Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes


Abstract
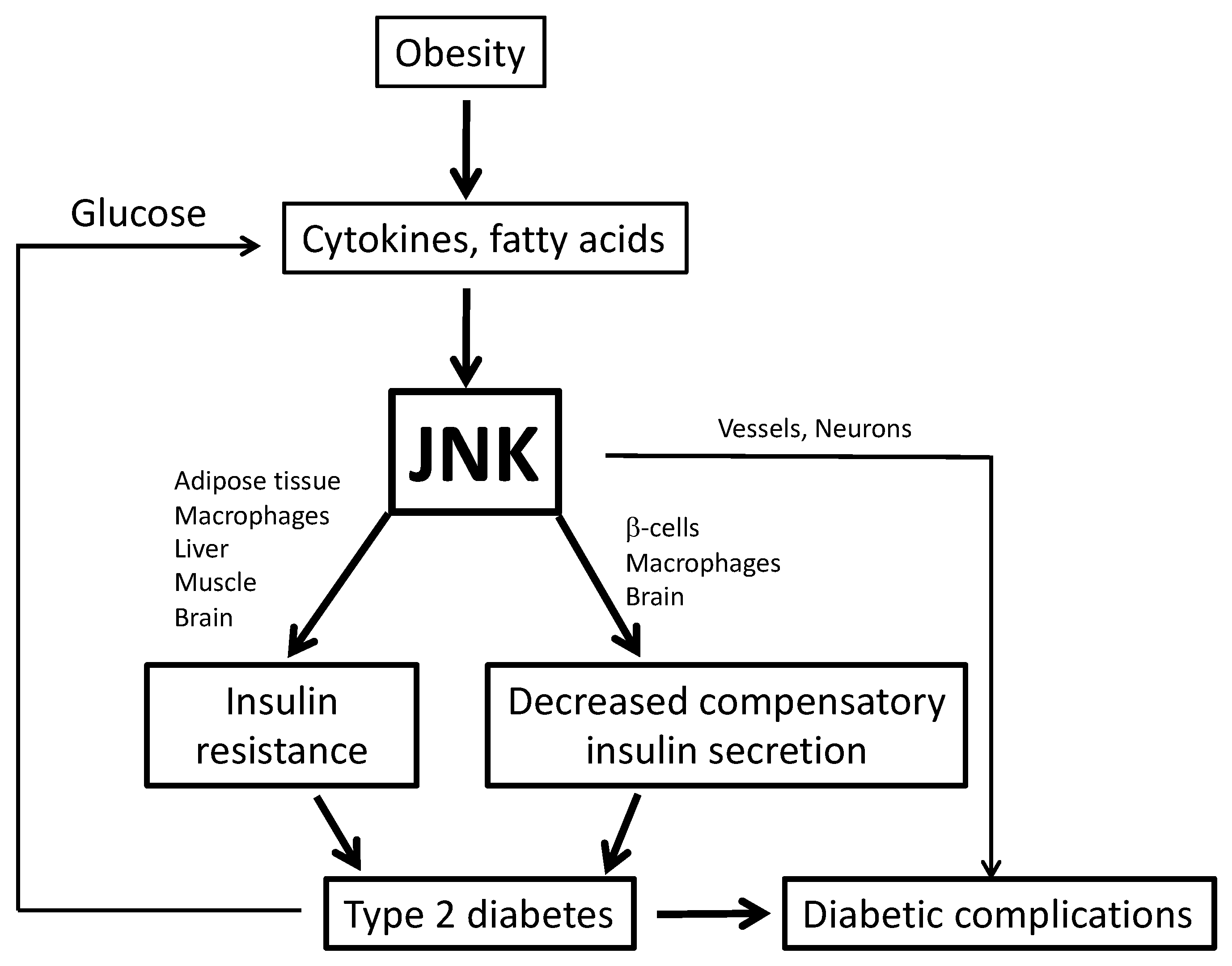
1. Obesity and Type 2 Diabetes
2. c-Jun N-terminal Kinase (JNK)
3. Role of Inflammation and JNK in Metabolic Disease
3.1. Adipose Tissue Inflammation
3.2. Ectopic Fat Accumulation on Insulin Resistance and ß-Cell Dysfunction
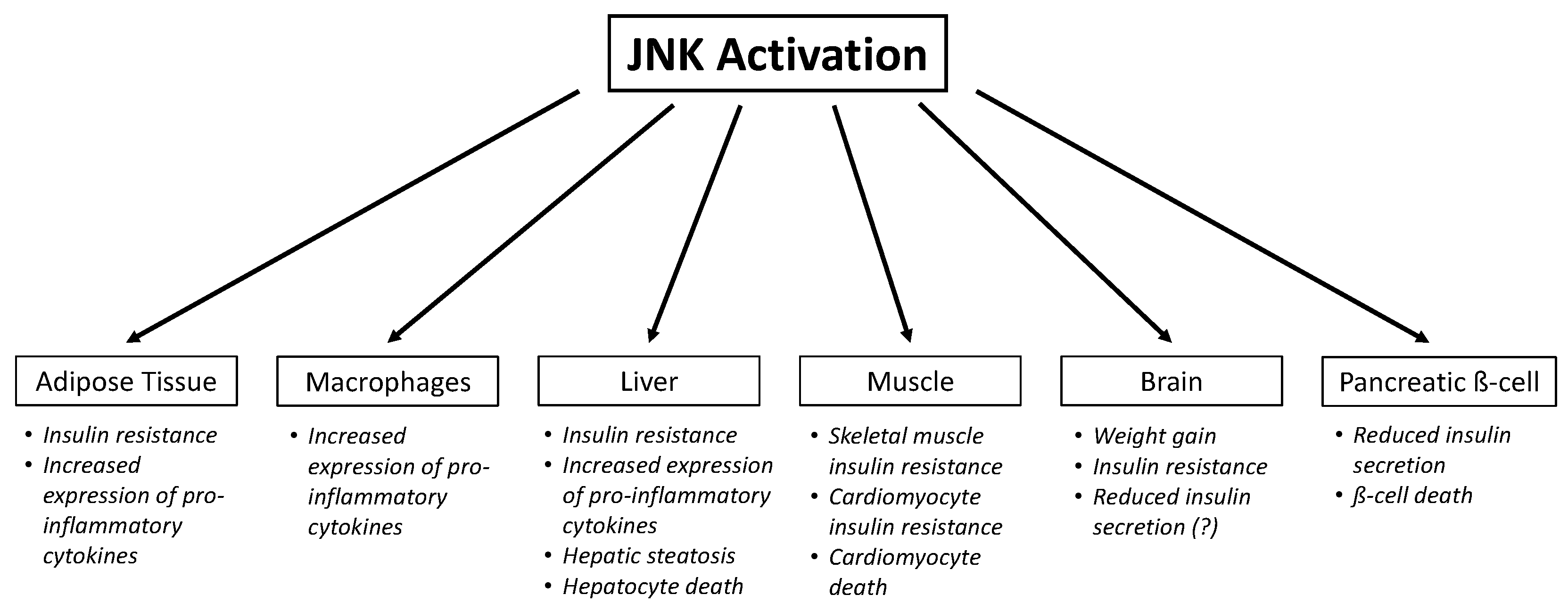
4. Role of JNK in Insulin Resistance
4.1. Adipose Tissue
4.2. Macrophages
4.3. Skeletal Muscle
4.4. Liver
4.5. Brain
5. Role of JNK in Insulin Secretion
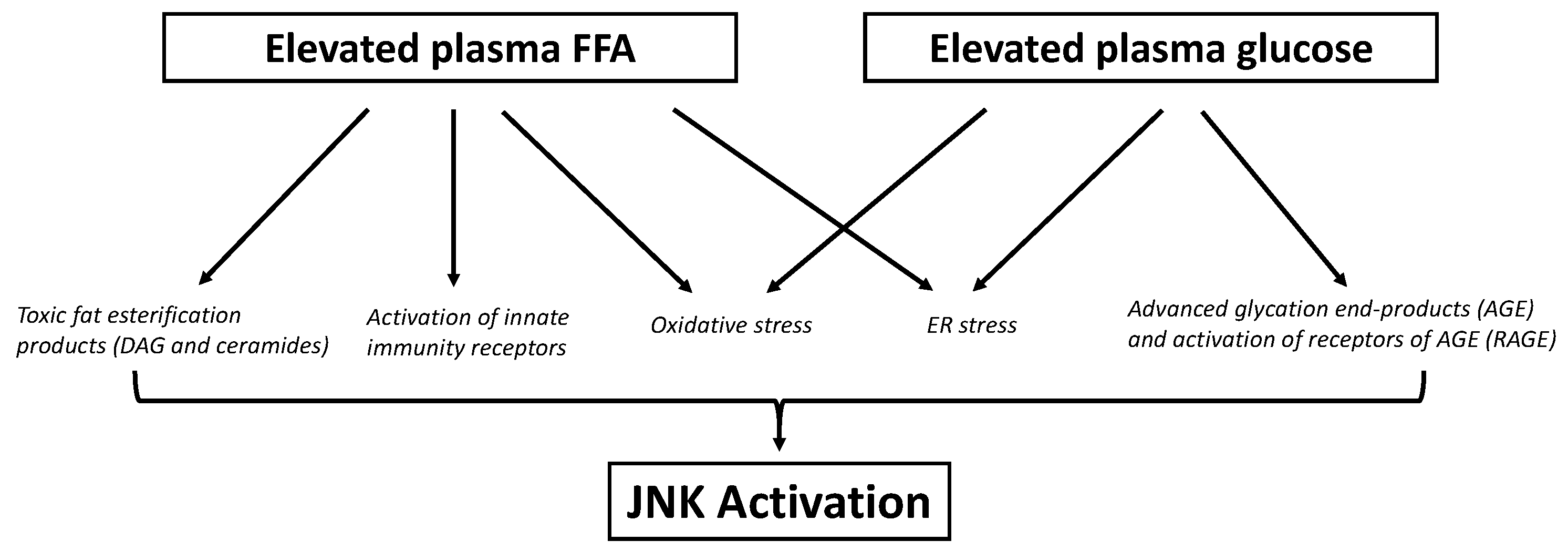
6. Role of JNK in Lipotoxicity
6.1. Skeletal Muscle Lipotoxicity
6.2. Hepatic Lipotoxicity
6.3. β-Cell Lipotoxicity
6.4. Hypothalamic Lipotoxicity
7. Role of JNK in Glucotoxicity
7.1. Adipose Tissue Glucotoxicity
7.2. Skeletal Muscle Glucotoxicity
7.3. Liver Glucotoxicity
7.4. β-Cell Glucotoxicity
7.5. Hypothalamic Glucotoxicity
8. Role of JNK in Diabetic Complications
9. JNK as a Therapeutic Target
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hill, J.O. Understanding and Addressing the Epidemic of Obesity: An Energy Balance Perspective. Endocr. Rev. 2006, 27, 750–761. [Google Scholar] [CrossRef]
- Jung, U.J.; Choi, M.-S. Obesity and Its Metabolic Complications: The Role of Adipokines and the Relationship between Obesity, Inflammation, Insulin Resistance, Dyslipidemia and Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2014, 15, 6184–6223. [Google Scholar] [CrossRef] [PubMed]
- Pi-Sunyer, X. The Medical Risks of Obesity. Postgrad. Med. 2009, 121, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Procaccini, C.; Santopaolo, M.; Faicchia, D.; Colamatteo, A.; Formisano, L.; de Candia, P.; Galgani, M.; De Rosa, V.; Matarese, G. Role of metabolism in neurodegenerative disorders. Metabolism 2016, 65, 1376–1390. [Google Scholar] [CrossRef] [PubMed]
- Mazon, J.N.; de Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28. [Google Scholar] [CrossRef]
- American Diabetes Association Diagnosis and Classification of Diabetes Mellitus. Diabetes Care 2013, 36, S67–S74. [CrossRef]
- Cerf, M.E. Beta Cell Dysfunction and Insulin Resistance. Front. Endocrinol. 2013, 4, 37. [Google Scholar] [CrossRef]
- Hirosumi, J.; Tuncman, G.; Chang, L.; Görgün, C.Z.; Uysal, K.T.; Maeda, K.; Karin, M.; Hotamisligil, G.S. A central role for JNK in obesity and insulin resistance. Nature 2002, 420, 333–336. [Google Scholar] [CrossRef]
- Bode, A.M.; Dong, Z. The Functional Contrariety of JNK. Mol. Carcinog. 2007, 46, 591–598. [Google Scholar] [CrossRef]
- Solinas, G.; Becattini, B. JNK at the crossroad of obesity, insulin resistance, and cell stress response. Mol. Metab. 2016, 6, 174–184. [Google Scholar] [CrossRef]
- Zeke, A.; Misheva, M.; Reményi, A.; Bogoyevitch, M.A. JNK Signaling: Regulation and Functions Based on Complex Protein-Protein Partnerships. Microbiol. Mol. Biol. Rev. 2016, 80, 793–835. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP Kinase Pathways. Cold Spring Harb. Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Karin, M.; Gallagher, E. From JNK to Pay Dirt: Jun Kinases, their Biochemistry, Physiology and Clinical Importance. IUBMB Life 2005, 57, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Muniyappa, H.; Das, K.C. Activation of c-Jun N-Terminal Kinase (JNK) by widely used specific p38 MAPK inhibitor SB202190 and SB203580: A MLK-3-MKK7-dependent mechanism. Cell Signal 2008, 20, 675–683. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Destrument, A.; Tournier, C. Physiological roles of MKK4 and MKK7: Insights from animal models. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2007, 1773, 1349–1357. [Google Scholar] [CrossRef] [PubMed]
- Tournier, C.; Dong, C.; Turner, T.K.; Jones, S.N.; Flavell, R.A.; Davis, R.J. MKK7 is an essential component of the JNK signal transduction pathway activated by proinflammatory cytokines. Genes Dev. 2001, 15, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Bergami, P.; Ronai, Z. Requirements for PKC-augmented JNK activation by MKK4/7. Int. J. Biochem. Cell Biol. 2008, 40, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, stress, and diabetes. J. Clin. Investig. 2005, 115, 1111–1119. [Google Scholar] [CrossRef]
- Engin, A. The Pathogenesis of Obesity-Associated Adipose Tissue Inflammation. In Obesity and Lipotoxicity; Engin, A.B., Engin, A., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2017; pp. 221–245. ISBN 978-3-319-48382-5. [Google Scholar]
- Ge, Q.; Brichard, S.; Yi, X.; Li, Q. microRNAs as a New Mechanism Regulating Adipose Tissue Inflammation in Obesity and as a Novel Therapeutic Strategy in the Metabolic Syndrome. J. Immunol. Res. 2014, 2014, 987285. [Google Scholar] [CrossRef]
- Fuster, J.J.; Ouchi, N.; Gokce, N.; Walsh, K. Obesity-Induced Changes in Adipose Tissue Microenvironment and Their Impact on Cardiovascular Disease. Circ. Res. 2016, 118, 1786–1807. [Google Scholar] [CrossRef]
- Sun, S.; Ji, Y.; Kersten, S.; Qi, L. Mechanisms of Inflammatory Responses in Obese Adipose Tissue. Annu. Rev. Nutr. 2012, 32, 261–286. [Google Scholar] [CrossRef] [PubMed]
- Revelo, X.S.; Luck, H.; Winer, S.; Winer, D.A. Morphological and Inflammatory Changes in Visceral Adipose Tissue During Obesity. Endocr. Pathol. 2014, 25, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Carrillo, J.L.M.; Campo, J.O.M.D.; Coronado, O.G.; Gutiérrez, P.T.V.; Cordero, J.F.C.; Juárez, J.V. Adipose Tissue and Inflammation. Adipose Tissue 2018. [Google Scholar] [CrossRef]
- Tajiri, Y.; Mimura, K.; Umeda, F. High-Sensitivity C-Reactive Protein in Japanese Patients with Type 2 Diabetes. Obes. Res. 2005, 13, 1810–1816. [Google Scholar] [CrossRef]
- Olefsky, J.M.; Glass, C.K. Macrophages, Inflammation, and Insulin Resistance. Annu. Rev. Physiol. 2010, 72, 219–246. [Google Scholar] [CrossRef]
- Makki, K.; Froguel, P.; Wolowczuk, I. Adipose Tissue in Obesity-Related Inflammation and Insulin Resistance: Cells, Cytokines, and Chemokines. ISRN Inflamm. 2013, 2013, 139239. [Google Scholar] [CrossRef]
- Lee, Y.S.; Li, P.; Huh, J.Y.; Hwang, I.J.; Lu, M.; Kim, J.I.; Ham, M.; Talukdar, S.; Chen, A.; Lu, W.J.; et al. Inflammation Is Necessary for Long-Term but Not Short-Term High-Fat Diet–Induced Insulin Resistance. Diabetes 2011, 60, 2474–2483. [Google Scholar] [CrossRef]
- Feng, B.; Jiao, P.; Nie, Y.; Kim, T.; Jun, D.; van Rooijen, N.; Yang, Z.; Xu, H. Clodronate Liposomes Improve Metabolic Profile and Reduce Visceral Adipose Macrophage Content in Diet-Induced Obese Mice. PLoS ONE 2011, 6, e24358. [Google Scholar] [CrossRef]
- Morigny, P.; Houssier, M.; Mouisel, E.; Langin, D. Adipocyte lipolysis and insulin resistance. Biochimie 2016, 125, 259–266. [Google Scholar] [CrossRef]
- Tamori, Y.; Masugi, J.; Nishino, N.; Kasuga, M. Role of Peroxisome Proliferator-Activated Receptor-γ in Maintenance of the Characteristics of Mature 3T3-L1 Adipocytes. Diabetes 2002, 51, 2045–2055. [Google Scholar] [CrossRef]
- Guilherme, A.; Virbasius, J.V.; Puri, V.; Czech, M.P. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell Biol. 2008, 9, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Boden, G. Obesity and Free Fatty Acids (FFA). Endocrinol. Metab. Clin. N. Am. 2008, 37, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Ajuwon, K.M.; Spurlock, M.E. Palmitate Activates the NF-κB Transcription Factor and Induces IL-6 and TNFα Expression in 3T3-L1 Adipocytes. J. Nutr. 2005, 135, 1841–1846. [Google Scholar] [CrossRef]
- Guo, W.; Wong, S.; Xie, W.; Lei, T.; Luo, Z. Palmitate modulates intracellular signaling, induces endoplasmic reticulum stress, and causes apoptosis in mouse 3T3-L1 and rat primary preadipocytes. Am. J. Physiol.-Endocrinol. Metab. 2007, 293, E576–E586. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Endoplasmic Reticulum Stress and the Inflammatory Basis of Metabolic Disease. Cell 2010, 140, 900. [Google Scholar] [CrossRef]
- Jiao, P.; Ma, J.; Feng, B.; Zhang, H.; Alan-Diehl, J.; Eugene-Chin, Y.; Yan, W.; Xu, H. FFA-Induced Adipocyte Inflammation and Insulin Resistance: Involvement of ER Stress and IKKβ Pathways. Obesity 2011, 19, 483–491. [Google Scholar] [CrossRef]
- Merrill, A.H. De Novo Sphingolipid Biosynthesis: A Necessary, but Dangerous, Pathway. J. Biol. Chem. 2002, 277, 25843–25846. [Google Scholar] [CrossRef]
- Holland, W.L.; Bikman, B.T.; Wang, L.-P.; Yuguang, G.; Sargent, K.M.; Bulchand, S.; Knotts, T.A.; Shui, G.; Clegg, D.J.; Wenk, M.R.; et al. Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid–induced ceramide biosynthesis in mice. J. Clin. Investig. 2011, 121, 1858–1870. [Google Scholar] [CrossRef]
- Kennedy, A.; Martinez, K.; Chuang, C.-C.; LaPoint, K.; McIntosh, M. Saturated Fatty Acid-Mediated Inflammation and Insulin Resistance in Adipose Tissue: Mechanisms of Action and Implications. J. Nutr. 2009, 139, 1–4. [Google Scholar] [CrossRef]
- Lyons, C.L.; Kennedy, E.B.; Roche, H.M. Metabolic Inflammation-Differential Modulation by Dietary Constituents. Nutrients 2016, 8, 247. [Google Scholar] [CrossRef]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like Receptor 4/NF-κB Pathway in Saturated Fatty Acid–Induced Inflammatory Changes in the Interaction Between Adipocytes and Macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Kim, K.H.; Yoon, J.M.; Kim, J.B. Activation of Toll-like receptor 4 is associated with insulin resistance in adipocytes. Biochem. Biophys. Res. Commun. 2006, 346, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Rutkowsky, J.M.; Snodgrass, R.G.; Ono-Moore, K.D.; Schneider, D.A.; Newman, J.W.; Adams, S.H.; Hwang, D.H. Saturated fatty acids activate TLR-mediated proinflammatory signaling pathways. J. Lipid Res. 2012, 53, 2002–2013. [Google Scholar] [CrossRef] [PubMed]
- Sparks Janet, D.; Sparks Charles, E. Adeli Khosrow Selective Hepatic Insulin Resistance, VLDL Overproduction, and Hypertriglyceridemia. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2104–2112. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, S.; Zhang, X.; Yi, Z.; Cichello, S. Skeletal intramyocellular lipid metabolism and insulin resistance. Biophys. Rep. 2015, 1, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Koves, T.R.; Ussher, J.R.; Noland, R.C.; Slentz, D.; Mosedale, M.; Ilkayeva, O.; Bain, J.; Stevens, R.; Dyck, J.R.B.; Newgard, C.B.; et al. Mitochondrial Overload and Incomplete Fatty Acid Oxidation Contribute to Skeletal Muscle Insulin Resistance. Cell Metab. 2008, 7, 45–56. [Google Scholar] [CrossRef]
- Aon, M.A.; Bhatt, N.; Cortassa, S.C. Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 2014, 5. [Google Scholar] [CrossRef]
- Sergi, D.; Naumovski, N.; Heilbronn, L.K.; Abeywardena, M.; O’Callaghan, N.; Lionetti, L.; Luscombe-Marsh, N. Mitochondrial (Dys)function and Insulin Resistance: From Pathophysiological Molecular Mechanisms to the Impact of Diet. Front. Physiol. 2019, 10, 532. [Google Scholar] [CrossRef]
- Kelley, D.E.; Goodpaster, B.; Wing, R.R.; Simoneau, J.-A. Skeletal muscle fatty acid metabolism in association with insulin resistance, obesity, and weight loss. Am. J. Physiol.-Endocrinol. Metab. 1999, 277, E1130–E1141. [Google Scholar] [CrossRef]
- Simoneau, J.-A.; Veerkamp, J.H.; Turcotte, L.P.; Kelley, D.E. Markers of capacity to utilize fatty acids in human skeletal muscle: Relation to insulin resistance and obesity and effects of weight loss. FASEB J. 1999, 13, 2051–2060. [Google Scholar] [CrossRef]
- Kim, J.-Y.; Hickner, R.C.; Cortright, R.L.; Dohm, G.L.; Houmard, J.A. Lipid oxidation is reduced in obese human skeletal muscle. Am. J. Physiol.-Endocrinol. Metab. 2000, 279, E1039–E1044. [Google Scholar] [CrossRef] [PubMed]
- Kelley, D.E.; He, J.; Menshikova, E.V.; Ritov, V.B. Dysfunction of Mitochondria in Human Skeletal Muscle in Type 2 Diabetes. Diabetes 2002, 51, 2944–2950. [Google Scholar] [CrossRef] [PubMed]
- Schrauwen-Hinderling, V.B.; Kooi, M.E.; Hesselink, M.K.C.; Jeneson, J.A.L.; Backes, W.H.; van Echteld, C.J.A.; van Engelshoven, J.M.A.; Mensink, M.; Schrauwen, P. Impaired in vivo mitochondrial function but similar intramyocellular lipid content in patients with type 2 diabetes mellitus and BMI-matched control subjects. Diabetologia 2007, 50, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Phielix, E.; Schrauwen-Hinderling, V.B.; Mensink, M.; Lenaers, E.; Meex, R.; Hoeks, J.; Kooi, M.E.; Moonen-Kornips, E.; Sels, J.-P.; Hesselink, M.K.C.; et al. Lower Intrinsic ADP-Stimulated Mitochondrial Respiration Underlies In Vivo Mitochondrial Dysfunction in Muscle of Male Type 2 Diabetic Patients. Diabetes 2008, 57, 2943–2949. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Ward, W.F. PGC-1α: A key regulator of energy metabolism. Adv. Physiol. Educ. 2006, 30, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.-F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstråle, M.; Laurila, E.; et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Tushuizen, M.E.; Bunck, M.C.; Pouwels, P.J.; Bontemps, S.; van Waesberghe, J.H.T.; Schindhelm, R.K.; Mari, A.; Heine, R.J.; Diamant, M. Pancreatic Fat Content and β-Cell Function in Men with and Without Type 2 Diabetes. Diabetes Care 2007, 30, 2916. [Google Scholar] [CrossRef]
- Godoy-Matos, A.F.; Valerio, C.M.; Moreira, R.O.; Momesso, D.P.; Bittencourt, L.K. Pancreatic fat deposition is increased and related to beta-cell function in women with familial partial lipodystrophy. Diabetol. Metab. Syndr. 2018, 10, 71. [Google Scholar] [CrossRef]
- Wilcox, G. Insulin and Insulin Resistance. Clin. Biochem. Rev. 2005, 26, 19–39. [Google Scholar]
- Petersen, M.C.; Shulman, G.I. Mechanisms of Insulin Action and Insulin Resistance. Physiol. Rev. 2018, 98, 2133–2223. [Google Scholar] [CrossRef] [PubMed]
- Dodd, G.T.; Tiganis, T. Insulin action in the brain: Roles in energy and glucose homeostasis. J. Neuroendocrinol. 2017, 29, e12513. [Google Scholar] [CrossRef] [PubMed]
- Kleinridders, A.; Ferris, H.A.; Cai, W.; Kahn, C.R. Insulin Action in Brain Regulates Systemic Metabolism and Brain Function. Diabetes 2014, 63, 2232–2243. [Google Scholar] [CrossRef] [PubMed]
- Ruud, J.; Steculorum, S.M.; Brüning, J.C. Neuronal control of peripheral insulin sensitivity and glucose metabolism. Nat. Commun. 2017, 8, 15259. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H. Central insulin-mediated regulation of hepatic glucose production. Endocrine J. 2016, 63, 1–7. [Google Scholar] [CrossRef]
- Könner, A.C.; Janoschek, R.; Plum, L.; Jordan, S.D.; Rother, E.; Ma, X.; Xu, C.; Enriori, P.; Hampel, B.; Barsh, G.S.; et al. Insulin Action in AgRP-Expressing Neurons Is Required for Suppression of Hepatic Glucose Production. Cell Metab. 2007, 5, 438–449. [Google Scholar] [CrossRef]
- Koch, L.; Wunderlich, F.T.; Seibler, J.; Könner, A.C.; Hampel, B.; Irlenbusch, S.; Brabant, G.; Kahn, C.R.; Schwenk, F.; Brüning, J.C. Central insulin action regulates peripheral glucose and fat metabolism in mice. J. Clin. Investig. 2008, 118, 2132–2147. [Google Scholar] [CrossRef]
- Kimura, K.; Tanida, M.; Nagata, N.; Inaba, Y.; Watanabe, H.; Nagashimada, M.; Ota, T.; Asahara, S.; Kido, Y.; Matsumoto, M.; et al. Central Insulin Action Activates Kupffer Cells by Suppressing Hepatic Vagal Activation via the Nicotinic Alpha 7 Acetylcholine Receptor. Cell Rep. 2016, 14, 2362–2374. [Google Scholar] [CrossRef]
- Inoue, H.; Ogawa, W.; Asakawa, A.; Okamoto, Y.; Nishizawa, A.; Matsumoto, M.; Teshigawara, K.; Matsuki, Y.; Watanabe, E.; Hiramatsu, R.; et al. Role of hepatic STAT3 in brain-insulin action on hepatic glucose production. Cell Metab. 2006, 3, 267–275. [Google Scholar] [CrossRef]
- García-Cáceres, C.; Quarta, C.; Varela, L.; Gao, Y.; Gruber, T.; Legutko, B.; Jastroch, M.; Johansson, P.; Ninkovic, J.; Yi, C.-X.; et al. Astrocytic Insulin Signaling Couples Brain Glucose Uptake with Nutrient Availability. Cell 2016, 166, 867–880. [Google Scholar] [CrossRef]
- Chen§, D.; Waters§, S.B.; Holt, K.H.; Pessin, J.E. SOS Phosphorylation and Disassociation of the Grb2-SOS Complex by the ERK and JNK Signaling Pathways. J. Biol. Chem. 1996, 271, 6328–6332. [Google Scholar] [CrossRef] [PubMed]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed]
- D’Oria, R.; Laviola, L.; Giorgino, F.; Unfer, V.; Bettocchi, S.; Scioscia, M. PKB/Akt and MAPK/ERK phosphorylation is highly induced by inositols: Novel potential insights in endothelial dysfunction in preeclampsia. Pregnancy Hypertens. 2017, 10, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Morino, K.; Maegawa, H.; Fujita, T.; Takahara, N.; Egawa, K.; Kashiwagi, A.; Kikkawa, R. Insulin-Induced c-Jun N-Terminal Kinase Activation Is Negatively Regulated by Protein Kinase C δ. Endocrinology 2001, 142, 2669–2676. [Google Scholar] [CrossRef][Green Version]
- Tan, S.-X.; Fisher-Wellman, K.H.; Fazakerley, D.J.; Ng, Y.; Pant, H.; Li, J.; Meoli, C.C.; Coster, A.C.F.; Stöckli, J.; James, D.E. Selective Insulin Resistance in Adipocytes. J. Biol. Chem. 2015, 290, 11337–11348. [Google Scholar] [CrossRef] [PubMed]
- Biddinger, S.B.; Hernandez-Ono, A.; Rask-Madsen, C.; Haas, J.T.; Alemán, J.O.; Suzuki, R.; Scapa, E.F.; Agarwal, C.; Carey, M.C.; Stephanopoulos, G.; et al. Hepatic Insulin Resistance is Sufficient to Produce Dyslipidemia and Susceptibility to Atherosclerosis. Cell Metab. 2008, 7, 125–134. [Google Scholar] [CrossRef]
- Ueno, M.; Carvalheira, J.B.C.; Tambascia, R.C.; Bezerra, R.M.N.; Amaral, M.E.; Carneiro, E.M.; Folli, F.; Franchini, K.G.; Saad, M.J.A. Regulation of insulin signalling by hyperinsulinaemia: Role of IRS-1/2 serine phosphorylation and the mTOR/p70 S6K pathway. Diabetologia 2005, 48, 506–518. [Google Scholar] [CrossRef]
- Draznin, B. Molecular Mechanisms of Insulin Resistance: Serine Phosphorylation of Insulin Receptor Substrate-1 and Increased Expression of p85: The Two Sides of a Coin. Diabetes 2006, 55, 2392–2397. [Google Scholar] [CrossRef]
- Ueki, K.; Kondo, T.; Kahn, C.R. Suppressor of Cytokine Signaling 1 (SOCS-1) and SOCS-3 Cause Insulin Resistance through Inhibition of Tyrosine Phosphorylation of Insulin Receptor Substrate Proteins by Discrete Mechanisms. Mol. Cell. Biol. 2004, 24, 5434–5446. [Google Scholar] [CrossRef] [PubMed]
- Cusi, K.; Maezono, K.; Osman, A.; Pendergrass, M.; Patti, M.E.; Pratipanawatr, T.; DeFronzo, R.A.; Kahn, C.R.; Mandarino, L.J. Insulin resistance differentially affects the PI 3-kinase– and MAP kinase–mediated signaling in human muscle. J. Clin. Investig. 2000, 105, 311–320. [Google Scholar] [CrossRef]
- Copps, K.D.; Hancer, N.J.; Opare-Ado, L.; Qiu, W.; Walsh, C.; White, M.F. Irs1 Serine 307 Promotes Insulin Sensitivity in Mice. Cell Metab. 2010, 11, 84–92. [Google Scholar] [CrossRef] [PubMed]
- Morino, K.; Neschen, S.; Bilz, S.; Sono, S.; Tsirigotis, D.; Reznick, R.M.; Moore, I.; Nagai, Y.; Samuel, V.; Sebastian, D.; et al. Muscle-Specific IRS-1 Ser→Ala Transgenic Mice Are Protected From Fat-Induced Insulin Resistance in Skeletal Muscle. Diabetes 2008, 57, 2644–2651. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.; Wayne Leitner, J.; Adochio, R.; Draznin, B. Knockdown of JNK rescues 3T3-L1 adipocytes from insulin resistance induced by mitochondrial dysfunction. Biochem. Biophys. Res. Commun. 2009, 378, 772–776. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.T.A.; Satoh, H.; Favelyukis, S.; Babendure, J.L.; Imamura, T.; Sbodio, J.I.; Zalevsky, J.; Dahiyat, B.I.; Chi, N.-W.; Olefsky, J.M. JNK and Tumor Necrosis Factor-α Mediate Free Fatty Acid-induced Insulin Resistance in 3T3-L1 Adipocytes. J. Biol. Chem. 2005, 280, 35361–35371. [Google Scholar] [CrossRef]
- Sabio, G.; Das, M.; Mora, A.; Zhang, Z.; Jun, J.Y.; Ko, H.J.; Barrett, T.; Kim, J.K.; Davis, R.J. A Stress Signaling Pathway in Adipose Tissue Regulates Hepatic Insulin Resistance. Science 2008, 322, 1539–1543. [Google Scholar] [CrossRef]
- Könner, A.C.; Brüning, J.C. Toll-like receptors: Linking inflammation to metabolism. Trends Endocrinol. Metab. 2011, 22, 16–23. [Google Scholar] [CrossRef]
- Seki, E.; Brenner, D.A.; Karin, M. A Liver Full of JNK: Signaling in Regulation of Cell Function and Disease Pathogenesis, and Clinical Approaches. Gastroenterology 2012, 143, 307–320. [Google Scholar] [CrossRef]
- Solinas, G.; Vilcu, C.; Neels, J.G.; Bandyopadhyay, G.K.; Luo, J.-L.; Naugler, W.; Grivennikov, S.; Wynshaw-Boris, A.; Scadeng, M.; Olefsky, J.M.; et al. JNK1 in Hematopoietically Derived Cells Contributes to Diet-Induced Inflammation and Insulin Resistance without Affecting Obesity. Cell Metab. 2007, 6, 386–397. [Google Scholar] [CrossRef]
- Han, M.S.; Jung, D.Y.; Morel, C.; Lakhani, S.A.; Kim, J.K.; Flavell, R.A.; Davis, R.J. JNK Expression by Macrophages Promotes Obesity-induced Insulin Resistance and Inflammation. Science 2013, 339, 218–222. [Google Scholar] [CrossRef]
- Sabio, G.; Kennedy, N.J.; Cavanagh-Kyros, J.; Jung, D.Y.; Ko, H.J.; Ong, H.; Barrett, T.; Kim, J.K.; Davis, R.J. Role of Muscle c-Jun NH2-Terminal Kinase 1 in Obesity-Induced Insulin Resistance. Mol. Cell. Biol. 2010, 30, 106–115. [Google Scholar] [CrossRef]
- Henstridge, D.C.; Bruce, C.R.; Pang, C.P.; Lancaster, G.I.; Allen, T.L.; Estevez, E.; Gardner, T.; Weir, J.M.; Meikle, P.J.; Lam, K.S.L.; et al. Skeletal muscle-specific overproduction of constitutively activated c-Jun N-terminal kinase (JNK) induces insulin resistance in mice. Diabetologia 2012, 55, 2769–2778. [Google Scholar] [CrossRef] [PubMed]
- Pal, M.; Wunderlich, C.M.; Spohn, G.; Brönneke, H.S.; Schmidt-Supprian, M.; Wunderlich, F.T. Alteration of JNK-1 Signaling in Skeletal Muscle Fails to Affect Glucose Homeostasis and Obesity-Associated Insulin Resistance in Mice. PLoS ONE 2013, 8, e54247. [Google Scholar] [CrossRef] [PubMed]
- Kitade, H.; Chen, G.; Ni, Y.; Ota, T. Nonalcoholic Fatty Liver Disease and Insulin Resistance: New Insights and Potential New Treatments. Nutrients 2017, 9, 387. [Google Scholar] [CrossRef] [PubMed]
- Ueki, K.; Kondo, T.; Tseng, Y.-H.; Kahn, C.R. Central role of suppressors of cytokine signaling proteins in hepatic steatosis, insulin resistance, and the metabolic syndrome in the mouse. Proc. Natl. Acad. Sci. USA 2004, 101, 10422–10427. [Google Scholar] [CrossRef]
- Brown, M.S.; Goldstein, J.L. Selective versus Total Insulin Resistance: A Pathogenic Paradox. Cell Metab. 2008, 7, 95–96. [Google Scholar] [CrossRef]
- Knebel, B.; Haas, J.; Hartwig, S.; Jacob, S.; Köllmer, C.; Nitzgen, U.; Muller–Wieland, D.; Kotzka, J. Liver-Specific Expression of Transcriptionally Active SREBP-1c Is Associated with Fatty Liver and Increased Visceral Fat Mass. PLoS ONE 2012, 7, e31812. [Google Scholar] [CrossRef]
- Sabio, G.; Cavanagh-Kyros, J.; Ko, H.J.; Jung, D.Y.; Gray, S.; Jun, J.Y.; Barrett, T.; Mora, A.; Kim, J.K.; Davis, R.J. Prevention of Steatosis by Hepatic JNK1. Cell Metab. 2009, 10, 491–498. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Garcia-Haro, L.; Sabio, G.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Xu, J.; Shulha, H.P.; Garber, M.; et al. The PPARα-FGF21 hormone axis contributes to metabolic regulation by the hepatic JNK signaling pathway. Cell Metab. 2014, 20, 512–525. [Google Scholar] [CrossRef]
- Singh, R.; Wang, Y.; Xiang, Y.; Tanaka, K.E.; Gaarde, W.A.; Czaja, M.J. Differential Effects of JNK1 and JNK2 Inhibition on Murine Steatohepatitis and Insulin Resistance. Hepatol. Baltim. Md 2009, 49, 87–96. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Barrett, T.; Tournier, C.; Davis, R.J. Fibroblast growth factor 21 mediates glycemic regulation by hepatic JNK. Cell Rep. 2016, 14, 2273–2280. [Google Scholar] [CrossRef]
- Lin, Z.; Tian, H.; Lam, K.S.L.; Lin, S.; Hoo, R.C.L.; Konishi, M.; Itoh, N.; Wang, Y.; Bornstein, S.R.; Xu, A.; et al. Adiponectin mediates the metabolic effects of FGF21 on glucose homeostasis and insulin sensitivity in mice. Cell Metab. 2013, 17, 779–789. [Google Scholar] [CrossRef] [PubMed]
- Xiang, M.; Wang, P.-X.; Wang, A.-B.; Zhang, X.-J.; Zhang, Y.; Zhang, P.; Mei, F.-H.; Chen, M.-H.; Li, H. Targeting hepatic TRAF1-ASK1 signaling to improve inflammation, insulin resistance, and hepatic steatosis. J. Hepatol. 2016, 64, 1365–1377. [Google Scholar] [CrossRef] [PubMed]
- Litwak, S.A.; Pang, L.; Galic, S.; Igoillo-Esteve, M.; Stanley, W.J.; Turatsinze, J.-V.; Loh, K.; Thomas, H.E.; Sharma, A.; Trepo, E.; et al. JNK Activation of BIM Promotes Hepatic Oxidative Stress, Steatosis, and Insulin Resistance in Obesity. Diabetes 2017, 66, 2973–2986. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, P.-X.; Zhang, Y.; Yu, C.-J.; Ji, Y.; Wang, X.; Zhang, P.; Jiang, X.; Jin, H.; Huang, Z.; et al. Tumor necrosis factor receptor-associated factor 5 (Traf5) acts as an essential negative regulator of hepatic steatosis. J. Hepatol. 2016, 65, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Belgardt, B.F.; Mauer, J.; Wunderlich, F.T.; Ernst, M.B.; Pal, M.; Spohn, G.; Brönneke, H.S.; Brodesser, S.; Hampel, B.; Schauss, A.C.; et al. Hypothalamic and pituitary c-Jun N-terminal kinase 1 signaling coordinately regulates glucose metabolism. Proc. Natl. Acad. Sci. USA 2010, 107, 6028–6033. [Google Scholar] [CrossRef] [PubMed]
- Jais, A.; Brüning, J.C. Hypothalamic inflammation in obesity and metabolic disease. J. Clin. Investig. 2017, 127, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Sabio, G.; Cavanagh-Kyros, J.; Barrett, T.; Jung, D.Y.; Ko, H.J.; Ong, H.; Morel, C.; Mora, A.; Reilly, J.; Kim, J.K.; et al. Role of the hypothalamic–pituitary–thyroid axis in metabolic regulation by JNK1. Genes Dev. 2010, 24, 256–264. [Google Scholar] [CrossRef]
- Vernia, S.; Cavanagh-Kyros, J.; Barrett, T.; Jung, D.Y.; Kim, J.K.; Davis, R.J. Diet-induced obesity mediated by the JNK/DIO2 signal transduction pathway. Genes Dev. 2013, 27, 2345–2355. [Google Scholar] [CrossRef] [PubMed]
- Guilherme, A.; Henriques, F.; Bedard, A.H.; Czech, M.P. Molecular pathways linking adipose innervation to insulin action in obesity and diabetes mellitus. Nat. Rev. Endocrinol. 2019, 15, 207–225. [Google Scholar] [CrossRef] [PubMed]
- Lanuza-Masdeu, J.; Arévalo, M.I.; Vila, C.; Barberà, A.; Gomis, R.; Caelles, C. In vivo JNK activation in pancreatic β-cells leads to glucose intolerance caused by insulin resistance in pancreas. Diabetes 2013, 62, 2308–2317. [Google Scholar] [CrossRef]
- Bennett, B.; Satoh, Y.; Lewis, A. JNK: A new therapeutic target for diabetes. Curr. Opin. Pharmacol. 2003, 3, 420–425. [Google Scholar] [CrossRef]
- Hagman, D.K.; Hays, L.B.; Parazzoli, S.D.; Poitout, V. Palmitate inhibits insulin gene expression by altering pdx-1 nuclear localization and reducing mafa expression in isolated rat islets of langerhans. J. Biol. Chem. 2005, 280, 32413–32418. [Google Scholar] [CrossRef] [PubMed]
- Henderson, E.; Stein, R. c-jun inhibits transcriptional activation by the insulin enhancer, and the insulin control element is the target of control. Mol. Cell. Biol. 1994, 14, 655–662. [Google Scholar] [CrossRef] [PubMed]
- Andreozzi, F.; D’Alessandris, C.; Federici, M.; Laratta, E.; Del Guerra, S.; Del Prato, S.; Marchetti, P.; Lauro, R.; Perticone, F.; Sesti, G. Activation of the Hexosamine Pathway Leads to Phosphorylation of Insulin Receptor Substrate-1 on Ser307 and Ser612 and Impairs the Phosphatidylinositol 3-Kinase/Akt/Mammalian Target of Rapamycin Insulin Biosynthetic Pathway in RIN Pancreatic β-Cells. Endocrinology 2004, 145, 2845–2857. [Google Scholar] [CrossRef] [PubMed]
- Melloul, D. Transcription Factors in Islet Development and Physiology: Role of PDX-1 in Beta-Cell Function. Ann. N. Y. Acad. Sci. 2004, 1014, 28–37. [Google Scholar] [CrossRef]
- Major, C.D.; Wolf, B.A. Interleukin-1β Stimulation of c-Jun NH2-Terminal Kinase Activity in Insulin-Secreting Cells: Evidence for Cytoplasmic Restriction. Diabetes 2001, 50, 2721–2728. [Google Scholar] [CrossRef][Green Version]
- Eguchi, K.; Nagai, R. Islet inflammation in type 2 diabetes and physiology. J. Clin. Investig. 2017, 127, 14–23. [Google Scholar] [CrossRef]
- Donath, M.Y.; Böni-Schnetzler, M.; Ellingsgaard, H.; Ehses, J.A. Islet Inflammation Impairs the Pancreatic β-Cell in Type 2 Diabetes. Physiology 2009, 24, 325–331. [Google Scholar] [CrossRef]
- Prause, M.; Mayer, C.M.; Brorsson, C.; Frederiksen, K.S.; Billestrup, N.; Størling, J.; Mandrup-Poulsen, T. JNK1 Deficient Insulin-Producing Cells Are Protected against Interleukin-1β-Induced Apoptosis Associated with Abrogated Myc Expression. Available online: https://www.hindawi.com/journals/jdr/2016/1312705/ (accessed on 3 November 2019).
- Dhanasekaran, D.N.; Reddy, E.P. JNK Signaling in Apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef]
- Abdelli, S.; Puyal, J.; Bielmann, C.; Buchillier, V.; Abderrahmani, A.; Clarke, P.G.H.; Beckmann, J.S.; Bonny, C. JNK3 is abundant in insulin-secreting cells and protects against cytokine-induced apoptosis. Diabetologia 2009, 52, 1871–1880. [Google Scholar] [CrossRef]
- Abdelli, S.; Bonny, C. JNK3 Maintains Expression of the Insulin Receptor Substrate 2 (IRS2) in Insulin-Secreting Cells: Functional Consequences for Insulin Signaling. PLoS ONE 2012, 7, e35997. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.J.; Biollaz, G.; et al. Increased Number of Islet-Associated Macrophages in Type 2 Diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.J.; Willcox, A.; Bone, A.J.; Foulis, A.K.; Morgan, N.G. Islet-associated macrophages in type 2 diabetes. Diabetologia 2009, 52, 1686–1688. [Google Scholar] [CrossRef] [PubMed]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; BaeSeo, J.; Yang, B.-H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting β Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474. [Google Scholar] [CrossRef]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated Fatty Acid and TLR Signaling Link β Cell Dysfunction and Islet Inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef]
- Paranjape, S.A.; Chan, O.; Zhu, W.; Horblitt, A.M.; Grillo, C.A.; Wilson, S.; Reagan, L.; Sherwin, R.S. Chronic reduction of insulin receptors in the ventromedial hypothalamus produces glucose intolerance and islet dysfunction in the absence of weight gain. Am. J. Physiol.-Endocrinol. Metab. 2011, 301, E978–E983. [Google Scholar] [CrossRef]
- Porte, D.; Smith, P.H.; Ensinck, J.W. Neurohumoral regulation of the pancreatic islet A and B cells. Metabolism 1976, 25, 1453–1456. [Google Scholar] [CrossRef]
- Blázquez, E.; Velázquez, E.; Hurtado-Carneiro, V.; Ruiz-Albusac, J.M. Insulin in the Brain: Its Pathophysiological Implications for States Related with Central Insulin Resistance, Type 2 Diabetes and Alzheimer’s Disease. Front. Endocrinol. 2014, 5, 161. [Google Scholar] [CrossRef]
- Arruda, A.P.; Milanski, M.; Coope, A.; Torsoni, A.S.; Ropelle, E.; Carvalho, D.P.; Carvalheira, J.B.; Velloso, L.A. Low-Grade Hypothalamic Inflammation Leads to Defective Thermogenesis, Insulin Resistance, and Impaired Insulin Secretion. Endocrinology 2011, 152, 1314–1326. [Google Scholar] [CrossRef]
- Ye, R.; Onodera, T.; Scherer, P.E. Lipotoxicity and β Cell Maintenance in Obesity and Type 2 Diabetes. J. Endocr. Soc. 2019, 3, 617–631. [Google Scholar] [CrossRef]
- Martínez de Morentin, P.B.; Varela, L.; Fernø, J.; Nogueiras, R.; Diéguez, C.; López, M. Hypothalamic lipotoxicity and the metabolic syndrome. Biochim. Biophys. Acta BBA-Mol. Cell Biol. Lipids 2010, 1801, 350–361. [Google Scholar] [CrossRef] [PubMed]
- Nemecz, M.; Constantin, A.; Dumitrescu, M.; Alexandru, N.; Filippi, A.; Tanko, G.; Georgescu, A. The Distinct Effects of Palmitic and Oleic Acid on Pancreatic Beta Cell Function: The Elucidation of Associated Mechanisms and Effector Molecules. Front. Pharmacol. 2019, 9, 1554. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.D.; Xu, S.; Choi, S.-K.; Ha, C.-M.; Thoudam, T.; Cha, S.-K.; Wiederkehr, A.; Wollheim, C.B.; Lee, I.-K.; Park, K.-S. Oxidative stress and calcium dysregulation by palmitate in type 2 diabetes. Exp. Mol. Med. 2017, 49, e291. [Google Scholar] [CrossRef]
- Gao, J.; Wu, D.; Guo, T.B.; Ruan, Q.; Li, T.; Lu, Z.; Xu, M.; Dai, W.; Lu, L. K+ channel activity and redox status are differentially required for JNK activation by UV and reactive oxygen species. Exp. Cell Res. 2004, 297, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Son, Y.; Kim, S.; Chung, H.-T.; Pae, H.-O. Chapter Two-Reactive Oxygen Species in the Activation of MAP Kinases. In Methods in Enzymology; Cadenas, E., Packer, L., Eds.; Hydrogen Peroxide and Cell Signaling, Part C; Academic Press: Cambridge, MA, USA, 2013; Volume 528, pp. 27–48. [Google Scholar]
- Shen, H.-M.; Liu, Z. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic. Biol. Med. 2006, 40, 928–939. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, U.; Moon, J.S.; Lee, H.W.; Won, K.C. CD36 initiated signaling mediates ceramide-induced TXNIP expression in pancreatic beta-cells. Biochim. Biophys. Acta BBA-Mol. Basis Dis. 2015, 1852, 2414–2422. [Google Scholar] [CrossRef][Green Version]
- Chen, C.-L.; Lin, C.-F.; Chang, W.-T.; Huang, W.-C.; Teng, C.-F.; Lin, Y.-S. Ceramide induces p38 MAPK and JNK activation through a mechanism involving a thioredoxin-interacting protein-mediated pathway. Blood 2008, 111, 4365–4374. [Google Scholar] [CrossRef]
- Saxena, G.; Chen, J.; Shalev, A. Intracellular Shuttling and Mitochondrial Function of Thioredoxin-interacting Protein. J. Biol. Chem. 2010, 285, 3997–4005. [Google Scholar] [CrossRef]
- Liu, Z.; Xia, Y.; Li, B.; Xu, H.; Wang, C.; Liu, Y.; Li, Y.; Li, C.; Gao, N.; Li, L. Induction of ER stress-mediated apoptosis by ceramide via disruption of ER Ca(2+) homeostasis in human adenoid cystic carcinoma cells. Cell Biosci. 2014, 4, 71. [Google Scholar] [CrossRef]
- Ruvolo, P.P. Ceramide regulates cellular homeostasis via diverse stress signaling pathways. Leukemia 2001, 15, 1153–1160. [Google Scholar] [CrossRef]
- Cunha, D.A.; Hekerman, P.; Ladrière, L.; Bazarra-Castro, A.; Ortis, F.; Wakeham, M.C.; Moore, F.; Rasschaert, J.; Cardozo, A.K.; Bellomo, E.; et al. Initiation and execution of lipotoxic ER stress in pancreatic β-cells. J. Cell Sci. 2008, 121, 2308–2318. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ge, M.; Ciani, L.; Kuriakose, G.; Westover, E.J.; Dura, M.; Covey, D.F.; Freed, J.H.; Maxfield, F.R.; Lytton, J.; et al. Enrichment of endoplasmic reticulum with cholesterol inhibits sarcoplasmic-endoplasmic reticulum calcium ATPase-2b activity in parallel with increased order of membrane lipids: Implications for depletion of endoplasmic reticulum calcium stores and apoptosis in cholesterol-loaded macrophages. J. Biol. Chem. 2004, 279, 37030–37039. [Google Scholar] [PubMed]
- Hara, T.; Mahadevan, J.; Kanekura, K.; Hara, M.; Lu, S.; Urano, F. Calcium Efflux from the Endoplasmic Reticulum Leads to β-Cell Death. Endocrinology 2014, 155, 758–768. [Google Scholar] [CrossRef]
- Volmer, R.; van der Ploeg, K.; Ron, D. Membrane lipid saturation activates endoplasmic reticulum unfolded protein response transducers through their transmembrane domains. Proc. Natl. Acad. Sci. USA 2013, 110, 4628–4633. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Montaño, P.; García-González, V. Effects of Dietary Fatty Acids in Pancreatic Beta Cell Metabolism, Implications in Homeostasis. Nutrients 2018, 10, 393. [Google Scholar] [CrossRef] [PubMed]
- Oh, Y.S.; Bae, G.D.; Baek, D.J.; Park, E.-Y.; Jun, H.-S. Fatty Acid-Induced Lipotoxicity in Pancreatic Beta-Cells During Development of Type 2 Diabetes. Front. Endocrinol. 2018, 9, 384. [Google Scholar] [CrossRef]
- Wang, Y.; Yamada, E.; Zong, H.; Pessin, J.E. Fyn Activation of mTORC1 Stimulates the IRE1α-JNK Pathway, Leading to Cell Death. J. Biol. Chem. 2015, 290, 24772–24783. [Google Scholar] [CrossRef]
- Urano, F.; Wang, X.; Bertolotti, A.; Zhang, Y.; Chung, P.; Harding, H.P.; Ron, D. Coupling of stress in the ER to activation of JNK protein kinases by transmembrane protein kinase IRE1. Science 2000, 287, 664–666. [Google Scholar] [CrossRef]
- Brozzi, F.; Gerlo, S.; Grieco, F.A.; Juusola, M.; Balhuizen, A.; Lievens, S.; Gysemans, C.; Bugliani, M.; Mathieu, C.; Marchetti, P.; et al. Ubiquitin D Regulates IRE1α/c-Jun N-terminal Kinase (JNK) Protein-dependent Apoptosis in Pancreatic Beta Cells. J. Biol. Chem. 2016, 291, 12040–12056. [Google Scholar] [CrossRef]
- Soustek, M.S.; Balsa, E.; Barrow, J.J.; Jedrychowski, M.; Vogel, R.; Smeitink, J.; Gygi, S.P.; Puigserver, P. Inhibition of the ER stress IRE1α inflammatory pathway protects against cell death in mitochondrial complex I mutant cells. Cell Death Dis. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Pal, D.; Dasgupta, S.; Kundu, R.; Maitra, S.; Das, G.; Mukhopadhyay, S.; Ray, S.; Majumdar, S.S.; Bhattacharya, S. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat. Med. 2012, 18, 1279–1285. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Chen, L.; Hu, L.; Liu, Y.; Sun, H.-Y.; Tang, J.; Hou, Y.-J.; Chang, Y.-X.; Tu, Q.-Q.; Feng, G.-S.; et al. Nuclear factor high-mobility group box1 mediating the activation of Toll-like receptor 4 signaling in hepatocytes in the early stage of nonalcoholic fatty liver disease in mice. Hepatol. Baltim. Md 2011, 54, 1620–1630. [Google Scholar] [CrossRef] [PubMed]
- Ehses, J.A.; Meier, D.T.; Wueest, S.; Rytka, J.; Boller, S.; Wielinga, P.Y.; Schraenen, A.; Lemaire, K.; Debray, S.; Van Lommel, L.; et al. Toll-like receptor 2-deficient mice are protected from insulin resistance and beta cell dysfunction induced by a high-fat diet. Diabetologia 2010, 53, 1795–1806. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid–induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [PubMed]
- Marijke Keestra-Gounder, A.; Tsolis, R.M. NOD1 and NOD2: Beyond peptidoglycan sensing. Trends Immunol. 2017, 38, 758–767. [Google Scholar] [CrossRef]
- Zhou, Y.; Tang, Y.; Song, Y.; Li, A.; Zhou, H.; Li, Y. Saturated Fatty Acid Induces Insulin Resistance Partially Through Nucleotide-binding Oligomerization Domain 1 Signaling Pathway in Adipocytes. Chin. Med. Sci. J. 2013, 28, 211–217. [Google Scholar] [CrossRef]
- Schertzer, J.D.; Tamrakar, A.K.; Magalhães, J.G.; Pereira, S.; Bilan, P.J.; Fullerton, M.D.; Liu, Z.; Steinberg, G.R.; Giacca, A.; Philpott, D.J.; et al. NOD1 Activators Link Innate Immunity to Insulin Resistance. Diabetes 2011, 60, 2206–2215. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid-induced NLRP3-PYCARD inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef]
- Sokolova, M.; Sahraoui, A.; Høyem, M.; Øgaard, J.; Lien, E.; Aukrust, P.; Yndestad, A.; Ranheim, T.; Scholz, H. NLRP3 inflammasome mediates oxidative stress-induced pancreatic islet dysfunction. Am. J. Physiol.-Endocrinol. Metab. 2018, 315, E912–E923. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Adijiang, A.; Vandanmagsar, B.; Burk, D.; Ravussin, A.; Dixit, V.D. Elimination of the NLRP3-ASC Inflammasome Protects against Chronic Obesity-Induced Pancreatic Damage. Endocrinology 2011, 152, 4039–4045. [Google Scholar] [CrossRef] [PubMed]
- Lambertucci, R.H.; Hirabara, S.M.; dos Silveira, L.R.; Levada-Pires, A.C.; Curi, R.; Pithon-Curi, T.C. Palmitate increases superoxide production through mitochondrial electron transport chain and NADPH oxidase activity in skeletal muscle cells. J. Cell. Physiol. 2008, 216, 796–804. [Google Scholar] [CrossRef]
- Yuzefovych, L.; Wilson, G.; Rachek, L. Different effects of oleate vs. palmitate on mitochondrial function, apoptosis, and insulin signaling in L6 skeletal muscle cells: Role of oxidative stress. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E1096–E1105. [Google Scholar] [CrossRef] [PubMed]
- Özcan, U.; Cao, Q.; Yilmaz, E.; Lee, A.-H.; Iwakoshi, N.N.; Özdelen, E.; Tuncman, G.; Görgün, C.; Glimcher, L.H.; Hotamisligil, G.S. Endoplasmic Reticulum Stress Links Obesity, Insulin Action, and Type 2 Diabetes. Science 2004, 306, 457–461. [Google Scholar] [CrossRef] [PubMed]
- Deldicque, L.; Cani, P.D.; Philp, A.; Raymackers, J.-M.; Meakin, P.J.; Ashford, M.L.J.; Delzenne, N.M.; Francaux, M.; Baar, K. The unfolded protein response is activated in skeletal muscle by high-fat feeding: Potential role in the downregulation of protein synthesis. Am. J. Physiol.-Endocrinol. Metab. 2010, 299, E695–E705. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C.; Craig, D.L.; Biden, T.J. Ceramide Generation Is Sufficient to Account for the Inhibition of the Insulin-stimulated PKB Pathway in C2C12 Skeletal Muscle Cells Pretreated with Palmitate. J. Biol. Chem. 1999, 274, 24202–24210. [Google Scholar] [CrossRef]
- Vlavcheski, F.; Tsiani, E. Attenuation of Free Fatty Acid-Induced Muscle Insulin Resistance by Rosemary Extract. Nutrients 2018, 10, 1623. [Google Scholar] [CrossRef]
- Cazzolli, R.; Carpenter, L.; Biden, T.J.; Schmitz-Peiffer, C. A Role for Protein Phosphatase 2A–Like Activity, but Not Atypical Protein Kinase Cζ, in the Inhibition of Protein Kinase B/Akt and Glycogen Synthesis by Palmitate. Diabetes 2001, 50, 2210–2218. [Google Scholar] [CrossRef]
- Pereira, S.; Park, E.; Mori, Y.; Haber, C.A.; Han, P.; Uchida, T.; Stavar, L.; Oprescu, A.I.; Koulajian, K.; Ivovic, A.; et al. FFA-induced hepatic insulin resistance in vivo is mediated by PKCδ, NADPH oxidase, and oxidative stress. Am. J. Physiol.-Endocrinol. Metab. 2014, 307, E34–E46. [Google Scholar] [CrossRef]
- Bezy, O.; Tran, T.T.; Pihlajamäki, J.; Suzuki, R.; Emanuelli, B.; Winnay, J.; Mori, M.A.; Haas, J.; Biddinger, S.B.; Leitges, M.; et al. PKCδ regulates hepatic insulin sensitivity and hepatosteatosis in mice and humans. J. Clin. Investig. 2011, 121, 2504–2517. [Google Scholar] [CrossRef]
- Samuel, V.T.; Liu, Z.-X.; Wang, A.; Beddow, S.A.; Geisler, J.G.; Kahn, M.; Zhang, X.; Monia, B.P.; Bhanot, S.; Shulman, G.I. Inhibition of protein kinase Cε prevents hepatic insulin resistance in nonalcoholic fatty liver disease. J. Clin. Investig. 2007, 117, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Liu, Z.-X.; Qu, X.; Elder, B.D.; Bilz, S.; Befroy, D.; Romanelli, A.J.; Shulman, G.I. Mechanism of Hepatic Insulin Resistance in Non-alcoholic Fatty Liver Disease. J. Biol. Chem. 2004, 279, 32345–32353. [Google Scholar] [CrossRef] [PubMed]
- Konstantynowicz-Nowicka, K.; Harasim, E.; Baranowski, M.; Chabowski, A. New Evidence for the Role of Ceramide in the Development of Hepatic Insulin Resistance. PLoS ONE 2015, 10, e0116858. [Google Scholar] [CrossRef] [PubMed]
- Longato, L.; Tong, M.; Wands, J.R.; de la Monte, S.M. High Fat Diet Induced Hepatic Steatosis And Insulin Resistance: Role Of Dysregulated Ceramide Metabolism. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2012, 42, 412–427. [Google Scholar] [CrossRef]
- Özcan, U.; Yilmaz, E.; Özcan, L.; Furuhashi, M.; Vaillancourt, E.; Smith, R.O.; Görgün, C.Z.; Hotamisligil, G.S. Chemical Chaperones Reduce ER Stress and Restore Glucose Homeostasis in a Mouse Model of Type 2 Diabetes. Science 2006, 313, 1137–1140. [Google Scholar] [CrossRef]
- Collins, Q.F.; Xiong, Y.; Lupo, E.G.; Liu, H.-Y.; Cao, W. p38 Mitogen-activated Protein Kinase Mediates Free Fatty Acid-induced Gluconeogenesis in Hepatocytes. J. Biol. Chem. 2006, 281, 24336–24344. [Google Scholar] [CrossRef]
- Pereira, S.; Yu, W.Q.; Moore, J.; Mori, Y.; Tsiani, E.; Giacca, A. Effect of a p38 MAPK inhibitor on FFA-induced hepatic insulin resistance in vivo. Nutr. Diabetes 2016, 6, e210. [Google Scholar] [CrossRef]
- Tang, P.; Low, H.B.; Png, C.W.; Torta, F.; Kumar, J.K.; Lim, H.Y.; Zhou, Y.; Yang, H.; Angeli, V.; Shabbir, A.; et al. Protective Function of Mitogen-Activated Protein Kinase Phosphatase 5 in Aging- and Diet-Induced Hepatic Steatosis and Steatohepatitis. Hepatol. Commun. 2019, 3, 748–762. [Google Scholar] [CrossRef]
- Xiong, Y.; Collins, Q.F.; An, J.; Lupo, E.; Liu, H.-Y.; Liu, D.; Robidoux, J.; Liu, Z.; Cao, W. p38 Mitogen-activated Protein Kinase Plays an Inhibitory Role in Hepatic Lipogenesis. J. Biol. Chem. 2007, 282, 4975–4982. [Google Scholar] [CrossRef]
- Kuo, L.-H.; Tsai, P.-J.; Jiang, M.-J.; Chuang, Y.-L.; Yu, L.; Lai, K.-T.A.; Tsai, Y.-S. Toll-like receptor 2 deficiency improves insulin sensitivity and hepatic insulin signalling in the mouse. Diabetologia 2011, 54, 168–179. [Google Scholar] [CrossRef]
- Jia, L.; Vianna, C.R.; Fukuda, M.; Berglund, E.D.; Liu, C.; Tao, C.; Sun, K.; Liu, T.; Harper, M.J.; Lee, C.E.; et al. Hepatocyte Toll-like receptor 4 regulates obesity-induced inflammation and insulin resistance. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef]
- Ibrahim, S.H.; Gores, G.J. Who pulls the trigger: JNK activation in liver lipotoxicity? J. Hepatol. 2012, 56, 17–19. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Malhi, H.; Bronk, S.F.; Werneburg, N.W.; Gores, G.J. Free Fatty Acids Induce JNK-dependent Hepatocyte Lipoapoptosis. J. Biol. Chem. 2006, 281, 12093–12101. [Google Scholar] [CrossRef] [PubMed]
- Wrede, C.E.; Dickson, L.M.; Lingohr, M.K.; Briaud, I.; Rhodes, C.J. Fatty acid and phorbol ester-mediated interference of mitogenic signaling via novel protein kinase C isoforms in pancreatic beta-cells (INS-1). J. Mol. Endocrinol. 2003, 30, 271–286. [Google Scholar] [CrossRef] [PubMed]
- Hennige, A.M.; Ranta, F.; Heinzelmann, I.; Düfer, M.; Michael, D.; Braumüller, H.; Lutz, S.Z.; Lammers, R.; Drews, G.; Bosch, F.; et al. Overexpression of Kinase-Negative Protein Kinase Cδ in Pancreatic β-Cells Protects Mice From Diet-Induced Glucose Intolerance and β-Cell Dysfunction. Diabetes 2010, 59, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Schmitz-Peiffer, C.; Laybutt, D.R.; Burchfield, J.G.; Gurisik, E.; Narasimhan, S.; Mitchell, C.J.; Pedersen, D.J.; Braun, U.; Cooney, G.J.; Leitges, M.; et al. Inhibition of PKCɛ Improves Glucose-Stimulated Insulin Secretion and Reduces Insulin Clearance. Cell Metab. 2007, 6, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Koulajian, K.; Ivovic, A.; Ye, K.; Desai, T.; Shah, A.; George Fantus, I.; Ran, Q.; Giacca, A. Overexpression of glutathione peroxidase 4 prevents β-cell dysfunction induced by prolonged elevation of lipids in vivo. Am. J. Physiol.-Endocrinol. Metab. 2013, 305, E254–E262. [Google Scholar] [CrossRef]
- Koulajian, K.; Desai, T.; Liu, G.C.; Ivovic, A.; Patterson, J.N.; Tang, C.; El-Benna, J.; Joseph, J.W.; Scholey, J.W.; Giacca, A. NADPH oxidase inhibition prevents beta cell dysfunction induced by prolonged elevation of oleate in rodents. Diabetologia 2013, 56, 1078–1087. [Google Scholar] [CrossRef]
- Drews, G.; Krippeit-Drews, P.; Düfer, M. Oxidative stress and beta-cell dysfunction. Pflugers Arch. 2010, 460, 703–718. [Google Scholar] [CrossRef]
- Solinas, G.; Naugler, W.; Galimi, F.; Lee, M.-S.; Karin, M. Saturated fatty acids inhibit induction of insulin gene transcription by JNK-mediated phosphorylation of insulin-receptor substrates. Proc. Natl. Acad. Sci. USA 2006, 103, 16454–16459. [Google Scholar] [CrossRef]
- Kelpe, C.L.; Moore, P.C.; Parazzoli, S.D.; Wicksteed, B.; Rhodes, C.J.; Poitout, V. Palmitate inhibition of insulin gene expression is mediated at the transcriptional level via ceramide synthesis. J. Biol. Chem. 2003, 278, 30015–30021. [Google Scholar] [CrossRef]
- Boslem, E.; Meikle, P.J.; Biden, T.J. Roles of ceramide and sphingolipids in pancreatic β-cell function and dysfunction. Islets 2012, 4, 177–187. [Google Scholar] [CrossRef]
- Plaisance, V.; Perret, V.; Favre, D.; Abderrahmani, A.; Yang, J.-Y.; Widmann, C.; Regazzi, R. Role of the transcriptional factor C/EBPβ in free fatty acid-elicited β-cell failure. Mol. Cell. Endocrinol. 2009, 305, 47–55. [Google Scholar] [CrossRef]
- Poitout, V.; Hagman, D.; Stein, R.; Artner, I.; Robertson, R.P.; Harmon, J.S. Regulation of the Insulin Gene by Glucose and Fatty Acids. J. Nutr. 2006, 136, 873–876. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, L.; Zhang, Y.; Zhang, W.J.; Xu, W.; Qin, Y.; Xu, J.; Zou, D. TLR4 is required for the obesity-induced pancreatic beta cell dysfunction. Acta Biochim. Biophys. Sin. 2013, 45, 1030–1038. [Google Scholar] [CrossRef] [PubMed]
- Benomar, Y.; Taouis, M. Molecular Mechanisms Underlying Obesity-Induced Hypothalamic Inflammation and Insulin Resistance: Pivotal Role of Resistin/TLR4 Pathways. Front. Endocrinol. 2019, 10. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.A.; Velloso, L.A. Consumption of a Fat-Rich Diet Activates a Proinflammatory Response and Induces Insulin Resistance in the Hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed]
- Tsaousidou, E.; Paeger, L.; Belgardt, B.F.; Pal, M.; Wunderlich, C.M.; Brönneke, H.; Collienne, U.; Hampel, B.; Wunderlich, F.T.; Schmidt-Supprian, M.; et al. Distinct Roles for JNK and IKK Activation in Agouti-Related Peptide Neurons in the Development of Obesity and Insulin Resistance. Cell Rep. 2014, 9, 1495–1506. [Google Scholar] [CrossRef]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol.-Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef]
- Cheng, L.; Yu, Y.; Szabo, A.; Wu, Y.; Wang, H.; Camer, D.; Huang, X.-F. Palmitic acid induces central leptin resistance and impairs hepatic glucose and lipid metabolism in male mice. J. Nutr. Biochem. 2015, 26, 541–548. [Google Scholar] [CrossRef]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.L.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated Fatty Acids Produce an Inflammatory Response Predominantly through the Activation of TLR4 Signaling in Hypothalamus: Implications for the Pathogenesis of Obesity. J. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Mayer, C.M.; Belsham, D.D. Palmitate Attenuates Insulin Signaling and Induces Endoplasmic Reticulum Stress and Apoptosis in Hypothalamic Neurons: Rescue of Resistance and Apoptosis through Adenosine 5′ Monophosphate-Activated Protein Kinase Activation. Endocrinology 2010, 151, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Won, J.C.; Jang, P.-G.; Namkoong, C.; Koh, E.H.; Kim, S.K.; Park, J.-Y.; Lee, K.-U.; Kim, M.-S. Central Administration of an Endoplasmic Reticulum Stress Inducer Inhibits the Anorexigenic Effects of Leptin and Insulin. Obesity 2009, 17, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Contreras, C.; González-García, I.; Martínez-Sánchez, N.; Seoane-Collazo, P.; Jacas, J.; Morgan, D.A.; Serra, D.; Gallego, R.; Gonzalez, F.; Casals, N.; et al. Central Ceramide-Induced Hypothalamic Lipotoxicity and ER Stress Regulate Energy Balance. Cell Rep. 2014, 9, 366–377. [Google Scholar] [CrossRef] [PubMed]
- Cintra, D.E.; Ropelle, E.R.; Moraes, J.C.; Pauli, J.R.; Morari, J.; de Souza, C.T.; Grimaldi, R.; Stahl, M.; Carvalheira, J.B.; Saad, M.J.; et al. Unsaturated Fatty Acids Revert Diet-Induced Hypothalamic Inflammation in Obesity. PLoS ONE 2012, 7, e30571. [Google Scholar] [CrossRef]
- Prato, S.D. Role of glucotoxicity and lipotoxicity in the pathophysiology of Type 2 diabetes mellitus and emerging treatment strategies. Diabet. Med. 2009, 26, 1185–1192. [Google Scholar] [CrossRef]
- Sivitz, W.I.; Yorek, M.A. Mitochondrial Dysfunction in Diabetes: From Molecular Mechanisms to Functional Significance and Therapeutic Opportunities. Antioxid. Redox Signal. 2010, 12, 537–577. [Google Scholar] [CrossRef]
- Volpe, C.M.O.; Villar-Delfino, P.H.; dos Anjos, P.M.F.; Nogueira-Machado, J.A. Cellular death, reactive oxygen species (ROS) and diabetic complications. Cell Death Dis. 2018, 9, 1–9. [Google Scholar] [CrossRef]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Chen, F.; Yu, Y.; Haigh, S.; Johnson, J.; Lucas, R.; Stepp, D.W.; Fulton, D.J.R. Regulation of NADPH Oxidase 5 by Protein Kinase C Isoforms. PLoS ONE 2014, 9, e88405. [Google Scholar] [CrossRef]
- Kaneto, H.; Matsuoka, T. Involvement of Oxidative Stress in Suppression of Insulin Biosynthesis under Diabetic Conditions. Int. J. Mol. Sci. 2012, 13, 13680–13690. [Google Scholar] [CrossRef] [PubMed]
- Back, S.H.; Kaufman, R.J. Endoplasmic Reticulum Stress and Type 2 Diabetes. Annu. Rev. Biochem. 2012, 81, 767–793. [Google Scholar] [CrossRef] [PubMed]
- Lipson, K.L.; Fonseca, S.G.; Ishigaki, S.; Nguyen, L.X.; Foss, E.; Bortell, R.; Rossini, A.A.; Urano, F. Regulation of insulin biosynthesis in pancreatic beta cells by an endoplasmic reticulum-resident protein kinase IRE1. Cell Metab. 2006, 4, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Sheikh-Ali, M.; Sultan, S.; Alamir, A.-R.; Haas, M.J.; Mooradian, A.D. Hyperglycemia-induced endoplasmic reticulum stress in endothelial cells. Nutrition 2010, 26, 1146–1150. [Google Scholar] [CrossRef]
- Lindenmeyer, M.T.; Rastaldi, M.P.; Ikehata, M.; Neusser, M.A.; Kretzler, M.; Cohen, C.D.; Schlöndorff, D. Proteinuria and Hyperglycemia Induce Endoplasmic Reticulum Stress. J. Am. Soc. Nephrol. JASN 2008, 19, 2225–2236. [Google Scholar] [CrossRef]
- Talior, I.; Yarkoni, M.; Bashan, N.; Eldar-Finkelman, H. Increased glucose uptake promotes oxidative stress and PKC-δ activation in adipocytes of obese, insulin-resistant mice. Am. J. Physiol.-Endocrinol. Metab. 2003, 285, E295–E302. [Google Scholar] [CrossRef]
- Lu, B.; Ennis, D.; Lai, R.; Bogdanovic, E.; Nikolov, R.; Salamon, L.; Fantus, C.; Le-Tien, H.; Fantus, I.G. Enhanced Sensitivity of Insulin-resistant Adipocytes to Vanadate Is Associated with Oxidative Stress and Decreased Reduction of Vanadate (+5) to Vanadyl (+4). J. Biol. Chem. 2001, 276, 35589–35598. [Google Scholar] [CrossRef]
- Gagnon, A.M.; Sorisky, A. The Effect of Glucose Concentration on Insulin-Induced 3T3-L1 Adipose Cell Differentiation. Obes. Res. 1998, 6, 157–163. [Google Scholar] [CrossRef]
- Longo, M.; Spinelli, R.; D’Esposito, V.; Zatterale, F.; Fiory, F.; Nigro, C.; Raciti, G.A.; Miele, C.; Formisano, P.; Beguinot, F.; et al. Pathologic endoplasmic reticulum stress induced by glucotoxic insults inhibits adipocyte differentiation and induces an inflammatory phenotype. Biochim. Biophys. Acta BBA-Mol. Cell Res. 2016, 1863, 1146–1156. [Google Scholar] [CrossRef]
- Alhusaini, S.; McGee, K.; Schisano, B.; Harte, A.; McTernan, P.; Kumar, S.; Tripathi, G. Lipopolysaccharide, high glucose and saturated fatty acids induce endoplasmic reticulum stress in cultured primary human adipocytes: Salicylate alleviates this stress. Biochem. Biophys. Res. Commun. 2010, 397, 472–478. [Google Scholar] [CrossRef]
- Tirosh, A.; Potashnik, R.; Bashan, N.; Rudich, A. Oxidative stress disrupts insulin-induced cellular redistribution of insulin receptor substrate-1 and phosphatidylinositol 3-kinase in 3T3-L1 adipocytes. A putative cellular mechanism for impaired protein kinase B activation and GLUT4 translocation. J. Biol. Chem. 1999, 274, 10595–10602. [Google Scholar] [CrossRef] [PubMed]
- Rudich, A.; Tirosh, A.; Potashnik, R.; Hemi, R.; Kanety, H.; Bashan, N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes 1998, 47, 1562–1569. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.; Bogdanovic, E.; Yu, Z.; Cho, C.; Liu, L.; Ho, K.; Guo, J.; Yeung, L.S.N.; Lehmann, R.; Hundal, H.S.; et al. Combined Hyperglycemia- and Hyperinsulinemia-Induced Insulin Resistance in Adipocytes Is Associated With Dual Signaling Defects Mediated by PKC-ζ. Endocrinology 2018, 159, 1658–1677. [Google Scholar] [CrossRef]
- Haber, C.A.; Lam, T.K.T.; Yu, Z.; Gupta, N.; Goh, T.; Bogdanovic, E.; Giacca, A.; Fantus, I.G. N-acetylcysteine and taurine prevent hyperglycemia-induced insulin resistance in vivo: Possible role of oxidative stress. Am. J. Physiol.-Endocrinol. Metab. 2003, 285, E744–E753. [Google Scholar] [CrossRef] [PubMed]
- Blair, A.S.; Hajduch, E.; Litherland, G.J.; Hundal, H.S. Regulation of Glucose Transport and Glycogen Synthesis in L6 Muscle Cells during Oxidative stress evidence for cross-talk between the insulin and sapk2/p38 mitogen-activated protein kinase signaling pathways. J. Biol. Chem. 1999, 274, 36293–36299. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.; Yeung, L.S.N.; Koulajian, K.; Zhang, L.; Tai, K.; Volchuk, A.; Giacca, A. Glucose-Induced β-Cell Dysfunction In Vivo: Evidence for a Causal Role of C-jun N-terminal Kinase Pathway. Endocrinology 2018, 159, 3643–3654. [Google Scholar] [CrossRef]
- Davidson, M.D.; Ballinger, K.R.; Khetani, S.R. Long-term exposure to abnormal glucose levels alters drug metabolism pathways and insulin sensitivity in primary human hepatocytes. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef]
- Du, D.; Shi, Y.-H.; Le, G.-W. Oxidative stress induced by high-glucose diet in liver of C57BL/6J mice and its underlying mechanism. Mol. Biol. Rep. 2010, 37, 3833–3839. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular Mechanisms of Lipotoxicity and Glucotoxicity in Nonalcoholic Fatty Liver Disease. Metabolism. 2016, 65, 1049–1061. [Google Scholar] [CrossRef]
- Tang, C.; Han, P.; Oprescu, A.I.; Lee, S.C.; Gyulkhandanyan, A.V.; Chan, G.N.Y.; Wheeler, M.B.; Giacca, A. Evidence for a role of superoxide generation in glucose-induced beta-cell dysfunction in vivo. Diabetes 2007, 56, 2722–2731. [Google Scholar] [CrossRef][Green Version]
- Tang, C.; Koulajian, K.; Schuiki, I.; Zhang, L.; Desai, T.; Ivovic, A.; Wang, P.; Robson-Doucette, C.; Wheeler, M.B.; Minassian, B.; et al. Glucose-induced beta cell dysfunction in vivo in rats: Link between oxidative stress and endoplasmic reticulum stress. Diabetologia 2012, 55, 1366–1379. [Google Scholar] [CrossRef]
- Kaneto, H.; Kajimoto, Y.; Miyagawa, J.; Matsuoka, T.; Fujitani, Y.; Umayahara, Y.; Hanafusa, T.; Matsuzawa, Y.; Yamasaki, Y.; Hori, M. Beneficial effects of antioxidants in diabetes: Possible protection of pancreatic beta-cells against glucose toxicity. Diabetes 1999, 48, 2398–2406. [Google Scholar] [CrossRef]
- Tanaka, Y.; Gleason, C.E.; Tran, P.O.; Harmon, J.S.; Robertson, R.P. Prevention of glucose toxicity in HIT-T15 cells and Zucker diabetic fatty rats by antioxidants. Proc. Natl. Acad. Sci. USA 1999, 96, 10857–10862. [Google Scholar] [CrossRef]
- Gao, Y.; Bielohuby, M.; Fleming, T.; Grabner, G.F.; Foppen, E.; Bernhard, W.; Guzmán-Ruiz, M.; Layritz, C.; Legutko, B.; Zinser, E.; et al. Dietary sugars, not lipids, drive hypothalamic inflammation. Mol. Metab. 2017, 6, 897–908. [Google Scholar] [CrossRef]
- Zhou, J.; Deo, B.K.; Hosoya, K.; Terasaki, T.; Obrosova, I.G.; Brosius, F.C.; Kumagai, A.K. Increased JNK Phosphorylation and Oxidative Stress in Response to Increased Glucose Flux through Increased GLUT1 Expression in Rat Retinal Endothelial Cells. Investig. Ophthalmol. Vis. Sci. 2005, 46, 3403–3410. [Google Scholar] [CrossRef]
- Hein, T.W.; Xu, W.; Xu, X.; Kuo, L. Acute and Chronic Hyperglycemia Elicit JIP1/JNK-Mediated Endothelial Vasodilator Dysfunction of Retinal Arterioles. Investig. Ophthalmol. Vis. Sci. 2016, 57, 4333–4340. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.-J.; Ji, R.-R. Activation of JNK pathway in persistent pain. Neurosci. Lett. 2008, 437, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Sheng, J.; Li, H.; Dai, Q.; Lu, C.; Xu, M.; Zhang, J.; Feng, J. DUSP1 recuses diabetic nephropathy via repressing JNK-Mff-mitochondrial fission pathways. J. Cell. Physiol. 2019, 234, 3043–3057. [Google Scholar] [CrossRef] [PubMed]
- Thallas-Bonke, V.; Thorpe, S.R.; Coughlan, M.T.; Fukami, K.; Yap, F.Y.T.; Sourris, K.C.; Penfold, S.A.; Bach, L.A.; Cooper, M.E.; Forbes, J.M. Inhibition of NADPH Oxidase Prevents Advanced Glycation End Product–Mediated Damage in Diabetic Nephropathy Through a Protein Kinase C-α–Dependent Pathway. Diabetes 2008, 57, 460–469. [Google Scholar] [CrossRef]
- Grynberg, K.; Ma, F.Y.; Nikolic-Paterson, D.J. The JNK Signaling Pathway in Renal Fibrosis. Front. Physiol. 2017, 8, 829. [Google Scholar] [CrossRef]
- Borst, M.D.; Prakash, J.; Melenhorst, W.; van den Heuvel, M.; Kok, R.J.; Navis, G.; van Goor, H. Glomerular and tubular induction of the transcription factor c-Jun in human renal disease. J. Pathol. 2007, 213, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.K.H.; Tesch, G.H. Inflammation in Diabetic Nephropathy. Mediators Inflamm. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Razani, B.; Chakravarthy, M.V.; Semenkovich, C.F. Insulin Resistance and Atherosclerosis. Endocrinol. Metab. Clin. N. Am. 2008, 37, 603–621. [Google Scholar] [CrossRef] [PubMed]
- Metzler, B.; Hu, Y.; Dietrich, H.; Xu, Q. Increased Expression and Activation of Stress-Activated Protein Kinases/c-Jun NH2-Terminal Protein Kinases in Atherosclerotic Lesions Coincide with p53. Am. J. Pathol. 2000, 156, 1875–1886. [Google Scholar] [CrossRef]
- Amini, N.; Boyle, J.J.; Moers, B.; Warboys, C.M.; Malik, T.H.; Zakkar, M.; Francis, S.E.; Mason, J.C.; Haskard, D.O.; Evans, P.C. Requirement of JNK1 for endothelial cell injury in atherogenesis. Atherosclerosis 2014, 235, 613–618. [Google Scholar] [CrossRef]
- Nishio, H.; Matsui, K.; Tsuji, H.; Tamura, A.; Suzuki, K. Immunohistochemical study of the phosphorylated and activated form of c-Jun NH2-terminal kinase in human aorta. Histochem. J. 2001, 33, 167–171. [Google Scholar] [CrossRef]
- Miller, T.A.; LeBrasseur, N.K.; Cote, G.M.; Trucillo, M.P.; Pimentel, D.R.; Ido, Y.; Ruderman, N.B.; Sawyer, D.B. Oleate prevents palmitate-induced cytotoxic stress in cardiac myocytes. Biochem. Biophys. Res. Commun. 2005, 336, 309–315. [Google Scholar] [CrossRef]
- Hreniuk, D.; Garay, M.; Gaarde, W.; Monia, B.P.; McKay, R.A.; Cioffi, C.L. Inhibition of C-Jun N-Terminal Kinase 1, but Not c-Jun N-Terminal Kinase 2, Suppresses Apoptosis Induced by Ischemia/Reoxygenation in Rat Cardiac Myocytes. Mol. Pharmacol. 2001, 59, 867–874. [Google Scholar] [CrossRef]
- Bellucci, P.N.; González Bagnes, M.F.; Di Girolamo, G.; González, C.D. Potential Effects of Nonsteroidal Anti-Inflammatory Drugs in the Prevention and Treatment of Type 2 Diabetes Mellitus. J. Pharm. Pract. 2017, 30, 549–556. [Google Scholar] [CrossRef]
- Pollack, R.M.; Donath, M.Y.; LeRoith, D.; Leibowitz, G. Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39, S244–S252. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y. Multiple benefits of targeting inflammation in the treatment of type 2 diabetes. Diabetologia 2016, 59, 679–682. [Google Scholar] [CrossRef] [PubMed]
- Esser, N.; Paquot, N.; Scheen, A.J. Anti-inflammatory agents to treat or prevent type 2 diabetes, metabolic syndrome and cardiovascular disease. Expert Opin. Investig. Drugs 2015, 24, 283–307. [Google Scholar] [CrossRef]
- Cui, J.; Zhang, M.; Zhang, Y.; Xu, Z. JNK pathway: Diseases and therapeutic potential. Acta Pharmacol. Sin. 2007, 28, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Falsig, J.; Pörzgen, P.; Lotharius, J.; Leist, M. Specific Modulation of Astrocyte Inflammation by Inhibition of Mixed Lineage Kinases with CEP-1347. J. Immunol. 2004, 173, 2762–2770. [Google Scholar] [CrossRef] [PubMed]
- Parkinson Study Group PRECEPT Investigators Mixed lineage kinase inhibitor CEP-1347 fails to delay disability in early Parkinson disease. Neurology 2007, 69, 1480–1490. [CrossRef]
- Messoussi, A.; Feneyrolles, C.; Bros, A.; Deroide, A.; Daydé-Cazals, B.; Chevé, G.; Van Hijfte, N.; Fauvel, B.; Bougrin, K.; Yasri, A. Recent Progress in the Design, Study, and Development of c-Jun N-Terminal Kinase Inhibitors as Anticancer Agents. Chem. Biol. 2014, 21, 1433–1443. [Google Scholar] [CrossRef]
- Halazy, S. Designing heterocyclic selective kinase inhibitors: From concept to new drug candidates. Arkivoc 2006, 2006, 496. [Google Scholar]
- Kennedy, N.J. Role of JNK in Tumor Development. Cell Cycle 2003, 2, 199–201. [Google Scholar]
- Deloche, C.; Lopez-Lazaro, L.; Mouz, S.; Perino, J.; Abadie, C.; Combette, J.-M. XG-102 administered to healthy male volunteers as a single intravenous infusion: A randomized, double-blind, placebo-controlled, dose-escalating study. Pharmacol. Res. Perspect. 2014, 2, e00020. [Google Scholar] [CrossRef]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. c-Jun N-terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef]
- Melino, M.; Hii, C.S.; McColl, S.R.; Ferrante, A. The Effect of the JNK Inhibitor, JIP Peptide, on Human T Lymphocyte Proliferation and Cytokine Production. J. Immunol. 2008, 181, 7300–7306. [Google Scholar] [CrossRef] [PubMed]
- Kaneto, H.; Nakatani, Y.; Miyatsuka, T.; Kawamori, D.; Matsuoka, T.; Matsuhisa, M.; Kajimoto, Y.; Ichijo, H.; Yamasaki, Y.; Hori, M. Possible novel therapy for diabetes with cell-permeable JNK-inhibitory peptide. Nat. Med. 2004, 10, 1128–1132. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Class | Drug | Mechanism of Action | Summary |
|---|---|---|---|
| ATP-competitive inhibitors | SP600125 | Directly inhibits all JNK isoforms at the ATP-binding site | Investigation of this compound is limited to pre-clinical studies |
| CEP-1347 | Inhibits MLK at the ATP-binding site | Stopped in Phase II/III clinical trials; was developed for Parkinson’s disease | |
| AS601245 (Bentamapimod) | Directly inhibits all JNK isoforms at the ATP-binding site, but with higher selectivity for JNK1 and JNK2 | Phase II clinical trial recently completed about the effects on endometriosis | |
| Peptide inhibitors of JNK | D-JNKI-1 peptide (XG-102/AM-111) | Specifically targets the JNK binding domain (JBD) to inhibit JNK activation | Phase III clinical trial recently completed about the effects on inflammation, post-operative pain, and hearing loss |
| TAT-JIP protein | Resembles JNK interacting protein (JIP) to inhibit JNK | Improves glucose tolerance and insulin sensitivity in mice and upregulates insulin signaling in insulin-sensitive tissues. No available data in humans |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yung, J.H.M.; Giacca, A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells 2020, 9, 706. https://doi.org/10.3390/cells9030706
Yung JHM, Giacca A. Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells. 2020; 9(3):706. https://doi.org/10.3390/cells9030706
Chicago/Turabian StyleYung, Justin Hou Ming, and Adria Giacca. 2020. "Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes" Cells 9, no. 3: 706. https://doi.org/10.3390/cells9030706
APA StyleYung, J. H. M., & Giacca, A. (2020). Role of c-Jun N-terminal Kinase (JNK) in Obesity and Type 2 Diabetes. Cells, 9(3), 706. https://doi.org/10.3390/cells9030706