.T.; Cao, L.; Roberson, E.D.O.; Pierson, K.C.; Yang, C.F.; Joyce, C.E.; Ryan, C.; Duan, S.; Helms, C.A.; Liu, Y.; et al. PSORS2 is due to mutations in CARD14. Am. J. Hum. Genet. 2012, 90, 784–795. [Google Scholar] [CrossRef]
- Tsoi, L.C.; Spain, S.L.; Knight, J.; Ellinghaus, E.; Stuart, P.E.; Capon, F.; Ding, J.; Li, Y.; Tejasvi, T.; Gudjonsson, J.E.; et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012, 44, 1341–1348. [Google Scholar] [CrossRef]
- Singh, S.; Pradhan, D.; Puri, P.; Ramesh, V.; Aggarwal, S.; Nayek, A.; Jain, A.K. Genomic Alterations Driving Psoriasis Pathogenesis; Elsevier B.V.: Amsterdam, The Netherlands, 2019; Volume 683, pp. 61–71. [Google Scholar]
- Wang, H.; Chan, H.H.; Ni, M.Y.; Lam, W.W.; Chan, W.M.M.; Pang, H. Bacteriophage of the Skin Microbiome in Patients with Psoriasis and Healthy Family Controls. J. Investig. Dermatol. 2020, 140, 182–190 e185. [Google Scholar] [CrossRef]
- Loesche, M.A.; Farahi, K.; Capone, K.; Fakharzadeh, S.; Blauvelt, A.; Duffin, K.C.; DePrimo, S.E.; Muñoz-Elías, E.J.; Brodmerkel, C.; Dasgupta, B.; et al. Longitudinal Study of the Psoriasis-Associated Skin Microbiome during Therapy with Ustekinumab in a Randomized Phase 3b Clinical Trial. J. Investig. Dermatol. 2018, 138, 1973–1981. [Google Scholar] [CrossRef]
- Afonina, I.S.; Van Nuffel, E.; Baudelet, G.; Driege, Y.; Kreike, M.; Staal, J.; Beyaert, R. The paracaspase MALT1 mediates CARD14-induced signaling in keratinocytes. EMBO Rep. 2016, 17, 914–927. [Google Scholar] [CrossRef]
- Blonska, M.; Lin, X. NF-κB Signaling Pathways Regulated by CARMA Family of Scaffold Proteins. Cell Res. 2011, 21, 55–70. [Google Scholar] [CrossRef]
- Hulpiau, P.; Driege, Y.; Staal, J.; Beyaert, R. MALT1 is not alone after all: Identification of novel paracaspases. Cell. Mol. Life Sci. 2016, 73, 1103–1116. [Google Scholar] [CrossRef] [PubMed]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of Regulatory T Cell Development by the Transcription Factor Foxp3. Science 2003, 299, 1057. [Google Scholar] [CrossRef] [PubMed]
- Mu, J.; Tai, X.; Iyer, S.S.; Weissman, J.D.; Singer, A.; Singer, D.S. Regulation of MHC Class I Expression by Foxp3 and Its Effect on Regulatory T Cell Function. J. Immunol. 2014, 192, 2892–2903. [Google Scholar] [CrossRef] [PubMed]
- Lopes, J.E.; Torgerson, T.R.; Schubert, L.A.; Anover, S.D.; Ocheltree, E.L.; Ochs, H.D.; Ziegler, S.F. Analysis of FOXP3 Reveals Multiple Domains Required for Its Function as a Transcriptional Repressor. J. Immunol. 2006, 177, 3133–3142. [Google Scholar] [CrossRef]
- Chen, L.; Wu, J.; Ren, W.; Yang, X.; Shen, Z. c-Jun N-terminal kinase (JNK)-phospho-c-JUN (ser63/73) pathway is essential for FOXP3 nuclear translocation in psoriasis. J. Dermatol. Sci. 2013, 69, 114–121. [Google Scholar] [CrossRef]
- Gao, L.; Li, K.; Li, F.; Li, H.; Liu, L.; Wang, L.; Zhang, Z.; Gao, T.; Liu, Y. Polymorphisms in the FOXP3 gene in Han Chinese psoriasis patients. J. Dermatol. Sci. 2010, 57, 51–56. [Google Scholar] [CrossRef]
- Wu, P.; Ma, G.; Zhu, X.; Gu, T.; Zhang, J.; Sun, Y.; Xu, H.; Huo, R.; Wang, B.; Shen, B.; et al. Cyr61/CCN1 is involved in the pathogenesis of psoriasis vulgaris via promoting IL-8 production by keratinocytes in a JNK/NF-κB pathway. Clin. Immunol. 2017, 174, 53–62. [Google Scholar] [CrossRef]
- Li, H.; Li, H.; Huo, R.; Wu, P.; Shen, Z.; Xu, H.; Shen, B.; Li, N. Cyr61/CCN1 induces CCL20 production by keratinocyte via activating p38 and JNK/AP-1 pathway in psoriasis. J. Dermatol. Sci. 2017, 88, 46–56. [Google Scholar] [CrossRef]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schröder, J.M.; Wang, J.M.; Howard, O.M.Z.; et al. β-Defensins: Linking innate and adaptive immunity through dendritic and T cell CCR6. Science 1999, 286, 525–528. [Google Scholar] [CrossRef]
- Niyonsaba, F.; Ushio, H.; Nakano, N.; Ng, W.; Sayama, K.; Hashimoto, K.; Nagaoka, I.; Okumura, K.; Ogawa, H. Antimicrobial peptides human β-defensins stimulate epidermal keratinocyte migration, proliferation and production of proinflammatory cytokines and chemokines. J. Investig. Dermatol. 2007, 127, 594–604. [Google Scholar] [CrossRef]
- Kanda, N.; Kamata, M.; Tada, Y.; Ishikawa, T.; Sato, S.; Watanabe, S. Human β-defensin-2 enhances IFN-γ and IL-10 production and suppresses IL-17 production in T cells. J. Leukoc. Biol. 2011, 89, 935–944. [Google Scholar] [CrossRef] [PubMed]
- Karakawa, M.; Komine, M.; Hanakawa, Y.; Tsuda, H.; Sayama, K.; Tamaki, K.; Ohtsuki, M. CCL27 is downregulated by interferon gamma via epidermal growth factor receptor in normal human epidermal keratinocytes. J. Cell. Physiol. 2014, 229, 1935–1945. [Google Scholar] [CrossRef] [PubMed]
- Arasa, J.; Terencio, M.C.; Andrés, R.M.; Marín-Castejón, A.; Valcuende-Cavero, F.; Payá, M.; Montesinos, M.C. Defective Induction of COX-2 Expression by Psoriatic Fibroblasts Promotes Pro-inflammatory Activation of Macrophages. Front. Immunol. 2019, 10, 536. [Google Scholar] [CrossRef] [PubMed]
- Lotti, T.; D’Erme, A.M.; Hercogová, J. The Role of Neuropeptides in the Control of Regional Immunity; Elsevier Inc.: Amsterdam, The Netherlands, 2014; Volume 32, pp. 633–645. [Google Scholar]
- Granstein, R.D.; Wagner, J.A.; Stohl, L.L.; Ding, W. Calcitonin gene-related peptide: Key regulator of cutaneous immunity. Acta Physiol. 2015, 213, 586–594. [Google Scholar] [CrossRef]
- Reich, A.; Orda, A.; Wiśnicka, B.; Szepietowski, J.C. Plasma concentration of selected neuropeptides in patients suffering from psoriasis. Exp. Dermatol. 2007, 16, 421–428. [Google Scholar] [CrossRef]
- Yu, X.J.; Li, C.Y.; Xu, Y.H.; Chen, L.M.; Zhou, C.L. Calcitonin gene-related peptide increases proliferation of human HaCaT keratinocytes by activation of MAP kinases. Cell Biol. Int. 2009, 33, 1144–1148. [Google Scholar] [CrossRef]
- Theoharides, T.C.; Zhang, B.; Kempuraj, D.; Tagen, M.; Vasiadi, M.; Angelidou, A.; Alysandratos, K.D.; Kalogeromitros, D.; Asadi, S.; Stavrianeas, N.; et al. IL-33 augments substance P-induced VEGF secretion from human mast cells and is increased in psoriatic skin. Proc. Natl. Acad. Sci. USA 2010, 107, 4448–4453. [Google Scholar] [CrossRef]
- Ostrowski, S.M.; Belkadi, A.; Loyd, C.M.; Diaconu, D.; Ward, N.L. Cutaneous denervation of psoriasiform mouse skin improves acanthosis and inflammation in a sensory neuropeptide-dependent manner. J. Investig. Dermatol. 2011, 131, 1530–1538. [Google Scholar] [CrossRef]
- Yu, X.J.; Ren, X.H.; Xu, Y.H.; Chen, L.M.; Zhou, C.L.; Li, C.Y. Vasoactive intestinal peptide induces vascular endothelial growth factor production in human HaCaT keratinocytes via MAPK pathway. Neuropeptides 2010, 44, 407–411. [Google Scholar] [CrossRef]
- Liang, J.; Chen, P.; Li, C.; Li, D.; Wang, J.; Xue, R.; Zhang, S.; Ruan, J.; Zhang, X. IL-22 Down-Regulates Cx43 Expression and Decreases Gap Junctional Intercellular Communication by Activating the JNK Pathway in Psoriasis. J. Investig. Dermatol. 2019, 139, 400–411. [Google Scholar] [CrossRef]
- Langlois, S.; Maher, A.C.; Manias, J.L.; Shao, Q.; Kidder, G.M.; Laird, D.W. Connexin levels regulate keratinocyte differentiation in the epidermis. J. Biol. Chem. 2007, 282, 30171–30180. [Google Scholar] [CrossRef]
- Kim, B.E.; Howell, M.D.; Guttman, E.; Gilleaudeau, P.M.; Cardinale, I.R.; Boguniewicz, M.; Krueger, J.G.; Leung, D.Y.M. TNF-α downregulates filaggrin and loricrin through c-Jun N-terminal kinase: Role for TNF-α antagonists to improve skin barrier. J. Investig. Dermatol. 2011, 131, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.R.; Tan, G.Z.; Cao, C.X.; Han, Y.F.; Meng, Z.; Man, X.Y.; Jiang, Z.X.; Zhang, Y.P.; Dang, N.N.; Wei, K.H.; et al. Decrease of galectin-3 in keratinocytes: A potential diagnostic marker and a critical contributor to the pathogenesis of psoriasis. J. Autoimmun. 2018, 89, 30–40. [Google Scholar] [CrossRef] [PubMed]
- Wynn, T.A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat. Rev. Immunol 2004, 4, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Thomas, R.M.; Worswick, S.; Aleshin, M. Retinoic acid for treatment of systemic sclerosis and morphea: A literature review. Dermatol. Ther. 2017, 30. [Google Scholar] [CrossRef]
- Sharma, A. Scleroderma-like Disorders. Curr. Rheumatol. Rev. 2018, 14, 22–27. [Google Scholar] [CrossRef]
- Reich, N.; Tomcik, M.; Zerr, P.; Lang, V.; Dees, C.; Avouac, J.; Palumbo, K.; Horn, A.; Akhmetshina, A.; Beyer, C.; et al. Jun N-terminal kinase as a potential molecular target for prevention and treatment of dermal fibrosis. Ann. Rheum. Dis. 2012, 71, 737–745. [Google Scholar] [CrossRef]
- Tamaki, Z.; Asano, Y.; Kubo, M.; Ihn, H.; Tada, Y.; Sugaya, M.; Kadono, T.; Sato, S. Effects of the immunosuppressant rapamycin on the expression of human alpha2(I) collagen and matrix metalloproteinase 1 genes in scleroderma dermal fibroblasts. J. Dermatol. Sci. 2014, 74, 251–259. [Google Scholar] [CrossRef]
- Brembilla, N.C.; Montanari, E.; Truchetet, M.E.; Raschi, E.; Meroni, P.; Chizzolini, C. Th17 cells favor inflammatory responses while inhibiting type I collagen deposition by dermal fibroblasts: Differential effects in healthy and systemic sclerosis fibroblasts. Arthritis Res. Ther. 2013, 15, R151. [Google Scholar] [CrossRef]
- Gilbane, A.J.; Denton, C.P.; Holmes, A.M. Scleroderma pathogenesis: A pivotal role for fibroblasts as effector cells. Arthritis Res. Ther. 2013, 15, 215. [Google Scholar] [CrossRef]
- Chakraborty, D.; Sumova, B.; Mallano, T.; Chen, C.W.; Distler, A.; Bergmann, C.; Ludolph, I.; Horch, R.E.; Gelse, K.; Ramming, A.; et al. Activation of STAT3 integrates common profibrotic pathways to promote fibroblast activation and tissue fibrosis. Nat. Commun. 2017, 8, 1130. [Google Scholar] [CrossRef]
- Hu, H.H.; Chen, D.Q.; Wang, Y.N.; Feng, Y.L.; Cao, G.; Vaziri, N.D.; Zhao, Y.Y. New insights into TGF-beta/Smad signaling in tissue fibrosis. Chem. -Biol. Interact. 2018, 292, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Meng, X.M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-beta: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef] [PubMed]
- Klinkhammer, B.M.; Floege, J.; Boor, P. PDGF in organ fibrosis. Mol. Asp. Med. 2018, 62, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Ying, H.Z.; Chen, Q.; Zhang, W.Y.; Zhang, H.H.; Ma, Y.; Zhang, S.Z.; Fang, J.; Yu, C.H. PDGF signaling pathway in hepatic fibrosis pathogenesis and therapeutics (Review). Mol. Med. Rep. 2017, 16, 7879–7889. [Google Scholar] [CrossRef] [PubMed]
- Finnson, K.W.; Almadani, Y.; Philip, A. Non-canonical (non-SMAD2/3) TGF-beta signaling in fibrosis: Mechanisms and targets. Semin. Cell Dev. Biol. 2019, in press. [Google Scholar] [CrossRef]
- Plantevin Krenitsky, V.; Nadolny, L.; Delgado, M.; Ayala, L.; Clareen, S.S.; Hilgraf, R.; Albers, R.; Hegde, S.; D’Sidocky, N.; Sapienza, J.; et al. Discovery of CC-930, an orally active anti-fibrotic JNK inhibitor. Bioorganic Med. Chem. Lett. 2012, 22, 1433–1438. [Google Scholar] [CrossRef]
- Sabapathy, K. Role of the JNK pathway in human diseases. Prog. Mol. Biol. Transl. Sci. 2012, 106, 145–169. [Google Scholar] [CrossRef]
- Avouac, J.; Palumbo, K.; Tomcik, M.; Zerr, P.; Dees, C.; Horn, A.; Maurer, B.; Akhmetshina, A.; Beyer, C.; Sadowski, A.; et al. Inhibition of activator protein 1 signaling abrogates transforming growth factor beta-mediated activation of fibroblasts and prevents experimental fibrosis. Arthritis Rheum. 2012, 64, 1642–1652. [Google Scholar] [CrossRef]
- Tourkina, E.; Richard, M.; Oates, J.; Hofbauer, A.; Bonner, M.; Gooz, P.; Visconti, R.; Zhang, J.; Znoyko, S.; Hatfield, C.M.; et al. Caveolin-1 regulates leucocyte behaviour in fibrotic lung disease. Ann. Rheum. Dis. 2010, 69, 1220–1226. [Google Scholar] [CrossRef]
- Beyer, C.; Reichert, H.; Akan, H.; Mallano, T.; Schramm, A.; Dees, C.; Palumbo-Zerr, K.; Lin, N.Y.; Distler, A.; Gelse, K.; et al. Blockade of canonical Wnt signalling ameliorates experimental dermal fibrosis. Ann. Rheum. Dis. 2013, 72, 1255–1258. [Google Scholar] [CrossRef] [PubMed]
- van der Velden, J.L.; Alcorn, J.F.; Chapman, D.G.; Lundblad, L.K.A.; Irvin, C.G.; Davis, R.J.; Butnor, K.; Janssen-Heininger, Y.M.W. Airway epithelial specific deletion of Jun-N-terminal kinase 1 attenuates pulmonary fibrosis in two independent mouse models. PLoS ONE 2020, 15, e0226904. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Cai, Y.; Soofi, A.; Dressler, G.R. Activation of Wnt11 by transforming growth factor-beta drives mesenchymal gene expression through non-canonical Wnt protein signaling in renal epithelial cells. J. Biol. Chem. 2012, 287, 21290–21302. [Google Scholar] [CrossRef] [PubMed]
- Distler, A.; Ziemer, C.; Beyer, C.; Lin, N.Y.; Chen, C.W.; Palumbo-Zerr, K.; Dees, C.; Weidemann, A.; Distler, O.; Schett, G.; et al. Inactivation of evenness interrupted (EVI) reduces experimental fibrosis by combined inhibition of canonical and non-canonical Wnt signalling. Ann. Rheum. Dis. 2014, 73, 624–627. [Google Scholar] [CrossRef] [PubMed]
- Akhmetshina, A.; Palumbo, K.; Dees, C.; Bergmann, C.; Venalis, P.; Zerr, P.; Horn, A.; Kireva, T.; Beyer, C.; Zwerina, J.; et al. Activation of canonical Wnt signalling is required for TGF-beta-mediated fibrosis. Nat. Commun. 2012, 3, 735. [Google Scholar] [CrossRef] [PubMed]
- Bartscherer, K.; Pelte, N.; Ingelfinger, D.; Boutros, M. Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell 2006, 125, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Fineschi, S.; Reith, W.; Guerne, P.A.; Dayer, J.M.; Chizzolini, C. Proteasome blockade exerts an antifibrotic activity by coordinately down-regulating type I collagen and tissue inhibitor of metalloproteinase-1 and up-regulating metalloproteinase-1 production in human dermal fibroblasts. FASEB J. 2006, 20, 562–564. [Google Scholar] [CrossRef]
- Zhou, B.; Zhu, H.; Luo, H.; Gao, S.; Dai, X.; Li, Y.; Zuo, X. MicroRNA-202-3p regulates scleroderma fibrosis by targeting matrix metalloproteinase 1. Biomed. Pharmacother. = Biomed. Pharmacother. 2017, 87, 412–418. [Google Scholar] [CrossRef]
- Arakaki, P.A.; Marques, M.R.; Santos, M.C. MMP-1 polymorphism and its relationship to pathological processes. J. Biosci. 2009, 34, 313–320. [Google Scholar] [CrossRef]
- Cortez, D.M.; Feldman, M.D.; Mummidi, S.; Valente, A.J.; Steffensen, B.; Vincenti, M.; Barnes, J.L.; Chandrasekar, B. IL-17 stimulates MMP-1 expression in primary human cardiac fibroblasts via p38 MAPK- and ERK1/2-dependent C/EBP-beta, NF-kappaB, and AP-1 activation. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3356–H3365. [Google Scholar] [CrossRef]
- Poulalhon, N.; Farge, D.; Roos, N.; Tacheau, C.; Neuzillet, C.; Michel, L.; Mauviel, A.; Verrecchia, F. Modulation of collagen and MMP-1 gene expression in fibroblasts by the immunosuppressive drug rapamycin. A direct role as an antifibrotic agent? J. Biol. Chem. 2006, 281, 33045–33052. [Google Scholar] [CrossRef] [PubMed]
- Murai, M.; Yamamura, K.; Hashimoto-Hachiya, A.; Tsuji, G.; Furue, M.; Mitoma, C. Tryptophan photo-product FICZ upregulates AHR/MEK/ERK-mediated MMP1 expression: Implications in anti-fibrotic phototherapy. J. Dermatol. Sci. 2018, 91, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Fineschi, S.; Goffin, L.; Rezzonico, R.; Cozzi, F.; Dayer, J.M.; Meroni, P.L.; Chizzolini, C. Antifibroblast antibodies in systemic sclerosis induce fibroblasts to produce profibrotic chemokines, with partial exploitation of toll-like receptor 4. Arthritis Rheum. 2008, 58, 3913–3923. [Google Scholar] [CrossRef] [PubMed]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK Signaling Pathway in Renal Fibrosis; Frontiers Media S.A.: Lausanne, Switzerland, 2017; Volume 8. [Google Scholar]
- Wang, Y.; Chen, L.; Wang, K.; Da, Y.; Zhou, M.; Yan, H.; Zheng, D.; Zhong, S.; Cai, S.; Zhu, H.; et al. Suppression of TRPM2 reduces renal fibrosis and inflammation through blocking TGF-beta1-regulated JNK activation. Biomed. Pharm. 2019, 120, 109556. [Google Scholar] [CrossRef] [PubMed]
- Bourlidou, E.; Vahtsevanos, K.; Kyrgidis, A.; Tilaveridis, I.; Patsatsi, A.; Andreadis, D.; Cheva, A.; Patrikidou, A.; Kitikidou, K.; Boboridis, K. Risk factors for local recurrence of basal cell carcinoma and cutaneous squamous cell carcinoma of the middle third of the face: A 15-year retrospective analysis based on a single centre. Eur. J. Dermatol. 2019, 29, 490–499. [Google Scholar] [CrossRef] [PubMed]
- Kansara, S.; Bell, D.; Weber, R. Surgical management of non melanoma skin cancer of the head and neck. Oral Oncol. 2020, 100, 104485. [Google Scholar] [CrossRef]
- Muzic, J.G.; Schmitt, A.R.; Wright, A.C.; Alniemi, D.T.; Zubair, A.S.; Olazagasti Lourido, J.M.; Sosa Seda, I.M.; Weaver, A.L.; Baum, C.L. Incidence and Trends of Basal Cell Carcinoma and Cutaneous Squamous Cell Carcinoma: A Population-Based Study in Olmsted County, Minnesota, 2000 to 2010. Mayo Clin. Proc. 2017, 92, 890–898. [Google Scholar] [CrossRef]
- Rogers, H.W.; Weinstock, M.A.; Feldman, S.R.; Coldiron, B.M. Incidence Estimate of Nonmelanoma Skin Cancer (Keratinocyte Carcinomas) in the US Population, 2012. JAMA Dermatol. 2015, 151, 1081–1086. [Google Scholar] [CrossRef]
- American Cancer Society. What Are Basal and Squamous Cell Skin Cancers?|Types of Skin Cancer 2019; American Cancer Society: Atlanta, GA, USA, 2019. [Google Scholar]
- American Cancer Society. Cancer Facts and Figures 2020; American Cancer Society: Atlanta, GA, USA, 2020. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA A Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef]
- Rubin, A.I.; Chen, E.H.; Ratner, D. Basal-Cell Carcinoma. N. Engl. J. Med. 2005, 353, 2262–2269. [Google Scholar] [CrossRef]
- Epstein, E.H. Basal cell carcinomas: Attack of the hedgehog. Nat. Rev. Cancer 2008, 8, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Kasper, M.; Jaks, V.; Are, A.; Bergstrom, A.; Schwager, A.; Svard, J.; Teglund, S.; Barker, N.; Toftgard, R. Wounding enhances epidermal tumorigenesis by recruiting hair follicle keratinocytes. Proc. Natl. Acad. Sci. USA 2011, 108, 4099–4104. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.Y.; Reiter, J.F. Wounding mobilizes hair follicle stem cells to form tumors. Proc. Natl. Acad. Sci. USA 2011, 108, 4093–4098. [Google Scholar] [CrossRef] [PubMed]
- Smola, S. Immunopathogenesis of HPV-Associated Cancers and Prospects for Immunotherapy; MDPI AG: Basel, Switzerland, 2017; Volume 9. [Google Scholar]
- Shaulian, E. AP-1—The Jun proteins: Oncogenes or tumor suppressors in disguise? Cell. Signal. 2010, 22, 894–899. [Google Scholar] [CrossRef]
- Kharman-Biz, A.; Gao, H.; Ghiasvand, R.; Zhao, C.; Zendehdel, K.; Dahlman-Wright, K. Expression of activator protein-1 (AP-1) family members in breast cancer. BMC Cancer 2013, 13, 441. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Adams, A.E.; Ridky, T.W.; Tao, S.; Khavari, P.A. Tumor Necrosis Factor Receptor 1/c-Jun-NH2-Kinase Signaling Promotes Human Neoplasia. Cancer Res. 2007, 67, 3827–3834. [Google Scholar] [CrossRef]
- Bubici, C.; Papa, S. JNK signalling in cancer: In need of new, smarter therapeutic targets. Br. J. Pharmacol. 2014, 171, 24–37. [Google Scholar] [CrossRef]
- Ke, H.; Harris, R.; Coloff, J.L.; Jin, J.Y.; Leshin, B.; Miliani de Marval, P.; Tao, S.; Rathmell, J.C.; Hall, R.P.; Zhang, J.Y. The c-Jun NH2-terminal kinase 2 plays a dominant role in human epidermal neoplasia. Cancer Res. 2010, 70, 3080–3088. [Google Scholar] [CrossRef]
- Chen, P.; O’Neal, J.F.; Ebelt, N.D.; Cantrell, M.A.; Mitra, S.; Nasrazadani, A.; Vandenbroek, T.L.; Heasley, L.E.; Van Den Berg, C.L. Jnk2 Effects on Tumor Development, Genetic Instability and Replicative Stress in an Oncogene-Driven Mouse Mammary Tumor Model. PLoS ONE 2010, 5, e10443. [Google Scholar] [CrossRef]
- Finegan, K.G.; Tournier, C. The mitogen-activated protein kinase kinase 4 has a pro-oncogenic role in skin cancer. Cancer Res. 2010, 70, 5797–5806. [Google Scholar] [CrossRef]
- She, Q.-B.; Chen, N.; Bode, A.M.; Flavell, R.A.; Dong, Z. Deficiency of c-Jun-NH(2)-terminal kinase-1 in mice enhances skin tumor development by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 2002, 62, 1343–1348. [Google Scholar] [PubMed]
- Jin, J.Y.; Ke, H.; Hall, R.P.; Zhang, J.Y. C-Jun promotes whereas JunB inhibits epidermal neoplasia. J. Investig. Dermatol. 2011, 131, 1149–1158. [Google Scholar] [CrossRef] [PubMed]
- Katagiri, C.; Nakanishi, J.; Kadoya, K.; Hibino, T. Serpin squamous cell carcinoma antigen inhibits UV-induced apoptosis via suppression of c-JUN NH2-terminal kinase. J. Cell Biol. 2006, 172, 983–990. [Google Scholar] [CrossRef] [PubMed]
- De Marval, P.M.; Lutfeali, S.; Jin, J.Y.; Leshin, B.; Angelica Selim, M.; Zhang, J.Y. CYLD inhibits tumorigenesis and metastasis by blocking JNK/AP1 signaling at multiple levels. Cancer Prev. Res. 2011, 4, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Pan, H.; Li, J.; Zhong, Q.; Chen, X.; Dry, S.M.; Wang, C.Y. Epigenetic activation of AP1 promotes squamous cell carcinoma metastasis. Sci. Signal. 2013, 6, ra28.1–ra28.13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wu, J.; Luo, S.; Lechler, T.; Zhang, J.Y. FRA1 promotes squamous cell carcinoma growth and metastasis through distinct AKT and c-Jun dependent mechanisms. Oncotarget 2016, 7, 34371–34383. [Google Scholar] [CrossRef] [PubMed]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The Role of the Hedgehog Signaling Pathway in Cancer: A Comprehensive Review. Bosn. J. Basic Med. J. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Laner-Plamberger, S.; Kaser, A.; Paulischta, M.; Hauser-Kronberger, C.; Eichberger, T.; Frischauf, A.M. Cooperation between GLI and JUN enhances transcription of JUN and selected GLI target genes. Oncogene 2009, 28, 1639–1651. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Selim, M.A. The role of the c-Jun N-terminal Kinase signaling pathway in skin cancer. Am. J. Cancer Res. 2012, 2, 691–698. [Google Scholar]
- Bigelow, R.L.H.; Jen, E.Y.; Delehedde, M.; Chari, N.S.; McDonnell, T.J. Sonic hedgehog induces epidermal growth factor dependent matrix infiltration in HaCat keratinocytes. J. Investig. Dermatol. 2005, 124, 457–465. [Google Scholar] [CrossRef]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Takeuchi, S.; Moroi, Y.; Hayashida, S.; Kido, M.; Chen, S.J.; Tomoeda, H.; Uenotsuchi, T.; Tu, Y.T.; Furue, M.; et al. Overexpression of Phosphorylated-ATF2 and STAT3 in Cutaneous Squamous Cell Carcinoma, Bowen’s Disease and Basal Cell Carcinoma. J. Dermatol. Sci. 2008, 51, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bergami, P.; Lau, E.; Ronai, Z. Emerging roles of ATF2 and the dynamic AP1 network in cancer. Nat. Rev. Cancer 2010, 10, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Maglic, D.; Schlegelmilch, K.; Dost, A.F.; Panero, R.; Dill, M.T.; Calogero, R.A.; Camargo, F.D. YAP-TEAD signaling promotes basal cell carcinoma development via a c-JUN/AP1 axis. EMBO J. 2018, 37. [Google Scholar] [CrossRef]
- Teh, M.-T.; Blaydon, D.; Ghali, L.R.; Edmunds, S.; Pantazi, E.; Barnes, M.R.; Leigh, I.M.; Kelsell, D.P.; Philpott, M.P. Role for WNT16B in human epidermal keratinocyte proliferation and differentiation. J. Cell Sci. 2007, 120, 330. [Google Scholar] [CrossRef]
- Jørgensen, K.; Davidson, B.; Flørenes, V.A. Activation of c-jun N-terminal kinase is associated with cell proliferation and shorter relapse-free period in superficial spreading malignant melanoma. Mod. Pathol 2006, 19, 1446–1455. [Google Scholar] [CrossRef]
- Kappelmann, M.; Bosserhoff, A.; Kuphal, S. AP-1/c-Jun transcription factors: Regulation and function in malignant melanoma. Eur. J. Cell Biol. 2014, 93, 76–81. [Google Scholar] [CrossRef]
- Lopez-Bergami, P.; Huang, C.; Goydos, J.S.; Yip, D.; Bar-Eli, M.; Herlyn, M.; Smalley, K.S.M.; Mahale, A.; Eroshkin, A.; Aaronson, S.; et al. Rewired ERK-JNK signaling pathways in melanoma. Cancer Cell 2007, 11, 447–460. [Google Scholar] [CrossRef]
- Kappelmann-Fenzl, M.; Gebhard, C.; Matthies, A.O.; Kuphal, S.; Rehli, M.; Bosserhoff, A.K. C-Jun drives melanoma progression in PTEN wild type melanoma cells. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef]
- Kogushi-Nishi, H.; Jinnin, M.; Kobayashi, Y.; Muchemwa, F.C.; Hirano, A.; Makino, T.; Fukushima, S.; Masuguchi, S.; Ishihara, T.; Inoue, Y.; et al. Role of c-Jun N-terminal kinase isoforms in the cellular activity of melanoma cell lines. Clin. Exp. Dermatol. 2013, 38, 890–896. [Google Scholar] [CrossRef]
- Alexaki, V.-I.; Javelaud, D.; Mauviel, A. JNK supports survival in melanoma cells by controlling cell cycle arrest and apoptosis. Pigm Cell Melanoma R 2008, 21, 429–438. [Google Scholar] [CrossRef] [PubMed]
- Ke, H.; Augustine, C.K.; Gandham, V.D.; Jin, J.Y.; Tyler, D.S.; Akiyama, S.K.; Hall, R.P.; Zhang, J.Y. CYLD Inhibits Melanoma Growth and Progression through Suppression of the JNK/AP-1 and β1-Integrin Signaling Pathways. J. Investig. Dermatol. 2013, 133, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, G.; Jin, J.; Degan, S.; Tameze, Y.; Zhang, J.Y. MALT1 promotes melanoma progression through JNK/c-Jun signaling. Oncogenesis 2017, 6, e365. [Google Scholar] [CrossRef] [PubMed]
- Kyula, J.N.; Khan, A.A.; Mansfield, D.; Karapanagiotou, E.M.; McLaughlin, M.; Roulstone, V.; Zaidi, S.; Pencavel, T.; Touchefeu, Y.; Seth, R.; et al. Synergistic cytotoxicity of radiation and oncolytic Lister strain vaccinia in (V600D/E)BRAF mutant melanoma depends on JNK and TNF-alpha signaling. Oncogene 2014, 33, 1700–1712. [Google Scholar] [CrossRef]
- Fallahi-Sichani, M.; Moerke, N.J.; Niepel, M.; Zhang, T.; Gray, N.S.; Sorger, P.K. Systematic analysis of BRAF V 600E melanomas reveals a role for JNK /c-Jun pathway in adaptive resistance to drug-induced apoptosis. Mol. Syst. Biol. 2015, 11, 797. [Google Scholar] [CrossRef]
- Bogoyevitch, M.A.; Ngoei, K.R.W.; Zhao, T.T.; Yeap, Y.Y.C.; Ng, D.C.H. c-Jun N-terminal kinase (JNK) signaling: Recent advances and challenges. Biochim. Biophys. Acta (BBA) - Proteins Proteom. 2010, 1804, 463–475. [Google Scholar] [CrossRef]
- Kumar, A.; Singh, U.K.; Kini, S.G.; Garg, V.; Agrawal, S.; Tomar, P.K.; Pathak, P.; Chaudhary, A.; Gupta, P.; Malik, A. JNK pathway signaling: A novel and smarter therapeutic targets for various biological diseases. Future Med. Chem. 2015, 7, 2065–2086. [Google Scholar] [CrossRef]
- Chen, T.; Kablaoui, N.; Little, J.; Timofeevski, S.; Tschantz, W.R.; Chen, P.; Feng, J.; Charlton, M.; Stanton, R.; Bauer, P. Identification of small-molecule inhibitors of the JIP–JNK interaction. Biochem. J. 2009, 420, 283–296. [Google Scholar] [CrossRef]
- Wong, C.H.; Iskandar, K.B.; Yadav, S.K.; Hirpara, J.L.; Loh, T.; Pervaiz, S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS ONE 2010, 5, e9996. [Google Scholar] [CrossRef]
- Gaillard, P.; Jeanclaude-Etter, I.; Ardissone, V.; Arkinstall, S.; Cambet, Y.; Camps, M.; Chabert, C.; Church, D.; Cirillo, R.; Gretener, D.; et al. Design and Synthesis of the First Generation of Novel Potent, Selective, and in Vivo Active (Benzothiazol-2-yl)acetonitrile Inhibitors of the c-Jun N-Terminal Kinase. J. Med. Chem. 2005, 48, 4596–4607. [Google Scholar] [CrossRef]
- Du, L.; Anderson, A.; Nguyen, K.; Ojeda, S.S.; Ortiz-Rivera, I.; Nguyen, T.N.; Zhang, T.; Kaoud, T.S.; Gray, N.S.; Dalby, K.N.; et al. JNK2 Is Required for the Tumorigenic Properties of Melanoma Cells. ACS Chem. Biol. 2019, 14, 1426–1435. [Google Scholar] [CrossRef] [PubMed]
- Riesenberg, S.; Groetchen, A.; Siddaway, R.; Bald, T.; Reinhardt, J.; Smorra, D.; Kohlmeyer, J.; Renn, M.; Phung, B.; Aymans, P.; et al. MITF and c-Jun antagonism interconnects melanoma dedifferentiation with pro-inflammatory cytokine responsiveness and myeloid cell recruitment. Nat. Commun. 2015, 6, 8755. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Hu, Y.; Sun, X.; Lu, G.; Li, Y.; Yang, J.; Tao, J. Suppression of c-FLIPL promotes JNK activation in malignant melanoma cells. Mol. Med. Rep. 2016, 13, 2904–2908. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.; Ekmekcioglu, S.; Liu, P.; Duncan, L.M.; Lizée, G.; Poindexter, N.; Grimm, E.A. Constitutive aberrant endogenous interleukin-1 facilitates inflammation and growth in human melanoma. Mol. Cancer Res. 2011, 9, 1537–1550. [Google Scholar] [CrossRef]
- Qin, Y.; Milton, D.R.; Oba, J.; Ding, Z.; Lizée, G.; Ekmekcioglu, S.; Grimm, E.A. Inflammatory IL-1β-driven JNK activation in stage III melanoma. Pigment. Cell Melanoma Res. 2015, 28, 236–239. [Google Scholar] [CrossRef]
- Kaoud, T.S.; Johnson, W.H.; Ebelt, N.D.; Piserchio, A.; Zamora-Olivares, D.; Van Ravenstein, S.X.; Pridgen, J.R.; Edupuganti, R.; Sammons, R.; Cano, M.; et al. Modulating multi-functional ERK complexes by covalent targeting of a recruitment site in vivo. Nat. Commun. 2019, 10, 5232. [Google Scholar] [CrossRef]
- Lu, H.; Liu, S.; Zhang, G.; Wu, B.; Zhu, Y.; Frederick, D.T.; Hu, Y.; Zhong, W.; Randell, S.; Sadek, N.; et al. PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 2017, 550, 133–136. [Google Scholar] [CrossRef]
- Szczepankiewicz, B.G.; Kosogof, C.; Nelson, L.T.J.; Liu, G.; Liu, B.; Zhao, H.; Serby, M.D.; Xin, Z.; Liu, M.; Gum, R.J.; et al. Aminopyridine-Based c-Jun N-Terminal Kinase Inhibitors with Cellular Activity and Minimal Cross-Kinase Activity †. J. Med. Chem. 2006, 49, 3563–3580. [Google Scholar] [CrossRef]
- LoGrasso, P.; Kamenecka, T. Inhibitors of c-jun-N-Terminal Kinase (JNK). Mini-Rev. Med. Chem. 2008, 8, 755–766. [Google Scholar] [CrossRef]
- Kamenecka, T.; Jiang, R.; Song, X.; Duckett, D.; Chen, W.; Ling, Y.Y.; Habel, J.; Laughlin, J.D.; Chambers, J.; Figuera-Losada, M.; et al. Synthesis, biological evaluation, X-ray structure, and pharmacokinetics of aminopyrimidine c-jun-N-terminal kinase (JNK) inhibitors. J. Med. Chem. 2010, 53, 419–431. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, M.; Zhang, Y.Q.; Xu, Z.H. JNK pathway: Diseases and therapeutic potential. Acta Pharmacol. Sin. 2007, 28, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Okada, M.; Takeda, H.; Sakaki, H.; Kuramoto, K.; Suzuki, S.; Sanomachi, T.; Togashi, K.; Seino, S.; Kitanaka, C. Repositioning CEP-1347, a chemical agent originally developed for the treatment of Parkinson’s disease, as an anti-cancer stem cell drug. Oncotarget 2017, 8, 94872–94882. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar] [CrossRef]
- Wang, W.; Shi, L.; Xie, Y.; Ma, C.; Li, W.; Su, X.; Huang, S.; Chen, R.; Zhu, Z.; Mao, Z.; et al. SP600125, a new JNK inhibitor, protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Neurosci. Res. 2004, 48, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Carboni, S.; Boschert, U.; Gaillard, P.; Gotteland, J.P.; Gillon, J.Y.; Vitte, P.A. AS601245, a c-Jun NH2-terminal kinase (JNK) inhibitor, reduces axon/dendrite damage and cognitive deficits after global cerebral ischaemia in gerbils. Br. J. Pharmacol. 2008, 153, 157–163. [Google Scholar] [CrossRef] [PubMed]
- Ferrandi, C.; Ballerio, R.; Gaillard, P.; Giachetti, C.; Carboni, S.; Vitte, P.-A.; Gotteland, J.-P.; Cirillo, R. Inhibition of c-Jun N-terminal kinase decreases cardiomyocyte apoptosis and infarct size after myocardial ischemia and reperfusion in anaesthetized rats. Br. J. Pharmacol. 2004, 142, 953–960. [Google Scholar] [CrossRef]
- Berwick, M. The Good, the Bad, and the Ugly of Sunscreens. Clin. Pharmacol. Ther. 2011, 89, 31–33. [Google Scholar] [CrossRef]
- van der Velden, J.L.J.; Ye, Y.; Nolin, J.D.; Hoffman, S.M.; Chapman, D.G.; Lahue, K.G.; Abdalla, S.; Chen, P.; Liu, Y.; Bennett, B.; et al. JNK inhibition reduces lung remodeling and pulmonary fibrotic systemic markers. Clin. Transl. Med. 2016, 5, 36. [Google Scholar] [CrossRef]
- Kukekov, N.V.; Xu, Z.; Greene, L.A. Direct Interaction of the Molecular Scaffolds POSH and JIP Is Required for Apoptotic Activation of JNKs. J. Biol. Chem. 2006, 281, 15517–15524. [Google Scholar] [CrossRef]
- Barr, R.K.; Bogoyevitch, M.A. The c-Jun N-terminal protein kinase family of mitogen-activated protein kinases (JNK MAPKs). Int. J. Biochem. Cell Biol 2001, 33, 1047–1063. [Google Scholar] [CrossRef]
- Barr, R.K.; Kendrick, T.S.; Bogoyevitch, M.A. Identification of the critical features of a small peptide inhibitor of JNK activity. J. Biol. Chem. 2002, 277, 10987–10997. [Google Scholar] [CrossRef] [PubMed]
- Barr, R.K.; Boehm, I.; Attwood, P.V.; Watt, P.M.; Bogoyevitch, M.A. The critical features and the mechanism of inhibition of a kinase interaction motif-based peptide inhibitor of JNK. J. Biol. Chem. 2004, 279, 36327–36338. [Google Scholar] [CrossRef] [PubMed]
- Gow, W.R.; Campbell, K.; Meade, A.J.; Watt, P.M.; Milech, N.; Knuckey, N.W.; Meloni, B.P. Lack of neuroprotection of inhibitory peptides targeting Jun/JNK after transient focal cerebral ischemia in spontaneously hypertensive rats. J. Cereb. Blood Flow Metab. 2011, 31, e1–e8. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; McLauchlan, H.; Elliott, M.; Cohen, P. The specificities of protein kinase inhibitors: An update. Biochem. J. 2003, 371, 199–204. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hammouda, M.B.; Ford, A.E.; Liu, Y.; Zhang, J.Y. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells 2020, 9, 857. https://doi.org/10.3390/cells9040857
Hammouda MB, Ford AE, Liu Y, Zhang JY. The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells. 2020; 9(4):857. https://doi.org/10.3390/cells9040857
Chicago/Turabian StyleHammouda, Manel B., Amy E. Ford, Yuan Liu, and Jennifer Y. Zhang. 2020. "The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer" Cells 9, no. 4: 857. https://doi.org/10.3390/cells9040857
APA StyleHammouda, M. B., Ford, A. E., Liu, Y., & Zhang, J. Y. (2020). The JNK Signaling Pathway in Inflammatory Skin Disorders and Cancer. Cells, 9(4), 857. https://doi.org/10.3390/cells9040857