The P2X7 Receptor and NLRP3 Axis in Non-Alcoholic Fatty Liver Disease: A Brief Review

 , , and
, , and
Abstract
1. Introduction
2. Extracellular Adenosine Tri-Phosphate (ATP) as a DAMP
3. ATP as an Extracellular Mediator of Inflammation
4. Ectonucleotidases
5. The NLRP3 Inflammasome
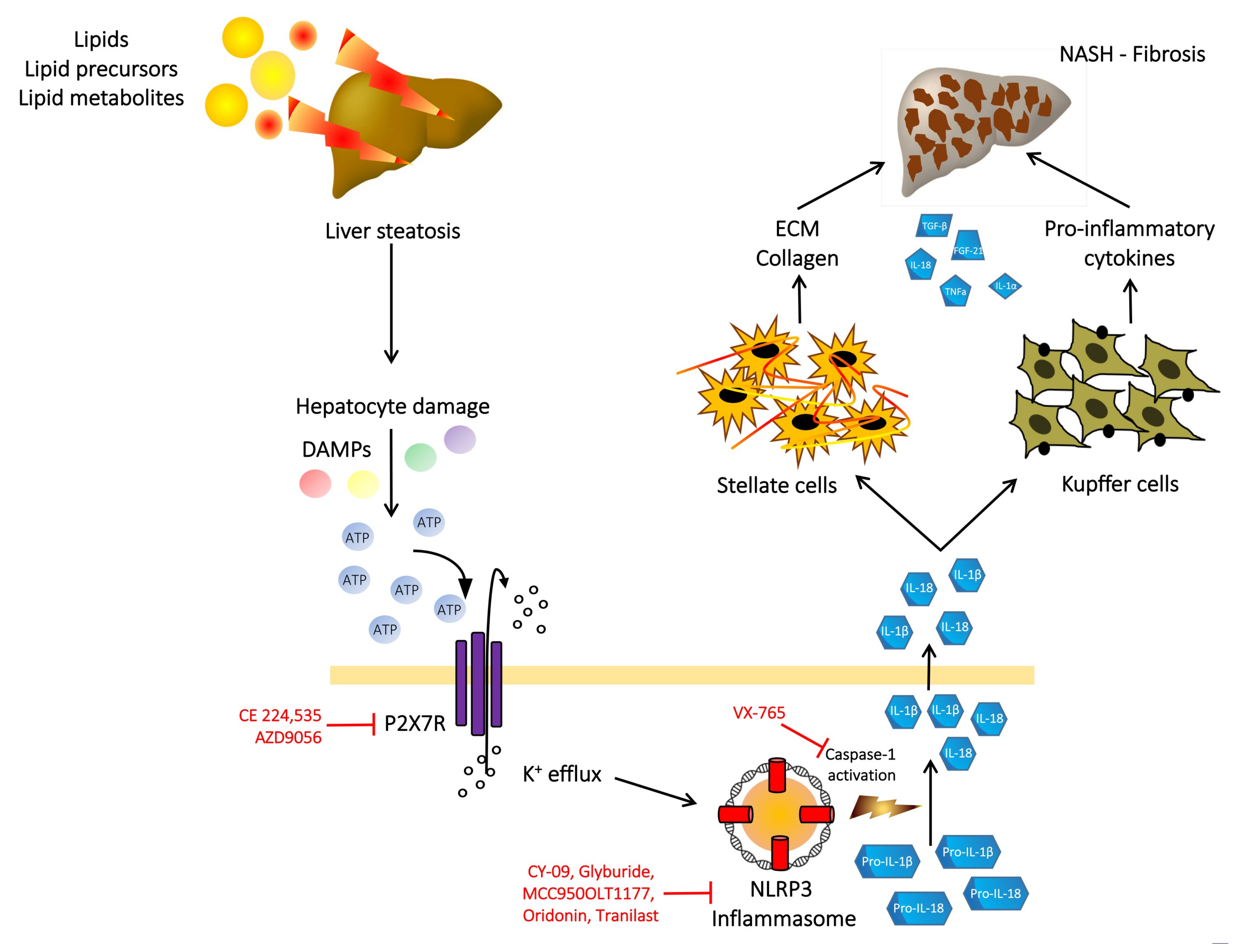
6. P2X7 Receptor, NLRP3 Inflammasome, NAFLD and Liver Fibrosis
7. P2X7 Receptor as a Therapeutic Target in NASH and Liver Fibrosis
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Stefan, N.; Haring, H.U.; Cusi, K. Non-alcoholic fatty liver disease: Causes, diagnosis, cardiometabolic consequences, and treatment strategies. Lancet Diabetes Endocrinol. 2019, 7, 313–324. [Google Scholar] [CrossRef]
- Angulo, P.; Machado, M.V.; Diehl, A.M. Fibrosis in nonalcoholic fatty liver disease: Mechanisms and clinical implications. Semin. Liver Dis. 2015, 35, 132–145. [Google Scholar] [CrossRef] [PubMed]
- Wong, C.R.; Nguyen, M.H.; Lim, J.K. Hepatocellular carcinoma in patients with nonalcoholic fatty liver disease. World J. Gasroenterol. 2016, 22, 8294–8303. [Google Scholar] [CrossRef] [PubMed]
- Diel, A.M.; Day, C. Cause, pathogenesis, and treatment of nonalcoholic steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Rafiq, N.; Makhlouf, H.; Younoszai, Z.; Agrawal, R.; Goodman, Z. Pathologic criteria for nonalcoholic steatohepatitis: Interprotocol agreement and ability to predict liver-related mortality. Hepatology 2011, 53, 1874–1882. [Google Scholar] [CrossRef]
- Heymann, F.; Tacke, F. Immunology in the liver—From homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef]
- Mehal, W.Z. The inflammasome in liver injury and non-alcoholic fatty liver disease. Dig. Dis. 2014, 32, 507–515. [Google Scholar] [CrossRef]
- Di Virgilio, F. Liaisons dangereuses: P2X(7) and the inflammasome. Trends Pharmacol. Sci. 2007, 28, 465–472. [Google Scholar] [CrossRef]
- Meylan, E.; Tschopp, J.; Karin, M. Intracellular pattern recognition receptors in the host response. Nature 2006, 442, 39–44. [Google Scholar] [CrossRef]
- Di Virgilio, F. Purinergic mechanism in the immune system: A signal of danger for dendritic cells. Purinergic Signal. 2005, 1, 205–209. [Google Scholar] [CrossRef]
- Burnstock, G. Purinergic nerves. Pharmacol. Rev. 1972, 24, 509–581. [Google Scholar] [PubMed]
- Di Virgilio, F.; Dal Ben, D.; Sarti, A.C.; Giuliani, A.L.; Falzoni, S. The P2X7 receptor in infection and inflammation. Immunity 2017, 47, 15–31. [Google Scholar] [CrossRef]
- Abudara, V.; Retamal, M.A.; Del Rio, M.; Orellana, J.A. Synaptic functions of hemichannels and pannexins: A double-edged sword. Front. Mol. Neurosci. 2018, 11, 435. [Google Scholar] [CrossRef]
- Bours, M.J.L.; Dagnelie, P.C.; Giuliani, A.L.; Wesselius, A.; Di Virgilio, F. P2 receptors and extracellular ATP: A novel homeostatic pathway in inflammation. Front. Biosci. 2011, 3, 1443–1456. [Google Scholar]
- Ralevic, V.; Burnstock, G. Receptors for purines and pyrimidines. Pharmacol. Rev. 1998, 50, 413–492. [Google Scholar] [PubMed]
- Abbracchio, M.P.; Burnstock, G. Purinoreceptors: Are there families of P2X and P2Y purinoceptors? Pharmacol. Ther. 1994, 64, 445–475. [Google Scholar] [CrossRef]
- Gicquel, T.; Le Dare, B.; Boichot, E.; Lagente, V. Purinergic receptors: New targets for the treatment of gout and fibrosis. Fundam. Clin. Pharmacol. 2017, 31, 136–146. [Google Scholar] [CrossRef]
- Adinolfi, E.; Giuliani, A.L.; De Marchi, E.; Pegoraro, A.; Orioli, E.; Di Virgilio, F. The P2X7 receptor: A main player in inflammation. Biochem. Pharmacol. 2018, 151, 234–244. [Google Scholar] [CrossRef]
- Von Kugelgen, I.; Harden, T.K. Molecular pharmacology of, physiology, and structure of the P2Y receptors. Adv. Pharmacol. 2011, 61, 373–415. [Google Scholar]
- Falzoni, S.; Munerati, M.; Ferrari, D.; Spisani, S.; Moretti, S.; Di Virgilio, F. The purinergi P2Z receptor of human macrophage cells. Characterization and possible physiological role. J. Clin. Investig. 1995, 95, 1207–1216. [Google Scholar] [CrossRef]
- Ferrari, D.; Pizzirani, C.; Adinolfi, E.; Lemoli, R.M.; Curti, A.; Idzko, M.; Panther, E.; Di Virgilio, F. The P2X7 receptor: A key player in IL-1 processing and release. J. Immunol. 2006, 176, 3877–3883. [Google Scholar] [CrossRef]
- Vuerich, M.; Robson, S.C.; Longhi, M.S. Ectonucleotidases in intestinal and hepatic inflammation. Front. Immunol. 2019, 10, 507–517. [Google Scholar] [CrossRef] [PubMed]
- Enjyoji, K.; Kotani, K.; Thukral, C.; Blumel, B.; Sun, X.; Wu, Y.; Imai, M.; Friedman, D.; Csizmadia, E.; Bleibel, W.; et al. Deletion of cd39/entpd1 results in hepatic insulin resistance. Diabetes 2008, 57, 2311–2320. [Google Scholar] [CrossRef] [PubMed]
- Fausther, M.; Sheung, N.; Saiman, Y.; Bansal, M.B.; Dranoff, J.A. Activated hepatic stellate cells upregulate transcription of ecto-5-nucleotidase/CD73 via specific SP1 and SMAD promoter elements. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G904–G914. [Google Scholar] [CrossRef]
- Feldbrugge, L.; Jiang, Z.G.; Csizmadia, E.; Mitsuhashi, S.; Tran, S.; Yee, E.U.; Rothweiler, S.; Vaid, K.A.; Sevigny, J.; Schmelzle, M.; et al. Distinct roles of ectonucleoside triphosphate diphosphohydrolase-2/NTPDase2) in liver regeneration and fibrosis. Purinergic Signal. 2018, 14, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Read, R.; Hansen, G.; Kramer, J.; Finch, R.; Li, L.; Vogel, P. Ectonucleoside triphosphate diphosphohydrolase type 5 (Entpd5)-deficient mice develop progressive hepatopathy, hepatocellular tumors, and spermatogenic arrest. Vet. Pathol. 2009, 46, 491–504. [Google Scholar] [CrossRef]
- Szabo, G.; Csak, T. Inflammasome in liver disease. J. Hepatol. 2012, 57, 642–654. [Google Scholar] [CrossRef]
- Negash, A.A.; Gale, M., Jr. Hepatitis regulation by the inflammasome pathway. Immunol. Rev. 2015, 265, 143–155. [Google Scholar] [CrossRef]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef]
- Dinarello, C.A. Immunologic and inflammatory functions of the interleukin-1 family. Ann. Rev. Immunol. 2009, 27, 519–550. [Google Scholar] [CrossRef]
- Ouyang, X.; Ghani, A.; Mehal, W.Z. Inflammasome biology in fibrogenesis. Biochim. Biophys. Acta 2013, 1832, 979–988. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.; Ruegg, A.; Werner, S.; Beer, H.D. Active caspase-1 is a regulator of unconventional protein secretion. Cell 2008, 132, 818–831. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Bronte, V.; Collavo, D.; Zanovello, P. Responses of mouse lymphocytes to extracellular adenosine 5′-triphosphate (ATP). Lymphocytes with cytotoxic activity are resistant to the permeabilizing effects of ATP. J. Immunol. 1989, 143, 1955–1960. [Google Scholar] [PubMed]
- Ganz, M.; Csak, T.; Nath, B.; Szabo, G. Lipopolysaccharide induces and activates the Nalp3 inflammasome in the liver. World J. Gastroenterol. 2011, 17, 4772–4778. [Google Scholar] [CrossRef] [PubMed]
- Hendrikx, T.; Bieghs, V.; Walenbergh, S.M.; van Gorp, P.J.; Verheyen, F.; Jeurissen, M.L.; Steinbush, M.M.; Vaes, N.; Binder, C.J.; Koek, G.H.; et al. Macrophage specific caspase-1/11 deficiency protects against cholesterol crystallization and hepatic inflammation in hyperlipemic mice. PLoS ONE 2013, 8, e78792. [Google Scholar] [CrossRef]
- Dixon, I.J.; Flask, C.A.; Papouchado, B.G.; Feldstein, A.E.; Nagy, L.E. Caspase-1 as a central regulator of high fat diet-induced non-alcoholic steatohepatitis. PLoS ONE 2013, 8, e56100. [Google Scholar] [CrossRef] [PubMed]
- Kamari, Y.; Shaish, A.; Vax, E.; Shemesh, S.; Kandel-Kfir, M.; Arbel, Y.; Olteanu, S.; Barshack, I.; Dotan, S.; Voronov, E.; et al. Lack of interleukin-1a or interleukin-1β inhibits transformation of steatosis to steatohepatitis and liver fibrosis in hypercholesterolemic mice. J. Hepatol. 2011, 55, 1086–1094. [Google Scholar] [CrossRef]
- Velazquez-Miranda, E.; Diaz-Munoz, M.; Vasquez-Cuevas, F.G. Purinergic signaling in hepatic disease. Purinergic Signal. 2019, 15, 477–489. [Google Scholar] [CrossRef]
- Rock, K.L.; Latz, E.; Ontiveros, F.; Kono, H. The sterile inflammatory response. Ann. Rev. Immunol. 2010, 28, 321–342. [Google Scholar] [CrossRef]
- Kahlenberg, J.M.; Dubyak, G.R. Mechanisms of caspase-1 activation by P2X7 receptor-mediated K+ release. Am. J. Physiol. Cell Physiol. 2004, 286, C1100–C1108. [Google Scholar] [CrossRef]
- Chatterjee, S.; Das, S. P2X7 receptor as a key player in oxidative stress-driven cell fate in non-alcoholic steatohepatitis. Oxidative Med. Cell. Longev. 2015, 172493, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F. Targeting hepatic macrophages to treat liver disease. J. Hepatol. 2017, 66, 1300–1312. [Google Scholar] [CrossRef]
- Postlewhite, A.E.; Raghow, R.; Stricklin, G.P.; Poppleton, H.; Seyer, J.M.; Kang, A.H. Modulation of fibroblast functions by interleukin 1: Increased steady-state accumulation of type 1 procollagen messenger RNAs and stimulation of other functions but not chemotaxis by human recombinant interleukin 1 alpha and beta. J. Cell Biol. 1998, 106, 311–318. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Friedman, S.L. Liver fibrogenesis and the role of hepatic stellate cells: New insights and prospects for therapy. J. Gastroenterol. Hepatol. 1999, 14, 618–633. [Google Scholar] [CrossRef] [PubMed]
- Gentile, D.; Natale, M.; Lazzerini, P.E.; Capecchi, P.L.; Laghi-Pasini, F. The role of P2X7 receptors in tissue fibrosis: A brief review. Purinergic Signal. 2015, 11, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Liver fibrosis—From bench to bedside. J. Hepatol. 2003, 38 (Suppl. 1), S38–S53. [Google Scholar] [CrossRef]
- Huang, C.; Yu, W.; Cui, H.; Wang, Y.; Zhang, L.; Ham, F.; Huang, T. P2X7 blockade attenuates mouse liver fibrosis. Mol. Med. Rep. 2014, 9, 57–62. [Google Scholar] [CrossRef]
- Tung, H.-C.; Lee, F.-Y.; Wang, S.-S.; Tsai, M.H.; Lee, J.Y.; Huo, T.I.; Huang, H.C.; Chuang, C.L.; Lin, H.C.; Lee, S.D. The beneficial effects of P2X7 antagonism in rats with bile duct ligation-induced cirrhosis. PLoS ONE 2015, 10, e0124654. [Google Scholar] [CrossRef]
- Das, S.; Seth, R.K.; Kumar, A.; Kadiiska, M.B.; Michelotti, G.; Diehl, A.M.; Chatterjee, S. Purinergic receptor X7 is a key modulator of metabolic oxidative stress-mediated autophagy and inflammation in experimental nonalcoholic steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2013, 305, G950–G963. [Google Scholar] [CrossRef]
- Hoque, R.; Sohail, M.A.; Salhanick, S.; Malik, A.F.; Ghani, A.; Robson, S.C.; Mehal, W.Z. P2X7 receptor-mediated purinergic signaling promotes liver injury in acetaminophen hepatotoxicity in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G1171–G1179. [Google Scholar] [CrossRef]
- Wree, A.; McGeough, M.D.; Pena, C.A.; Schlattjan, M.; Li, H.; Inzaugarat, M.E.; Messer, K.; Canbay, A.; Hoffman, H.M.; Feldstein, A.E. NLRP3 inflammasome activation is required for fibrosis development in NAFLD. J. Mol. Med. 2014, 92, 1069–1082. [Google Scholar] [CrossRef] [PubMed]
- Mridha, A.R.; Wree, A.; Robertson, A.A.B.; Yeh, M.M.; Johnson, C.D.; Van Rooyen, D.M.; Haczeyni, F.; Teoh, N.C.; Savard, C.; Ioannou, G.N.; et al. NLRP3 inflammasome blockade reduces liver inflammation and fibrosis in experimental NASH in mice. J. Hepatol. 2017, 66, 1037–1046. [Google Scholar] [CrossRef] [PubMed]
- Blasetti Fantauzzi, C.; Menini, S.; Iacobimi, C.; Rossi, C.; Santini, E.; Solini, A.; Pugliese, G. Deficiency of the purinergic receptor 2X7 attenuates nonalcoholic steatohepatitis induced by high-fat diet: Possible role of the NLRP3 inflammasome. Oxidative Med. Cell. Longev. 2017, 2017, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Hepatic stellate cells: Protean, multifunctional, and enigmatic cells of the liver. Physiol. Rev. 2008, 88, 125–172. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Zhang, Y.; Zheng, J.H.; Li, X.; Yao, Y.L.; Wu, Y.L.; Song, S.Z.; Sun, P.; Nan, J.X.; Lian, L.H. Potentiation of hepatic stellate cell activation by extracellular ATP is dependent on P2X7R-mediated NLRP3 inflammasome activation. Pharmacol. Res. 2017, 117, 82–93. [Google Scholar] [CrossRef]
- Chandrashekaran, V.; Das, S.; Seth, R.K.; Dattaroy, D.; Alhasson, F.; Michelotti, G.; Nagarkatti, M.; Nagarkatti, P.; Diehl, A.M.; Chatterjee, S. Purinergic receptor X7 mediates leptin induced GLUT4 function in stellate cells in nonalcoholic steatohepatitis. Biochim. Biophys. Acta 2016, 1862, 32–45. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10. [Google Scholar] [CrossRef]
- Zahid, A.; Bofeng, L.; Kombe Kombe, A.J.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef]
- Shao, B.Z.; Xu, Z.Q.; Han, B.Z.; Su, D.F.; Liu, C. NLRP3 inflammasome and its inhibitors: A review. Front. Pharmacol. 2015, 6, 262. [Google Scholar] [CrossRef]
- Fuller, S.J.; Stokes, L.; Skarratt, K.K.; Gu, B.J.; Wiley, J.S. Genetics of the P2X7 receptor and human disease. Purinergic Signal. 2009, 5, 257–262. [Google Scholar] [CrossRef]
- Karasawa, A.; Kawate, T. Structural basis for subtype-specific inhibition of the P2X7 receptor. eLife 2016, 5, e22153. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Purinergic Receptor | Endogenous Agonist | Expression in Normal Liver | |
|---|---|---|---|
| P1 receptors | A1 | Adenosine | Yes |
| A2a | Adenosine | Yes | |
| A2b | Adenosine | Yes | |
| A3 | Adenosine | Yes | |
| P2X receptors | P2X1 | ATP | Yes |
| P2X2 | ATP | Yes | |
| P2X3 | ATP | Yes | |
| P2X4 | ATP | Yes | |
| P2X5 | ATP | No | |
| P2X6 | ATP | Yes | |
| P2X7 | ATP | Yes | |
| P2Y receptors | P2Y1 | ADP | Low |
| P2Y2 | ATP, UTP | Yes | |
| P2Y4 | ATP, UTP | Yes | |
| P2Y6 | UDP | Yes | |
| P2Y11 | ATP | Yes | |
| P2Y12 | ADP | Low | |
| P2Y13 | ADP | Low | |
| P2Y14 | UDP | Low |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossato, M.; Di Vincenzo, A.; Pagano, C.; Hadi, H.E.; Vettor, R. The P2X7 Receptor and NLRP3 Axis in Non-Alcoholic Fatty Liver Disease: A Brief Review. Cells 2020, 9, 1047. https://doi.org/10.3390/cells9041047
Rossato M, Di Vincenzo A, Pagano C, Hadi HE, Vettor R. The P2X7 Receptor and NLRP3 Axis in Non-Alcoholic Fatty Liver Disease: A Brief Review. Cells. 2020; 9(4):1047. https://doi.org/10.3390/cells9041047
Chicago/Turabian StyleRossato, Marco, Angelo Di Vincenzo, Claudio Pagano, Hamza El Hadi, and Roberto Vettor. 2020. "The P2X7 Receptor and NLRP3 Axis in Non-Alcoholic Fatty Liver Disease: A Brief Review" Cells 9, no. 4: 1047. https://doi.org/10.3390/cells9041047
APA StyleRossato, M., Di Vincenzo, A., Pagano, C., Hadi, H. E., & Vettor, R. (2020). The P2X7 Receptor and NLRP3 Axis in Non-Alcoholic Fatty Liver Disease: A Brief Review. Cells, 9(4), 1047. https://doi.org/10.3390/cells9041047