Autophagy Modulators Profoundly Alter the Astrocyte Cellular Proteome

Abstract
1. Introduction
2. Materials and Methods
2.1. Cells
2.2. Pharmacologic Treatments
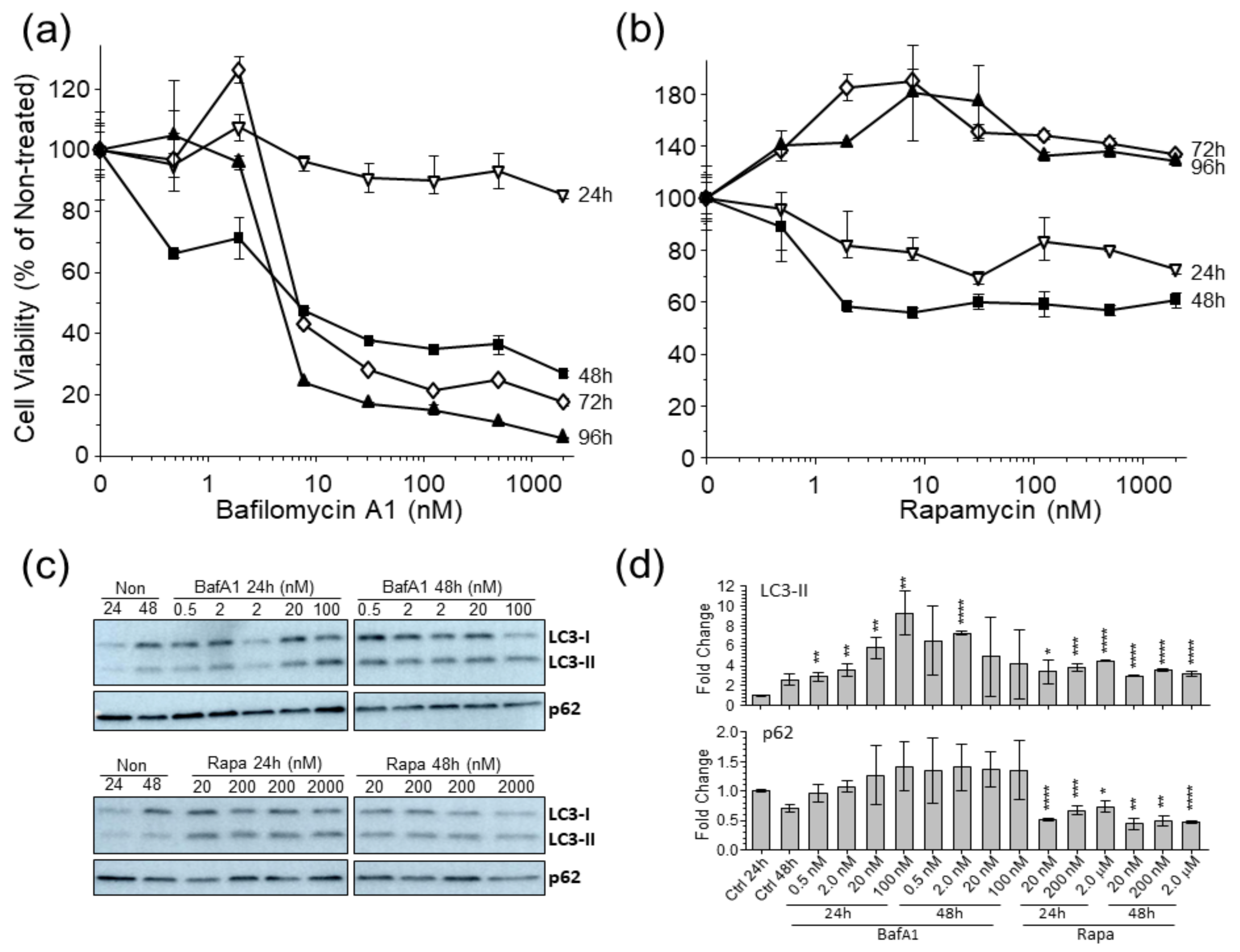
2.3. Cell Viability
2.4. Protein Quantification
2.5. SOMAscan® Analyses
2.6. Western Blots
2.7. Cytokine Arrays
2.8. Statistical and Bioinformatic Analyses
3. Results
3.1. Autophagy Markers Are Affected in U-251 Astrocytoma Cells by Bafilomycina1 and Rapamycin
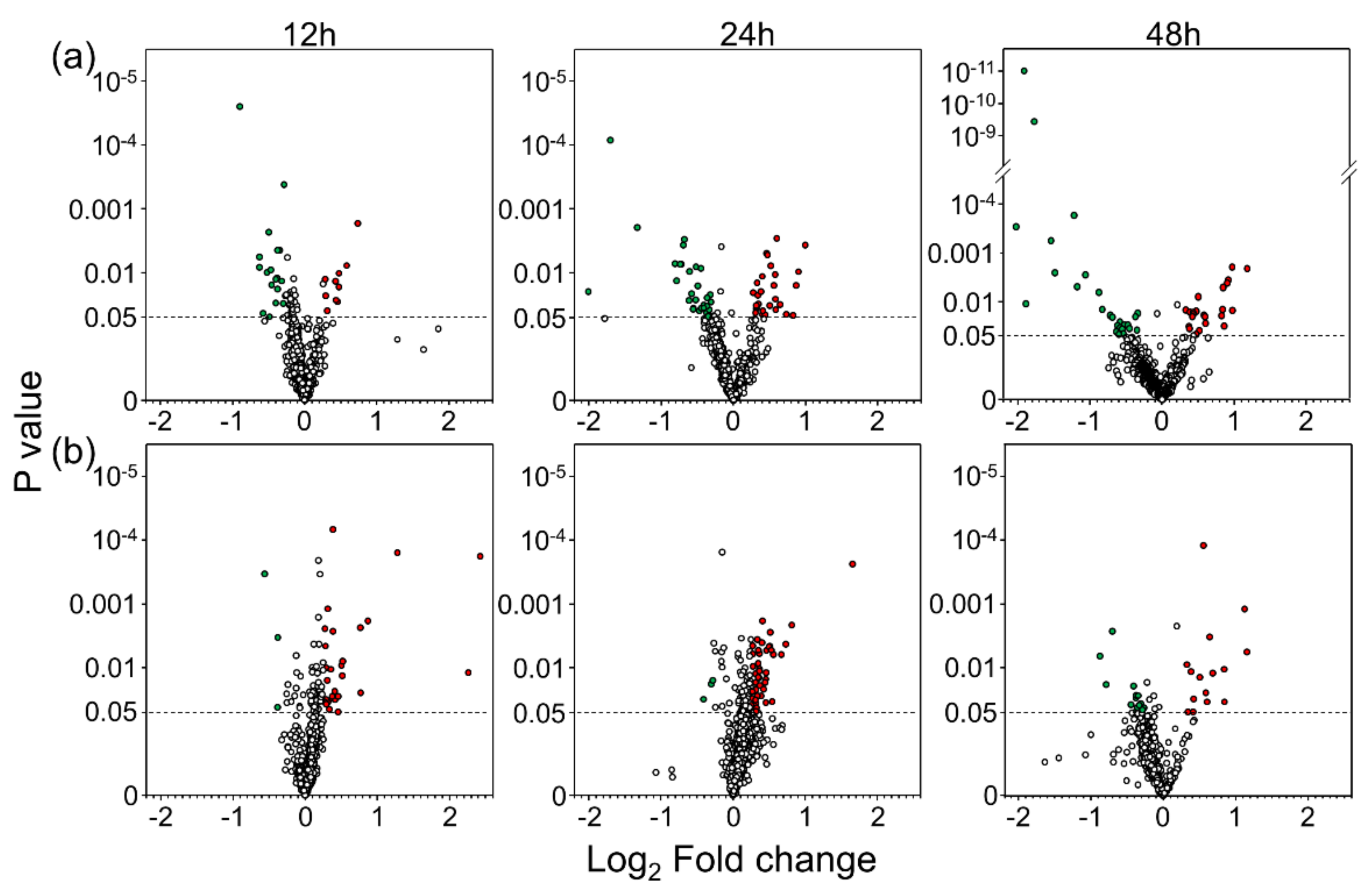
3.2. Bafa1 and Rapa Induce Dysregulation of Numerous U-251 Proteins
3.3. Bafa1 Generally Activates the U-251 Proteome and Rapa Generally Inhibits the U-251 Proteome
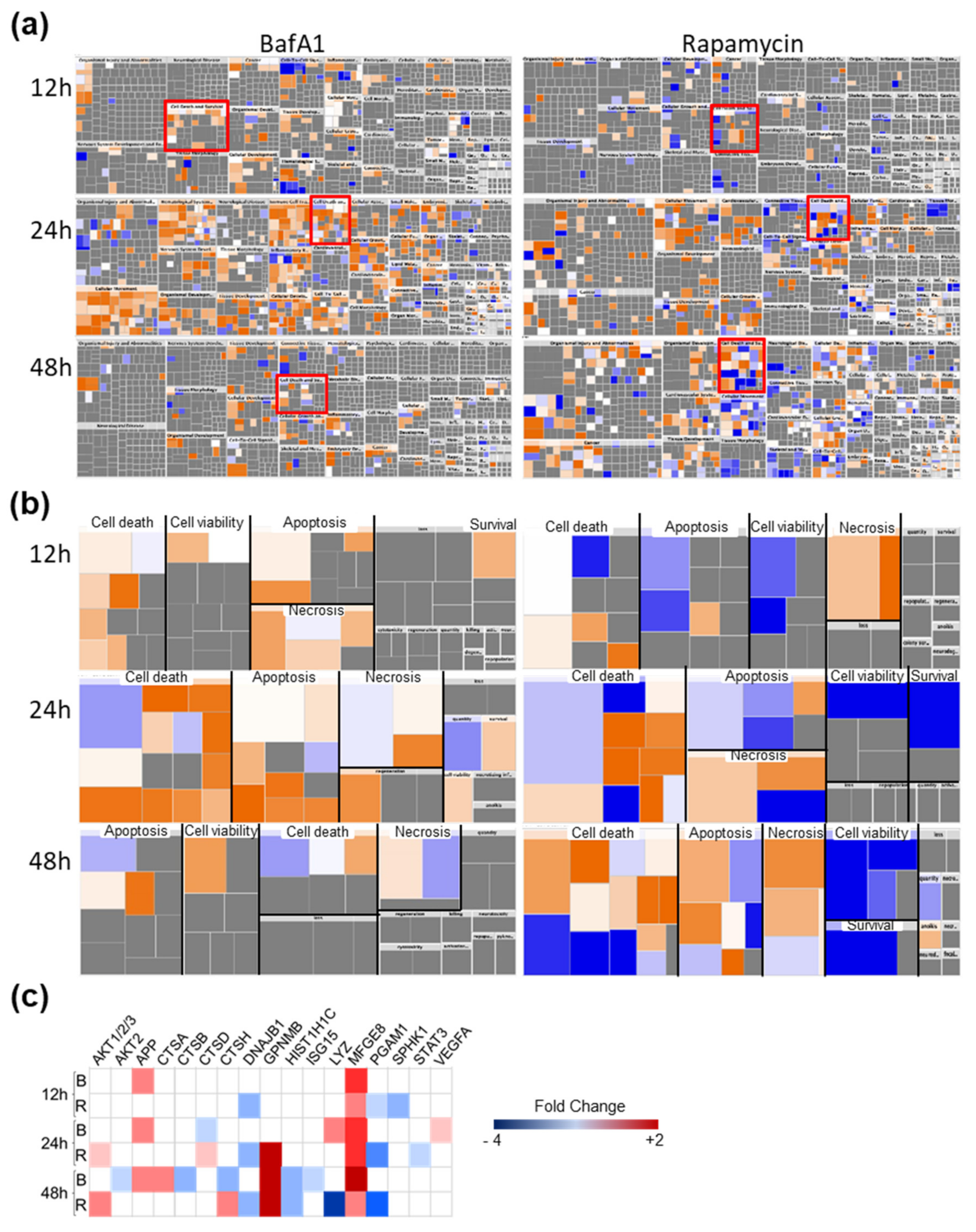
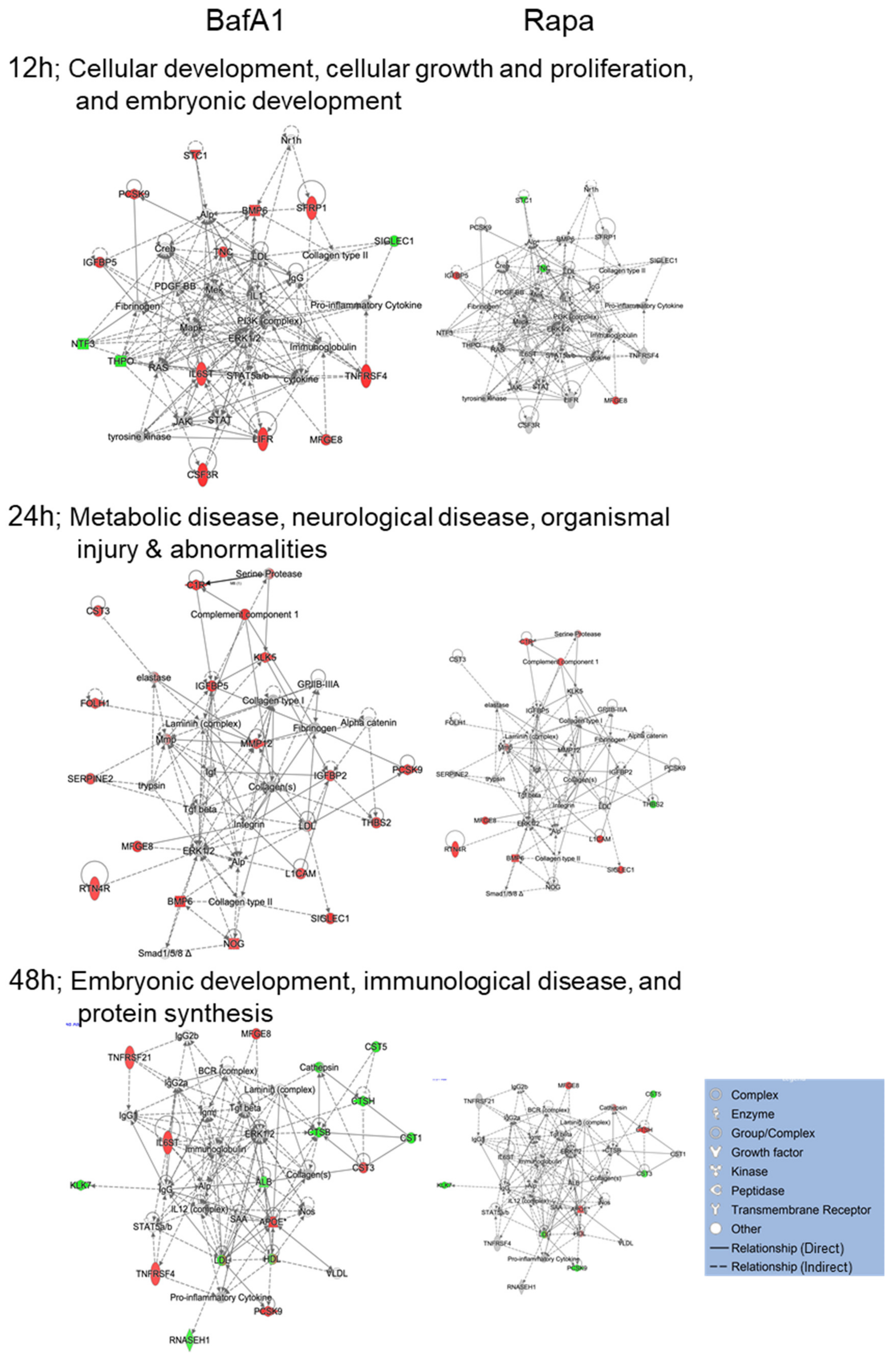
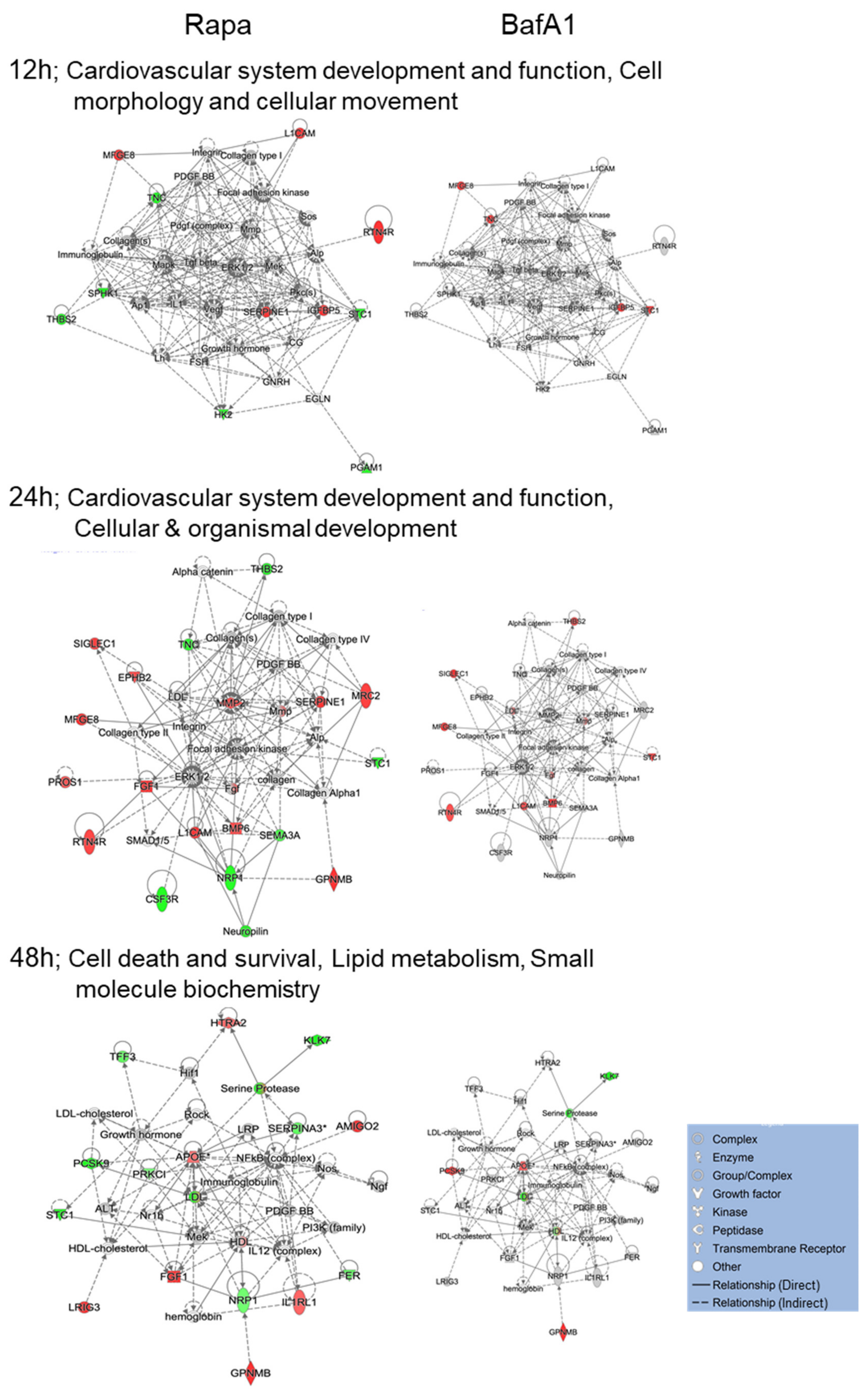
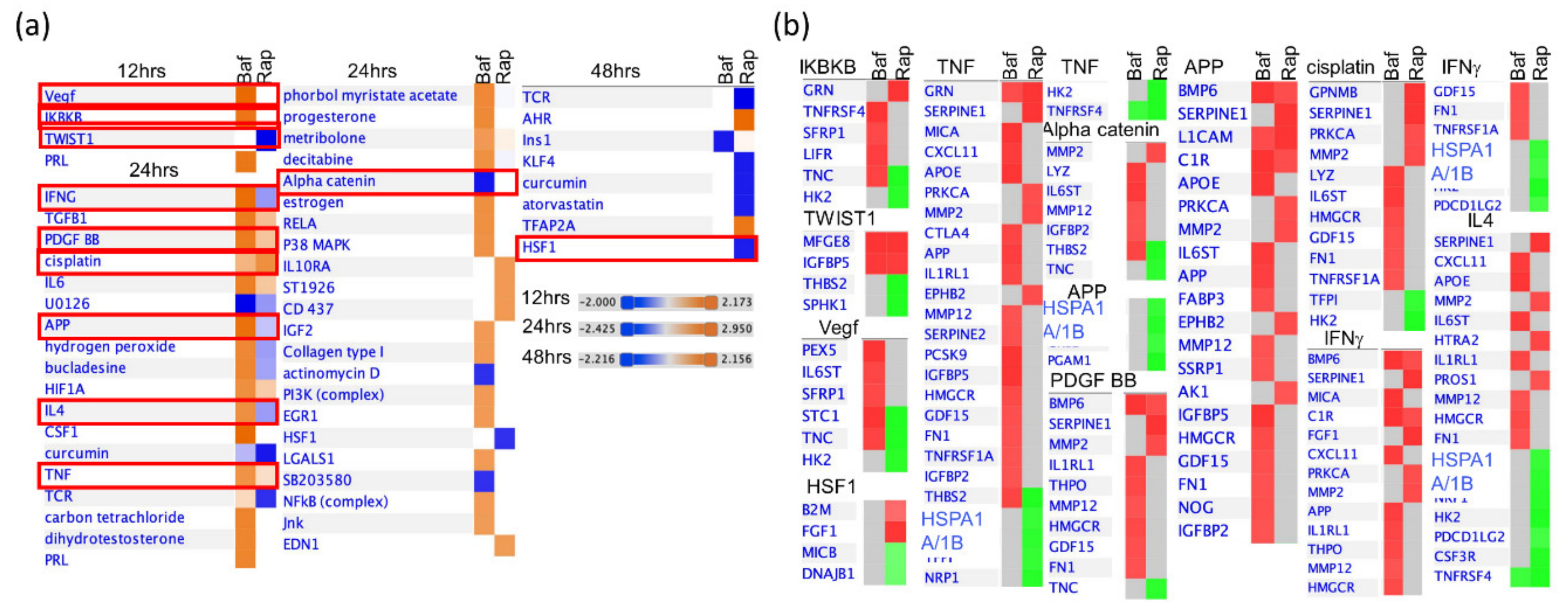
3.4. Bafa1 and Rapamycin Differentially Affect U-251 Bio-Functions
3.5. Bafilomycin and Rapamycin Differentially Affect Levels of Cytokine Secretion
4. Discussion
4.1. Implications of Bafa1/Rapa-Dysregulated Intracellular Astrocytic Proteins
4.2. Implications of Bafa1/Rapa-Dysregulated Secreted Cytokines
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- He, C.; Klionsky, D.J. Regulation mechanisms and signaling pathways of autophagy. Annu. Rev. Genet. 2009, 43, 67–93. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K. Organellophagy: Eliminating cellular building blocks via selective autophagy. J. Cell Biol. 2014, 205, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Acevedo-Arozena, A.; Adachi, H.; Adams, C.M.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Ravikumar, B.; Futter, M.; Jahreiss, L.; Korolchuk, V.I.; Lichtenberg, M.; Luo, S.; Massey, D.C.O.; Menzies, F.M.; Narayanan, U.; Renna, M.; et al. Mammalian macroautophagy at a glance. J. Cell Sci. 2009, 122, 1707–1711. [Google Scholar] [CrossRef] [PubMed]
- Li, W.-W.; Li, J.; Bao, J. Microautophagy: Lesser-known self-eating. Cell. Mol. Life Sci. 2011, 69, 1125–1136. [Google Scholar] [CrossRef]
- Schneider, J.L.; Suh, Y.; Cuervo, A.M. Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab. 2014, 20, 417–432. [Google Scholar] [CrossRef]
- Kaushik, S.; Bandyopadhyay, U.; Sridhar, S.; Kiffin, R.; Martinez-Vicente, M.; Kon, M.; Orenstein, S.J.; Wong, E.; Cuervo, A.M. Chaperone-mediated autophagy at a glance. J. Cell Sci. 2011, 124, 495–499. [Google Scholar] [CrossRef]
- Bandyopadhyay, U.; Kaushik, S.; Varticovski, L.; Cuervo, A.M. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol. Cell Biol. 2008, 28, 5747–5763. [Google Scholar] [CrossRef]
- Babak, S.; Mason, W.P. mTOR inhibition in glioblastoma: Requiem for a dream? Neuro-Oncology 2018, 20, 584–585. [Google Scholar] [CrossRef]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in glioblastoma: Rationale and preclinical/clinical evidence. Dis. Mark. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Li, B.; Zhou, C.; Yi, L.; Xu, L.; Xu, M.-H. Effect and molecular mechanism of mTOR inhibitor rapamycin on temozolomide-induced autophagic death of U251 glioma cells. Oncol. Lett. 2017, 15, 2477–2484. [Google Scholar] [CrossRef] [PubMed]
- Wojton, J.; Elder, J.; Kaur, B. How efficient are autophagy inhibitors as treatment for glioblastoma? CNS Oncol. 2014, 3, 5–7. [Google Scholar] [CrossRef]
- Cloughesy, T.F.; Yoshimoto, K.; Nghiemphu, P.; Brown, K.; Dang, J.; Zhu, S.; Hsueh, T.; Chen, Y.; Wang, W.; Youngkin, D.; et al. Antitumor activity of rapamycin in a phase I trial for patients with recurrent PTEN-deficient glioblastoma. PLoS Med. 2008, 5, e8. [Google Scholar] [CrossRef] [PubMed]
- Dolcetta, D.; Dominici, R. The local mammalian target of rapamycin (mTOR) modulation: A promising strategy to counteract neurodegeneration. Neural Regen. Res. 2019, 14, 1711–1712. [Google Scholar] [CrossRef] [PubMed]
- Pivtoraiko, V.N.; Harrington, A.J.; Mader, B.J.; Luker, A.M.; Caldwell, G.A.; Caldwell, K.A.; Roth, K.A.; Shacka, J.J. Low-dose bafilomycin attenuates neuronal cell death associated with autophagy-lysosome pathway dysfunction. J. Neurochem. 2010, 114, 1193–1204. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.; Ayers, D.; Bertino, J.; Bock, C.; Bock, A.; Brody, E.N.; Carter, J.; Dalby, A.B.; Eaton, B.E.; Fitzwater, T.; et al. Aptamer-based multiplexed proteomic technology for biomarker discovery. Nat. Proced. 2010, 5, e15004. [Google Scholar]
- SOMAScan Proteomic Assay; Technical White Paper; SSM-002; SomaLogic: Boulder, CO, USA, 2017.
- Jiang, G.; Tan, Y.; Wang, H.; Peng, L.; Chen, H.-T.; Meng, X.-J.; Li, L.-L.; Liu, Y.; Li, W.-F.; Shan, H. The relationship between autophagy and the immune system and its applications for tumor immunotherapy. Mol. Cancer 2019, 18, 17. [Google Scholar] [CrossRef]
- Jang, Y.J.; Kim, J.H.; Byun, S. Modulation of autophagy for controlling immunity. Cells 2019, 8, 138. [Google Scholar] [CrossRef]
- Rosello, A.; Warnes, G.; Meier, U.-C. Cell death pathways and autophagy in the central nervous system and its involvement in neurodegeneration, immunity and central nervous system infection: To die or not to die—that is the question. Clin. Exp. Immunol. 2012, 168, 52–57. [Google Scholar] [CrossRef]
- Alirezaei, M.; Kemball, C.C.; Whitton, J.L. Autophagy, inflammation and neurodegenerative disease. Eur. J. Neurosci. 2010, 33, 197–204. [Google Scholar] [CrossRef]
- Su, P.; Zhang, J.; Wang, D.; Zhao, F.; Cao, Z.; Aschner, M.; Luo, W. The role of autophagy in modulation of neuroinflammation in microglia. Neuroscience 2016, 319, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Glover, K.K.M.; Gao, A.; Zahedi-Amiri, A.; Coombs, K.M. Vero cell proteomic changes induced by Zika virus infection. Proteomics 2019, 19, 1800309. [Google Scholar] [CrossRef]
- Sher, A.A.; Glover, K.K.M.; Coombs, K.M. Zika virus infection disrupts astrocytic proteins involved in synapse control and axon guidance. Front. Microbiol. 2019, 10, 596. [Google Scholar] [CrossRef] [PubMed]
- Coombs, K.M.; Berard, A.; Xu, W.; Krokhin, O.; Meng, X.; Cortens, J.P.; Kobasa, D.; Wilkins, J.; Brown, E.G. Quantitative proteomic analyses of influenza virus-infected cultured human lung cells. J. Virol. 2010, 84, 10888–10906. [Google Scholar] [CrossRef]
- Le Bot, N. Autophagy: A new regulator of development. Nat. Cell Biol. 2007, 9, 741. [Google Scholar] [CrossRef]
- Cecconi, F.; Levine, B. The role of autophagy in mammalian development: Cell makeover rather than cell death. Dev. Cell 2008, 15, 344–357. [Google Scholar] [CrossRef] [PubMed]
- Fitzwalter, B.; Towers, C.G.; Sullivan, K.; Andrysik, Z.; Hoh, M.; Ludwig, M.; O’Prey, J.; Ryan, K.M.; Espinosa, J.M.; Morgan, M.J.; et al. Autophagy inhibition mediates apoptosis sensitization in cancer therapy by relieving FOXO3a turnover. Dev. Cell 2018, 44, 555–565.e3. [Google Scholar] [CrossRef]
- Wang, R.C.; Levine, B. Autophagy in cellular growth control. FEBS Lett. 2010, 584, 1417–1426. [Google Scholar] [CrossRef]
- Mauvezin, C.; Neufeld, T.P. Bafilomycin A1 disrupts autophagic flux by inhibiting both V-ATPase-dependent acidification and Ca-P60A/SERCA-dependent autophagosome-lysosome fusion. Autophagy 2015, 11, 1437–1438. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, Q.; Ma, X.; Zhang, Z.; Liu, N.; Wang, M. Role of mammalian target of rapamycin signaling in autophagy and the neurodegenerative process using a senescence accelerated mouse-prone 8 model. Exp. Ther. Med. 2017, 14, 1051–1057. [Google Scholar] [CrossRef]
- Moulding, H.D.; Martuza, R.L.; Rabkin, S. Clinical mutations in the L1 neural cell adhesion molecule affect cell-surface expression. J. Neurosci. 2000, 20, 5696–5702. [Google Scholar] [CrossRef] [PubMed]
- Linneberg, C.; Toft, C.L.F.; Kjaer-Sorensen, K.; Laursen, L.S. L1cam-mediated developmental processes of the nervous system are differentially regulated by proteolytic processing. Sci. Rep. 2019, 9, 3716. [Google Scholar] [CrossRef] [PubMed]
- Cahoy, J.D.; Emery, B.; Kaushal, A.; Foo, L.C.; Zamanian, J.; Christopherson, K.S.; Xing, Y.; Lubischer, J.L.; Krieg, P.A.; Krupenko, S.A.; et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J. Neurosci. 2008, 28, 264–278. [Google Scholar] [CrossRef]
- Xu, X.T.; Zhang, A.W.; Zhu, Y.T.; He, W.; Di, W.; Fang, Y.N.; Shi, X.L. MFG-E8 reverses microglial-induced neurotoxic astrocyte (A1) via NF-kappa B and PI3K-Akt pathways. J. Cell. Physiol. 2019, 234, 904–914. [Google Scholar] [CrossRef] [PubMed]
- Cheyuo, C.; Aziz, M.; Wang, P. Neurogenesis in neurodegenerative diseases: Role of MFG-E8. Front. Mol. Neurosci. 2019, 13, 569. [Google Scholar] [CrossRef]
- Ernst, A.-S.; Böhler, L.-I.; Hagenston, A.M.; Hoffmann, A.; Heiland, S.; Sticht, C.; Bendszus, M.; Hecker, M.; Bading, H.; Marti, H.H.; et al. EphB2-dependent signaling promotes neuronal excitotoxicity and inflammation in the acute phase of ischemic stroke. Acta Neuropathol. Commun. 2019, 7, 15. [Google Scholar] [CrossRef]
- Bundesen, L.Q.; Scheel, T.A.; Bregman, B.S.; Kromer, L.F. Ephrin-B2 and EphB2 Regulation of astrocyte-meningeal fibroblast interactions in response to spinal cord lesions in adult rats. J. Neurosci. 2003, 23, 7789–7800. [Google Scholar] [CrossRef]
- Cassina, P.; Pehar, M.; Vargas, M.R.; Castellanos, R.; Barbeito, A.G.; Estévez, A.G.; Thompson, J.A.; Beckman, J.S.; Barbeito, L. Astrocyte activation by fibroblast growth factor-1 and motor neuron apoptosis: Implications for amyotrophic lateral sclerosis. J. Neurochem. 2005, 93, 38–46. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef]
- Burnside, M.N.; Pyatt, R.E.; Hughes, A.; Baker, P.B.; Pierson, C.R. Complex brain malformations associated with chromosome 6q27 gain that includes THBS2, which encodes thrombospondin 2, an astrocyte-derived protein of the extracellular matrix. Pediatr. Dev. Pathol. 2015, 18, 59–65. [Google Scholar] [CrossRef]
- Rousselet, E.; Marcinkiewicz, J.; Kriz, J.; Zhou, A.; Hatten, M.E.; Prat, A.; Seidah, N.G. PCSK9 reduces the protein levels of the LDL receptor in mouse brain during development and after ischemic stroke. J. Lipid Res. 2011, 52, 1383–1391. [Google Scholar] [CrossRef]
- Picard, C.; Poirier, A.; Belanger, S.; Labonte, A.; Auld, D.; Poirier, J.; Breitner, J.C.S.; Poirier, J.; Etienne, P.; Villeneuve, S.; et al. Proprotein convertase subtilisin/kexin type 9 (PCSK9) in Alzheimer’s disease: A genetic and proteomic multi-cohort study. PLoS ONE 2019, 14, e0220254. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yao, L.; Zhao, S.; Zhang, X.; Yin, J.; Zhang, Y.; Chen, X.; Gao, M.; Ling, E.-A.; Hao, A.; et al. Granulocyte-colony stimulating factor promotes proliferation, migration and invasion in glioma cells. Cancer Biol. Ther. 2012, 13, 389–400. [Google Scholar] [CrossRef] [PubMed]
- Tranque, P.A.; Calle, R.; Naftolin, F.; Robbins, R. Involvement of protein-kinase-C in the mitogenic effect of insulin-like growth factor-I on rat astrocytes. Endocrinology 1992, 131, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.C.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2013, 33, 5225–5237. [Google Scholar] [CrossRef]
- Scholze, A.R.; Foo, L.C.; Mulinyawe, S.; Barres, B.A. BMP signaling in astrocytes downregulates EGFR to modulate survival and maturation. PLoS ONE 2014, 9, e110668. [Google Scholar] [CrossRef]
- Qi, J.; Esfahani, D.R.; Huang, T.; Ozark, P.A.; Bartom, E.T.; Hashizume, R.; Bonner, E.; An, S.; Horbinski, C.; James, C.; et al. Tenascin-C expression contributes to pediatric brainstem glioma tumor phenotype and represents a novel biomarker of disease. Acta Neuropathol. Commun. 2019, 7, 75. [Google Scholar] [CrossRef]
- Soriano-Carot, M.; Quilis, I.; Bañó, C.; Igual, J.C. Protein kinase C controls activation of the DNA integrity checkpoint. Nucleic Acids Res. 2014, 42, 7084–7095. [Google Scholar] [CrossRef]
- Mani, N.; Khaibullina, A.; Krum, J.M.; Rosenstein, J.M. Astrocyte growth effects of vascular endothelial growth factor (VEGF) application to perinatal neocortical explants: Receptor mediation and signal transduction pathways. Exp. Neurol. 2005, 192, 394–406. [Google Scholar] [CrossRef]
- Argaw, A.T.; Asp, L.; Zhang, J.; Navrazhina, K.; Pham, T.; Mariani, J.N.; Mahase, S.; Dutta, D.J.; Seto, J.; Kramer, E.G.; et al. Astrocyte-derived VEGF-A drives blood-brain barrier disruption in CNS inflammatory disease. J. Clin. Investig. 2012, 122, 2454–2468. [Google Scholar] [CrossRef]
- Bethel-Brown, C.; Yao, H.; Hu, G.; Buch, S. Platelet-derived growth factor (PDGF)-BB-mediated induction of monocyte chemoattractant protein 1 in human astrocytes: Implications for HIV-associated neuroinflammation. J. Neuroinflammation 2012, 9, 262. [Google Scholar] [CrossRef] [PubMed]
- Hindinger, C.; Bergmann, C.; Hinton, D.R.; Phares, T.W.; Parra, G.I.; Hussain, S.; Savarin, C.; Atkinson, R.D.; Stohlman, S.A. IFN-γ signaling to astrocytes protects from autoimmune mediated neurological disability. PLoS ONE 2012, 7, e42088. [Google Scholar] [CrossRef]
- Douglass, J.D.; Dorfman, M.D.; Fasnacht, R.; Shaffer, L.D.; Thaler, J.P. Astrocyte IKKβ/NF-κB signaling is required for diet-induced obesity and hypothalamic inflammation. Mol. Metab. 2017, 6, 366–373. [Google Scholar] [CrossRef]
- Zhang, Y.L.; Reichel, J.M.; Han, C.; Zuniga-Hertz, J.P.; Cai, D.S. Astrocytic process plasticity and IKKβ/NF-κB in central control of blood glucose, blood pressure, and body weight. Cell Metab. 2017, 25, 1091–1102. [Google Scholar] [CrossRef]
- Funa, K.; Sasahara, M. The roles of PDGF in development and during neurogenesis in the normal and diseased nervous system. J. Neuroimmune Pharmacol. 2013, 9, 168–181. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.L.; Yan, Y.P.; Li, X.; Li, K.; Ciric, B.; Yang, J.X.; Zhang, Y.; Wu, S.; Xu, H.; Chen, W.J.; et al. Silencing IFN-γ binding/signaling in astrocytes versus microglia leads to opposite effects on central nervous system autoimmunity. J. Immunol. 2015, 194, 4251–4264. [Google Scholar] [CrossRef] [PubMed]
- Engelhardt, B. Regulation of immune cell entry into the central nervous system. Adv. Exp. Med. Biol. 2006, 43, 259–280. [Google Scholar]
- Griffin, D.E.; Hess, J.L.; Moench, T.R. Immune responses in the central nervous system. Toxicol. Pathol. 1987, 15, 294–302. [Google Scholar] [CrossRef]
- Klein, R.S.; Garber, C.; Howard, N. Infectious immunity in the central nervous system and brain function. Nat. Immunol. 2017, 18, 132–141. [Google Scholar] [CrossRef]
- Galic, M.A.; Riazi, K.; Pittman, Q.J. Cytokines and brain excitability. Front. Neuroendocr. 2011, 33, 116–125. [Google Scholar] [CrossRef]
- Rempel, J.D.; Quina, L.A.; Blakely-Gonzales, P.K.; Buchmeier, M.J.; Gruol, D.L. Viral induction of central nervous system innate immune responses. J. Virol. 2005, 79, 4369–4381. [Google Scholar] [CrossRef]
- Wraith, D.C.; Nicholson, L.B. The adaptive immune system in diseases of the central nervous system. J. Clin. Investig. 2012, 122, 1172–1179. [Google Scholar] [CrossRef] [PubMed]
- Garay, P.A.; McAllister, A.K. Novel roles for immune molecules in neural development: Implications for neurodevelopmental disorders. Front. Synaptic Neurosci. 2010, 2, 136. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Wesselingh, R.; Butzkueven, H.; Buzzard, K.; Tarlinton, D.; O’Brien, T.J.; Monif, M. Innate immunity in the central nervous system: A missing piece of the autoimmune encephalitis puzzle? Front. Immunol. 2019, 10, 2066. [Google Scholar] [CrossRef]
- Rider, P.; Carmi, Y.; Voronov, E.; Apte, R.N. Interleukin-1α. Semin. Immunol. 2013, 25, 430–438. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive astrocytes: Production, function, and therapeutic potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef]
- Wang, L.-Y.; Tu, Y.-F.; Lin, Y.-C.; Huang, C.-C. CXCL5 signaling is a shared pathway of neuroinflammation and blood-brain barrier injury contributing to white matter injury in the immature brain. J. Neuroinflammation 2016, 13, 6. [Google Scholar] [CrossRef]
- Guha, D.; Klamar, C.R.; Reinhart, T.; Ayyavoo, V. Transcriptional regulation of CXCL5 in HIV-1-infected macrophages and its functional consequences on CNS pathology. J. Interferon Cytokine Res. 2015, 35, 373–384. [Google Scholar]
- Rivest, S. Regulation of innate immune responses in the brain. Nat. Rev. Immunol. 2009, 9, 429–439. [Google Scholar] [CrossRef]
- Blatt, N.L.; Khaiboullin, T.I.; Lombardi, V.C.; Rizvanov, A.; Khaiboullina, S.F. The skin–brain connection hypothesis, bringing together CCL27-mediated T-cell activation in the skin and neural cell damage in the adult brain. Front. Immunol. 2017, 7, 544. [Google Scholar] [CrossRef] [PubMed]
- Khaibullin, Т.; Ivanova, V.; Martynova, E.; Cherepnev, G.; Khabirov, F.; Granatov, E.; Rizvanov, A.; Khaiboullina, S. Elevated levels of proinflammatory cytokines in cerebrospinal fluid of multiple sclerosis patients. Front. Immunol. 2017, 8, 531. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and related cytokines in the regulation of inflammation and immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Olmos, G.; Llado, J. Tumor necrosis factor alpha: A link between neuroinflammation and excitotoxicity. Mediat. Inflamm. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Francisco, N.M.; Hsu, N.-J.; Keeton, R.; Randall, P.; Sebesho, B.; Allie, N.; Govender, D.; Quesniaux, V.F.J.; Ryffel, B.; Kellaway, L.A.; et al. TNF-dependent regulation and activation of innate immune cells are essential for host protection against cerebral tuberculosis. J. Neuroinflammation 2015, 12, 125. [Google Scholar] [CrossRef]
- Ding, X.; Cao, F.; Cui, L.; Ciric, B.; Zhang, G.-X.; Rostami, A. IL-9 signaling affects central nervous system resident cells during inflammatory stimuli. Exp. Mol. Pathol. 2015, 99, 570–574. [Google Scholar] [CrossRef]
- Gaceb, A.; Özen, I.; Padel, T.; Barbariga, M.; Paul, G. Pericytes secrete pro-regenerative molecules in response to platelet-derived growth factor-BB. Br. J. Pharmacol. 2017, 38, 45–57. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number that Are Significant | Total Unique | BafilomycinA1 | Rapamycin | ||||
|---|---|---|---|---|---|---|---|
| 12 h | 24 h | 48 h | 12 h | 24 h | 48 h | ||
| and fold-change > 1.000 | 309 | 73 | 11 | 139 | 28 | 16 | 25 |
| and fold-change < 1.000 | 15 | 51 | 14 | 38 | 22 | 27 | |
| and fold-change > 1.150 | 256 | 40 | 76 | 15 | 11 | 28 | 25 |
| and fold-change < 0.870 | 6 | 6 | 18 | 27 | 35 | 26 | |
| and fold-change > 1.250 | 118 | 20 | 30 | 15 | 7 | 24 | 24 |
| and fold-change < 0.800 | 3 | 1 | 12 | 15 | 30 | 26 | |
| and fold-change > 1.333 | 76 | 9 | 16 | 11 | 7 | 15 | 20 |
| and fold-change < 0.750 | 1 | 0 | 4 | 8 | 19 | 23 | |
| and fold-change > 1.500 | 44 | 4 | 4 | 8 | 1 | 8 | 11 |
| and fold-change < 0.667 | 0 | 0 | 3 | 3 | 10 | 16 | |
| and fold-change > 2.000 | 14 | 0 | 0 | 2 | 0 | 0 | 1 |
| and fold-change < 0.500 | 0 | 0 | 0 | 0 | 2 | 9 | |
| EntrezGene Symbol | Protein | Bafilomycin A1 | Rapamycin | ||||
|---|---|---|---|---|---|---|---|
| 12 h | 24 h | 48 h | 12 h | 24 h | 48 h | ||
| Up-regulated by drug treatment * | |||||||
| PCSK9 | Proprotein convertase subtilisin/kexin type 9 | 1.83 | 3.15 | 2.22 | 0.92 | 0.93 | 0.25 |
| STC1 | Stanniocalcin-1 | 1.70 | 1.46 | 1.28 | 0.67 | 0.31 | 0.34 |
| MFGE8 | Lactadherin | 1.70 | 1.76 | 1.79 | 1.50 | 1.56 | 1.39 |
| TNFRSF4 | Tumor necrosis factor receptor superfamily member 4 | 1.43 | 0.75 | 1.27 | 1.01 | 0.58 | 1.03 |
| PEX5 | Peroxisomal targeting signal 1 receptor | 1.43 | 0.81 | 1.02 | 1.08 | 0.70 | 0.95 |
| APP | Amyloid beta A4 protein | 1.43 | 1.41 | 1.50 | 1.08 | 1.06 | 1.09 |
| CSF3R | Granulocyte colony-stimulating factor receptor | 1.42 | 0.82 | 1.12 | 1.04 | 0.67 | 1.03 |
| FTH1 FTL | Ferritin | 1.37 | 1.46 | 2.17 | 1.21 | 1.37 | 1.43 |
| IGFBP5 | Insulin-like growth factor-binding protein 5 | 1.37 | 1.60 | 0.99 | 1.34 | 1.00 | 0.43 |
| BMP6 | Bone morphogenetic protein 6 | 1.30 | 1.66 | 1.56 | 1.22 | 1.30 | 1.21 |
| LAMA1 LAMB1 LAMC1 | Laminin | 1.31 | 1.59 | 1.19 | 1.14 | 1.02 | 0.56 |
| APOE | Apolipoprotein E (isoform E4) | 1.12 | 1.47 | 1.42 | 1.04 | 1.14 | 1.25 |
| IL6ST | Interleukin-6 receptor subunit beta | 1.28 | 1.45 | 1.33 | 1.06 | 1.03 | 0.98 |
| CXCL11 | C-X-C motif chemokine 11 | 0.89 | 1.43 | 0.92 | 0.92 | 1.23 | 0.76 |
| LYZ | Lysozyme C | 0.98 | 1.42 | 0.32 | 0.95 | 1.12 | 0.27 |
| CST3 | Cystatin-C | 1.33 | 1.38 | 1.33 | 1.17 | 1.09 | 0.77 |
| CTLA4 | Cytotoxic T-lymphocyte protein 4 | 0.94 | 1.37 | 0.91 | 0.84 | 1.10 | 1.07 |
| THBS2 | Thrombospondin-2 | 1.21 | 1.37 | 1.08 | 0.73 | 0.65 | 0.48 |
| KLK5 | Kallikrein-5 | 0.93 | 1.37 | 0.80 | 0.97 | 1.08 | 0.65 |
| ROR1 | Tyrosine-protein kinase transmembrane receptor ROR1 | 1.03 | 1.36 | 0.96 | 1.02 | 1.32 | 1.09 |
| FGF20 | Fibroblast growth factor 20 | 1.08 | 1.34 | 1.03 | 0.98 | 1.05 | 0.92 |
| GPNMB | Transmembrane glycoprotein NMB | 1.08 | 1.18 | 1.79 | 1.16 | 1.87 | 1.82 |
| GRN | Granulins | 1.17 | 1.31 | 1.61 | 1.67 | 1.99 | 1.87 |
| IGFBP2 | Insulin-like growth factor-binding protein 2 | 1.24 | 1.27 | 1.52 | 0.89 | 0.74 | 0.54 |
| CTSA | Lysosomal protective protein | 1.02 | 0.97 | 1.47 | 1.19 | 1.22 | 1.27 |
| PSMD7 | 26S proteasome non-ATPase regulatory subunit 7 | 1.04 | 1.14 | 1.00 | 1.39 | 1.42 | 1.06 |
| AMIGO2 | Amphoterin-induced protein 2 | 1.29 | 1.01 | 1.23 | 1.39 | 1.14 | 1.96 |
| L1CAM | Neural cell adhesion molecule L1 | 1.09 | 1.32 | 1.07 | 1.37 | 1.50 | 0.79 |
| RTN4R | Reticulon-4 receptor | 1.19 | 1.27 | 1.12 | 1.35 | 1.34 | 1.01 |
| C4A C4B | Complement C4 | 0.89 | 1.48 | 0.86 | 0.98 | 1.83 | 1.33 |
| FSTL3 | Follistatin-related protein 3 | 1.01 | 1.00 | 0.94 | 1.17 | 1.78 | 1.89 |
| SERPINE1 | Plasminogen activator inhibitor 1 | 1.19 | 1.26 | 1.01 | 1.31 | 1.66 | 1.52 |
| FGF1 | Fibroblast growth factor 1 | 0.96 | 1.18 | 0.85 | 1.00 | 1.52 | 1.77 |
| LRIG3 | Leucine-rich repeats and immunoglobulin-like domains protein 3 | 1.21 | 1.20 | 1.08 | 1.17 | 1.50 | 2.26 |
| ICAM5 | Intercellular adhesion molecule 5 | 1.06 | 1.25 | 1.16 | 1.15 | 1.49 | 1.25 |
| ACY1 | Aminoacylase-1 | 0.96 | 1.02 | 0.85 | 1.23 | 1.48 | 1.56 |
| GSN | Gelsolin | 1.03 | 1.12 | 1.01 | 1.15 | 1.43 | 1.11 |
| MRC2 | C-type mannose receptor 2 | 1.01 | 1.18 | 1.11 | 1.14 | 1.38 | 1.35 |
| HTRA2 | Serine protease HTRA2, mitochondrial | 1.09 | 1.12 | 1.00 | 1.20 | 1.36 | 1.35 |
| IDUA | Alpha-L-iduronidase | 1.02 | 1.06 | 0.82 | 1.06 | 1.26 | 1.97 |
| MMP2 | 72 kDa type IV collagenase | 1.13 | 1.23 | 1.23 | 1.12 | 1.32 | 1.80 |
| EFNA2 | Ephrin-A2 | 1.14 | 1.06 | 1.03 | 1.14 | 1.22 | 1.50 |
| GAS1 | Growth arrest-specific protein 1 | 1.10 | 1.09 | 1.01 | 1.10 | 1.20 | 1.41 |
| EPHB2 | Ephrin type-B receptor 2 | 1.08 | 1.04 | 0.96 | 1.22 | 1.31 | 1.39 |
| AKT1 AKT2 AKT3 | RAC-alpha/beta/gamma serine/threonine-protein kinase | 0.94 | 1.02 | 0.92 | 1.13 | 1.24 | 1.35 |
| PROS1 | Vitamin K-dependent protein S | 0.97 | 1.19 | 0.94 | 1.09 | 1.27 | 1.34 |
| Down-regulated by drug treatment * | |||||||
| SIGLEC1 | Sialoadhesin | 0.68 | 1.56 | 1.01 | 0.87 | 1.39 | 0.92 |
| CTSB | Cathepsin B | 0.90 | 0.89 | 0.55 | 1.12 | 1.23 | 1.12 |
| CTSH | Cathepsin H | 0.92 | 0.84 | 0.58 | 1.08 | 1.22 | 1.51 |
| HIST1H1C | Histone H1.2 | 1.02 | 1.01 | 0.61 | 1.05 | 0.96 | 0.62 |
| CKB | Creatine kinase B-type | 0.96 | 0.91 | 0.73 | 0.93 | 0.78 | 0.83 |
| NACA | Nascent polypeptide-associated complex subunit alpha | 1.02 | 0.98 | 0.75 | 0.54 | 0.62 | 0.44 |
| CDC2 CCNB1 | Cyclin-dependent kinase 1:G2/mitotic-specific cyclin-B1 complex | 0.92 | 0.94 | 0.81 | 0.65 | 0.60 | 1.05 |
| PFDN5 | Prefoldin subunit 5 | 0.91 | 0.97 | 0.93 | 0.65 | 0.68 | 1.02 |
| SPHK1 | Sphingosine kinase 1 | 0.94 | 0.98 | 0.85 | 0.70 | 0.75 | 0.77 |
| EIF4G2 | Eukaryotic translation initiation factor 4 gamma 2 | 0.94 | 1.04 | 0.92 | 0.71 | 0.71 | 0.87 |
| DNAJB1 | DnaJ homolog subfamily B member 1 | 1.07 | 1.04 | 0.79 | 0.71 | 0.62 | 0.66 |
| TNC | Tenascin | 1.30 | 1.21 | 0.95 | 0.72 | 0.70 | 1.01 |
| PGAM1 | Phosphoglycerate mutase 1 | 0.95 | 0.89 | 0.84 | 0.77 | 0.40 | 0.36 |
| CDK2 CCNA2 | Cyclin-dependent kinase 2:Cyclin-A2 complex | 0.92 | 0.83 | 0.98 | 0.68 | 0.57 | 1.79 |
| HK2 | Hexokinase-2 | 1.04 | 1.04 | 0.81 | 0.76 | 0.61 | 0.64 |
| KPNA2 | Importin subunit alpha-1 | 1.00 | 0.95 | 0.80 | 0.85 | 0.66 | 0.87 |
| NRP1 | Neuropilin-1 | 1.13 | 1.13 | 0.86 | 0.88 | 0.68 | 0.68 |
| TFPI | Tissue factor pathway inhibitor | 1.11 | 1.11 | 0.93 | 0.88 | 0.72 | 0.77 |
| HSPA1A | Heat shock 70 kDa protein 1A | 1.14 | 1.19 | 0.98 | 0.80 | 0.73 | 0.77 |
| CFI | Complement factor I | 1.08 | 1.11 | 0.86 | 0.78 | 0.73 | 0.76 |
| TGM3 | Protein-glutamine gamma-glutamyltransferase E | 0.85 | 1.33 | 0.50 | 0.83 | 1.05 | 0.27 |
| KLK7 | Kallikrein-7 | 0.97 | 1.18 | 0.37 | 0.99 | 0.86 | 0.29 |
| SERPINA3 | Alpha-1-antichymotrypsin | 0.98 | 1.31 | 0.84 | 1.05 | 1.23 | 0.61 |
| ANG | Angiogenin | 1.09 | 1.29 | 0.92 | 1.13 | 0.97 | 0.65 |
| MICB | MHC class I polypeptide-related sequence B | 1.11 | 1.14 | 0.94 | 0.96 | 0.81 | 0.67 |
| TNFRSF1A | Tumor necrosis factor receptor superfamily member 1A | 1.22 | 1.26 | 1.01 | 1.04 | 0.90 | 0.67 |
| PES1 | Pescadillo homolog | 1.02 | 0.99 | 0.69 | 0.79 | 0.75 | 0.69 |
| GFRA1 | GDNF family receptor alpha-1 | 1.12 | 1.03 | 0.90 | 0.93 | 0.75 | 0.70 |
| RPS3 | 40S ribosomal protein S3 | 0.98 | 1.04 | 0.98 | 1.02 | 0.81 | 0.72 |
| PRKCI | Protein kinase C iota type | 1.00 | 1.08 | 0.88 | 0.86 | 0.82 | 0.73 |
| BafilomycinA1/Mock | Rapamycin/Mock | |||||||
|---|---|---|---|---|---|---|---|---|
| 16 h | 40 h | 16 h | 40 h | |||||
| Baf/Untr * | p-Value | Baf/Untr | p-Value | Rap/Untr | p-Value | Rap/Untr | p-Value | |
| Up-Regulated Proteins | ||||||||
| IL-1a | 738 | 0.008 | 9714 | 0.001 | 0.40 | 0.071 | 1.47 | 0.113 |
| TPO | 7.72 | 1.8∙10−4 | 5.34 | 0.014 | 3.85 | 0.001 | 9.03 | 4.6∙10−4 |
| Eotaxin | 7.25 | 0.022 | 2.50 | 0.039 | 4.89 | 2.4∙10−4 | 2.95 | 0.002 |
| MIP-1a | 5.54 | 0.026 | 2.78 | 0.147 | 5.40 | 0.005 | 4.50 | 0.045 |
| MCP-4 | 5.39 | 0.002 | 5.62 | 0.001 | 5.30 | 0.002 | 5.90 | 0.001 |
| IL-12P70 | 4.34 | 0.003 | 11.35 | 0.036 | 7.56 | 0.078 | 8.92 | 0.085 |
| MCP-3 | 4.18 | 0.001 | 7.29 | 7.6∙10−5 | 3.00 | 0.001 | 3.60 | 0.007 |
| IFNa2 | 4.05 | 0.030 | 1.38 | 0.480 | 2.60 | 0.083 | 3.49 | 0.027 |
| Fractalkine | 3.36 | 0.001 | 0.88 | 0.155 | 1.98 | 0.005 | 2.36 | 0.013 |
| IL-1RA | 3.18 | 0.017 | 2.22 | 0.090 | 2.79 | 0.012 | 4.86 | 0.260 |
| IL-13 | 2.95 | 0.011 | 1.16 | 0.465 | 1.94 | 0.075 | 3.78 | 0.003 |
| IL-8 | 2.36 | 0.008 | 7.21 | 0.019 | 0.81 | 0.0503 | 1.89 | 0.001 |
| IL-5 | 2.32 | 0.003 | 1.80 | 0.009 | 1.78 | 0.010 | 3.12 | 0.027 |
| MCP-2 | 2.31 | 4.3∙10−4 | 3.99 | 0.001 | 2.83 | 0.026 | 5.03 | 0.001 |
| BCA-1 | 2.27 | 0.010 | 1.04 | 0.447 | 2.26 | 0.001 | 2.51 | 9.9∙10−5 |
| MCP-1 | 2.24 | 0.023 | 1.70 | 0.039 | 1.45 | 0.029 | 3.20 | 0.001 |
| IL-3 | 2.22 | 0.037 | 1.19 | 0.119 | 0.96 | 0.845 | 2.69 | 0.340 |
| IL-6 | 2.19 | 0.001 | 1.60 | 0.012 | 1.71 | 0.027 | 3.17 | 0.005 |
| GRO alpha | 2.03 | 0.002 | 6.70 | 1.6∙10−4 | 1.17 | 0.080 | 1.85 | 0.004 |
| IL−9 | 44.31 | 0.195 | 47.07 | 8.9∙10−5 | 65.08 | 0.018 | 23.63 | 0.006 |
| CTACK | 30.10 | 0.102 | 45.12 | 0.002 | 31.66 | 0.180 | 21.95 | 0.303 |
| TNFa | 1.76 | 0.005 | 12.10 | 0.001 | 0.69 | 0.029 | 2.80 | 0.003 |
| IL-1B | 4.56 | 0.203 | 6.36 | 0.043 | 1.69 | 0.137 | 5.01 | 0.024 |
| G-CSF | 1.53 | 0.038 | 5.48 | 5.9∙10−5 | 5.13 | 0.001 | 3.66 | 0.003 |
| IL-4 | 1.73 | 0.066 | 5.43 | 0.001 | 2.39 | 0.052 | 1.66 | 0.422 |
| Eotaxin-2 | 1.30 | 0.026 | 3.96 | 0.002 | 0.97 | 0.561 | 1.83 | 0.004 |
| ENA-78 | 2.88 | 0.180 | 2.94 | 0.020 | 1.38 | 0.605 | 6.15 | 0.004 |
| IL-2 | 3.39 | 0.058 | 2.78 | 0.005 | 2.29 | 0.089 | 2.79 | 0.035 |
| IP-10 | 0.73 | 0.0497 | 2.16 | 0.009 | 0.69 | 0.033 | 1.84 | 0.006 |
| TNFB | 2.66 | 0.180 | 1.88 | 0.190 | 2.16 | 0.334 | 7.80 | 0.037 |
| PDGF-AA | 1.96 | 0.008 | 1.50 | 0.015 | 1.42 | 0.021 | 3.17 | 0.006 |
| EGF | 1.74 | 0.005 | 1.30 | 0.023 | 0.92 | 0.238 | 2.79 | 0.005 |
| IL-10 | 1.56 | 0.118 | 1.37 | 0.594 | 0.88 | 0.742 | 2.68 | 0.038 |
| IL-28A | 2.19 | 0.093 | 1.97 | 0.316 | 2.04 | 0.064 | 2.68 | 0.017 |
| IL-12P40 | 1.31 | 0.061 | 2.49 | 0.113 | 1.67 | 0.127 | 2.33 | 0.043 |
| IL-23 | 1.95 | 0.297 | 0.78 | 0.309 | 1.20 | 0.747 | 2.30 | 0.012 |
| FGF-2 | 0.52 | 2.2∙10−6 | 1.03 | 0.006 | 0.93 | 0.003 | 2.26 | 4.9∙10−4 |
| IL-7 | 1.00 | 0.945 | 1.23 | 0.026 | 0.62 | 0.011 | 2.26 | 0.012 |
| Flt-3L | 1.00 | 0.958 | 1.33 | 0.017 | 0.73 | 0.011 | 2.22 | 4.0∙10−4 |
| Down-Regulated Proteins | ||||||||
| LIF | 0.04 | 0.001 | 0.38 | 0.002 | 0.05 | 0.001 | 0.13 | 0.001 |
| PDGF-BB | 0.04 | 1.2∙10−4 | 0.33 | 3.0∙10−4 | 0.04 | 1.2∙10−4 | 1.23 | 0.003 |
| TGF-a | 0.16 | 0.004 | 0.46 | 0.010 | 0.17 | 0.003 | 1.42 | 0.025 |
| GM-CSF | 0.20 | 2.6∙10−4 | 6.34 | 0.005 | 0.07 | 2.0∙10−4 | 2.45 | 0.006 |
| IL-18 | 0.30 | 0.001 | 0.39 | 3.8∙10−4 | 0.42 | 0.001 | 4.48 | 4.0∙10−4 |
| Eotaxin-3 | 0.35 | 0.033 | 0.76 | 0.197 | 0.26 | 0.025 | 1.86 | 0.020 |
| SDF-1a+B | 0.61 | 0.008 | 0.27 | 0.002 | 0.44 | 0.005 | 0.76 | 0.018 |
| SCF | 0.77 | 0.054 | 1.09 | 0.324 | 0.46 | 0.011 | 1.55 | 0.053 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sher, A.A.; Gao, A.; Coombs, K.M. Autophagy Modulators Profoundly Alter the Astrocyte Cellular Proteome. Cells 2020, 9, 805. https://doi.org/10.3390/cells9040805
Sher AA, Gao A, Coombs KM. Autophagy Modulators Profoundly Alter the Astrocyte Cellular Proteome. Cells. 2020; 9(4):805. https://doi.org/10.3390/cells9040805
Chicago/Turabian StyleSher, Affan Ali, Ang Gao, and Kevin M. Coombs. 2020. "Autophagy Modulators Profoundly Alter the Astrocyte Cellular Proteome" Cells 9, no. 4: 805. https://doi.org/10.3390/cells9040805
APA StyleSher, A. A., Gao, A., & Coombs, K. M. (2020). Autophagy Modulators Profoundly Alter the Astrocyte Cellular Proteome. Cells, 9(4), 805. https://doi.org/10.3390/cells9040805