E-Cadherin in Pancreatic Ductal Adenocarcinoma: A Multifaceted Actor during EMT



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
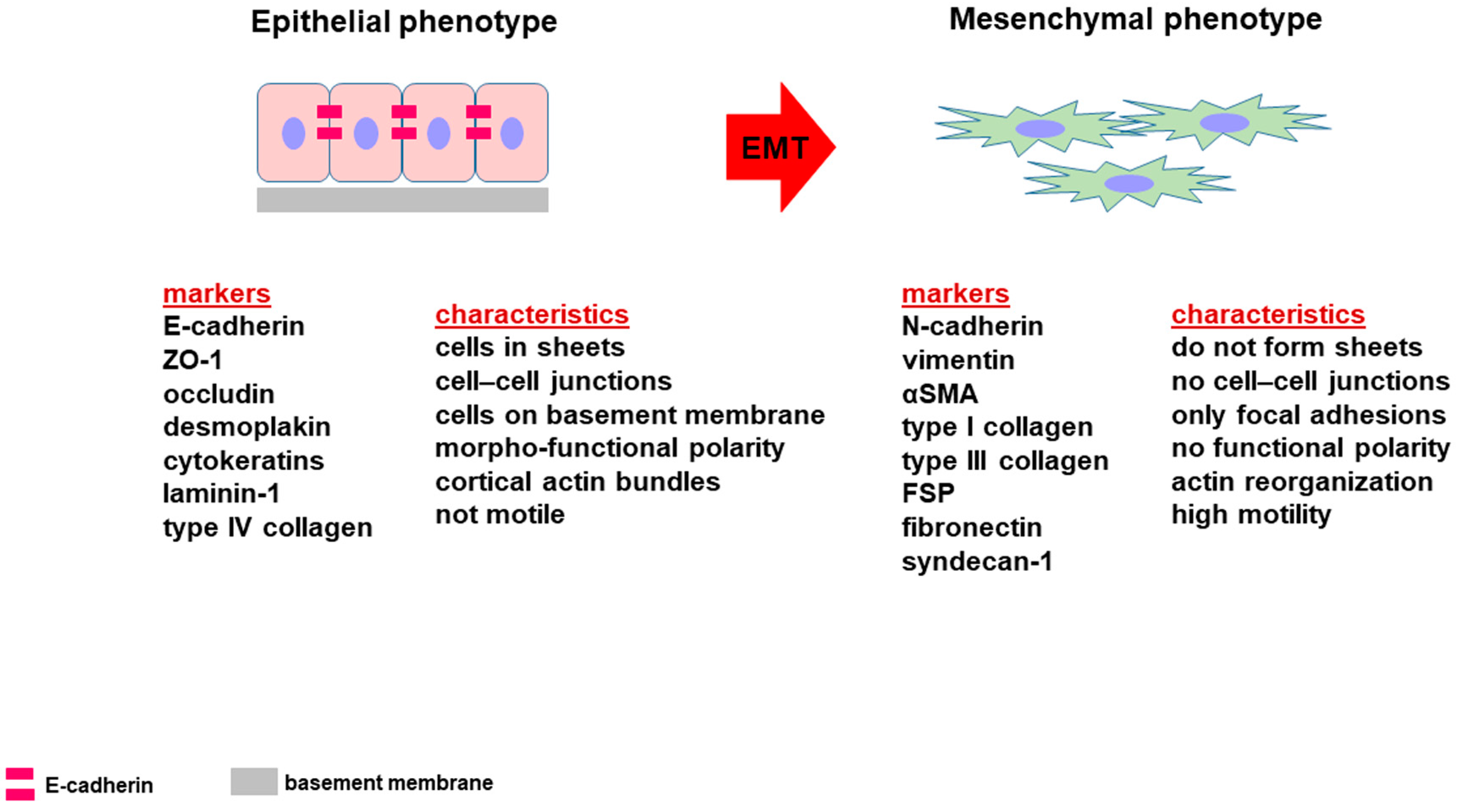
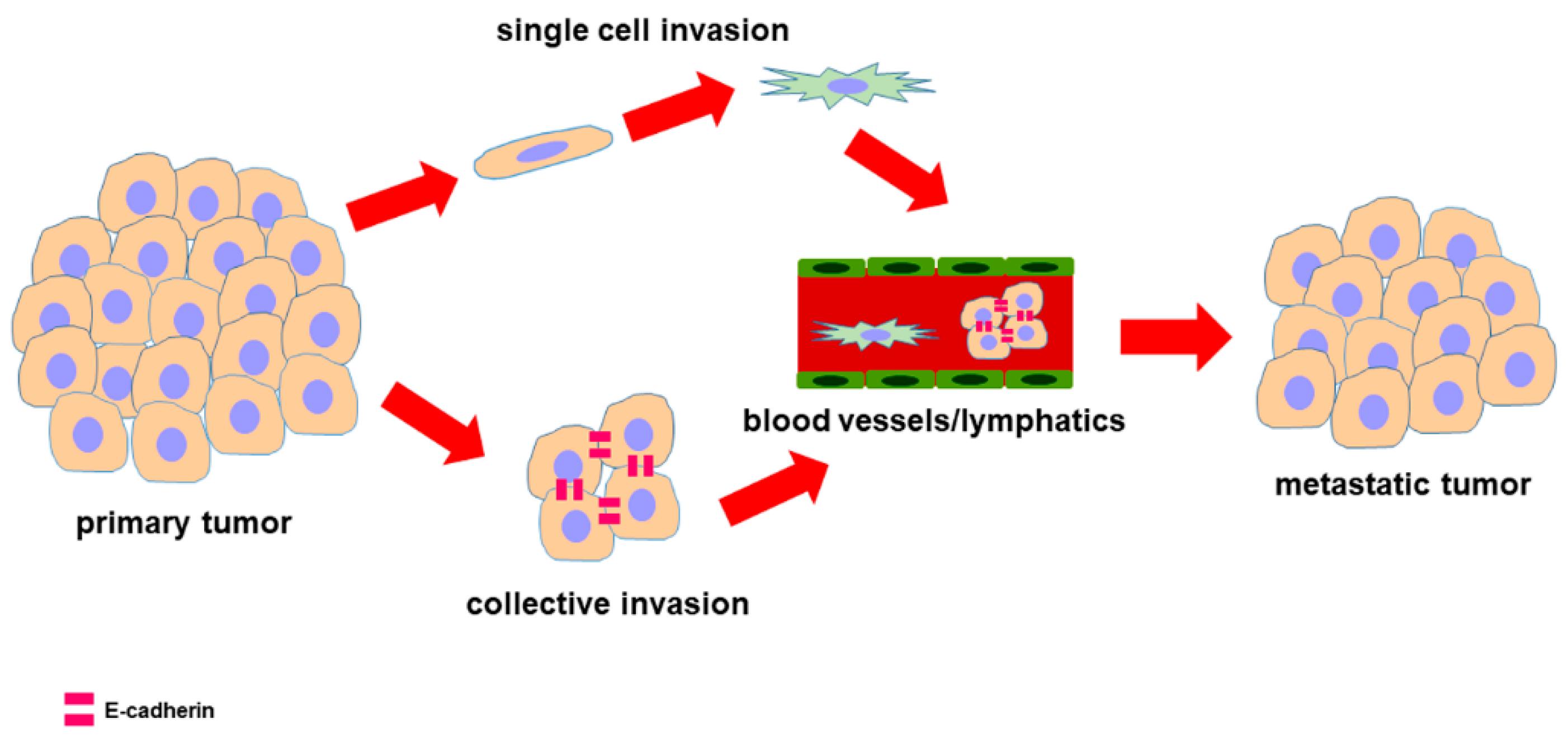
2. Epithelial-to-Mesenchymal Transition
3. E-Cadherin and EMT
4. PDAC and E-Cad Expression
5. Effect of 3D Arrangement on E-Cadherin Expression
6. Effect of ECM on E-Cadherin Expression
7. EMT-Related Phenotypes and Hybrid Phenotypes
8. E-Cadherin and the Immune System
9. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Nieto, M.A.; Huang, R.Y.; Jackson, R.A.; Thiery, J.P. EMT: 2016. Cell 2016, 166, 21–45. [Google Scholar] [CrossRef]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Xie, K.; Wolff, R.; Abbruzzese, J.L. Pancreatic cancer. Lancet 2004, 363, 1049–1057. [Google Scholar] [CrossRef]
- GBD 2017 Pancreatic Cancer Collaborators. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947. [Google Scholar] [CrossRef]
- Ghaneh, P.; Costello, E.; Neoptolemos, J.P. Biology and management of pancreatic cancer. Gut 2007, 56, 1134–1152. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Sleeman, J.P. Complex networks orchestrate epithelial-mesenchymal transitions. Nat. Rev. Mol. Cell Biol. 2006, 7, 131–142. [Google Scholar] [CrossRef]
- Thiery, J.P. Epithelial-mesenchymal transitions in tumour progression. Nat. Rev. Cancer 2002, 2, 442–454. [Google Scholar] [CrossRef]
- Yang, J.; Weinberg, R.A. Epithelial-mesenchymal transition: At the crossroads of development and tumor metastasis. Dev. Cell 2008, 14, 818–829. [Google Scholar] [CrossRef]
- Zeisberg, M.; Neilson, E.G. Biomarkers for Epithelial-Mesenchymal Transitions. J. Clin. Investig. 2009, 119, 1429–1437. [Google Scholar] [CrossRef]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef]
- Olmeda, D.; Jorda, M.; Peinado, H.; Fabra, A.; Cano, A. Snail silencing effectively suppresses tumour growth and invasiveness. Oncogene 2007, 26, 1862–1874. [Google Scholar] [CrossRef]
- Peinado, H.; Olmeda, D.; Cano, A. Snail, Zeb and bHLH factors in tumour progression: An alliance against the epithelial phenotype? Nat. Rev. Cancer 2007, 7, 415–428. [Google Scholar] [CrossRef]
- Nakajima, S.; Doi, R.; Toyoda, E.; Tsuji, S.; Wada, M.; Koizumi, M.; Tulachan, S.S.; Ito, D.; Kami, K.; Mori, T.; et al. N-cadherin expression and epithelial mesenchymal transition in pancreatic carcinoma. Clin. Cancer Res. 2004, 10, 4125–4133. [Google Scholar] [CrossRef]
- Joo, Y.E.; Rew, J.S.; Park, C.S.; Kim, S.J. Expression of E-cadherin, alpha- and beta-catenins in patients with pancreatic adenocarcinoma. Pancreatology 2002, 2, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Loh, C.-Y.; Chai, J.Y.; Tang, T.F.; Wong, W.F.; Sethi, G.; Shanmugam, M.K.; Chong, P.P.; Looi, C.Y. The E-Cadherin and N-Cadherin Switch in Epithelial-to-Mesenchymal Transition: Signaling, Therapeutic Implications, and Challenges. Cells 2019, 8, 1118. [Google Scholar] [CrossRef]
- Wang, M.; Ren, D.; Guo, W.; Huang, S.; Wang, Z.; Li, Q.; Du, H.; Song, L.; Peng, X. N-cadherin promotes epithelial-mesenchymal transition and cancer stem cell-like traits via ErbB signaling in prostate cancer cells. Int. J. Oncol. 2016, 48, 595–606. [Google Scholar] [CrossRef]
- Jennbacken, K.; Tešan, T.; Wang, W.; Gustavsson, H.; Damber, J.-E.; Welen, K. N-cadherin increases after androgen deprivation and is associated with metastasis in prostate cancer. Endocr. Relat. Cancer 2010, 17, 469–479. [Google Scholar] [CrossRef]
- Hulit, J.; Suyama, K.; Chung, S.; Keren, R.; Agiostratidou, G.; Shan, W.; Dong, X.; Williams, T.M.; Lisanti, M.P.; Knudsen, K. N-cadherin signaling potentiates mammary tumor metastasis via enhanced extracellular signal-regulated kinase activation. Cancer Res. 2007, 67, 3106–3116. [Google Scholar] [CrossRef]
- Hui, L.; Zhang, S.; Dong, X.; Tian, D.; Cui, Z.; Qiu, X. Prognostic significance of twist and N-cadherin expression in NSCLC. PLoS ONE 2013, 8, e62171. [Google Scholar] [CrossRef]
- Cano, C.E.; Motoo, Y.; Iovanna, J.L. Epithelial-to-mesenchymal transition in pancreatic adenocarcinoma. Sci. World J. 2010, 10, 1947–1957. [Google Scholar] [CrossRef]
- Shintani, Y.; Hollingsworth, M.A.; Wheelock, M.; Johnson, K.J. Collagen I promotes metastasis in pancreatic cancer by activating c-Jun NH2-terminal kinase 1 and up-regulating N−cadherin expression. Cancer Res. 2006, 66, 11745–11753. [Google Scholar] [CrossRef]
- Shintani, Y.; Fukumoto, Y.; Chaika, N.; Grandgenett, P.M.; Hollingsworth, M.A.; Wheelock, M.J.; Johnson, K.R. ADH-1 suppresses N-cadherin-dependent pancreatic cancer progression. Int. J. Cancer 2008, 122, 71–77. [Google Scholar] [CrossRef]
- Su, Y.; Li, J.; Witkiewicz, A.K.; Brennan, D.; Neill, T.; Talarico, J.; Radice, G.L. N-cadherin haploinsufficiency increases survival in a mouse model of pancreatic cancer. Oncogene 2012, 31, 4484–4489. [Google Scholar] [CrossRef][Green Version]
- Meng, W.; Takeichi, M. Adherens junction: Molecular architecture and regulation. Cold Spring Harb. Perspect. Biol. 2009, 1, a002899. [Google Scholar] [CrossRef]
- St Croix, B.; Sheehan, C.; Rak, J.W.; Florenes, V.A.; Slingerland, J.M.; Kerbel, R.S. E-Cadherin-dependent growth suppression is mediated by the cyclin-dependent kinase inhibitor p27(KIP1). J. Cell. Biol. 1998, 142, 557–571. [Google Scholar] [CrossRef]
- Mendonsa, A.M.; Na, T.Y.; Gumbiner, B.M. E-cadherin in contact inhibition and cancer. Oncogene 2018, 37, 4769–4780. [Google Scholar] [CrossRef]
- Lecuit, T.; Yap, A.S. E-cadherin junctions as active mechanical integrators in tissue dynamics. Nat. Cell. Biol. 2015, 17, 533–539. [Google Scholar] [CrossRef]
- Hotz, B.; Arndt, M.; Dullat, S.; Bhargava, S.; Buhr, H.-J.; Hotz, H.G. Epithelial to mesenchymal transition: Expression of the regulators snail, slug, and twist in pancreatic cancer. Clin. Cancer Res. 2007, 13, 4769–4776. [Google Scholar] [CrossRef]
- Celesti, G.; Di Caro, G.; Bianchi, P.; Grizzi, F.; Basso, G.; Marchesi, F.; Doni, A.; Marra, G.; Roncalli, M.; Mantovani, A.; et al. Presence of Twist1-positive neoplastic cells in the stroma of chromosome-unstable colorectal tumors. Gastroenterology 2013, 145, 647–657. [Google Scholar] [CrossRef]
- Beuran, M.; Negoi, I.; Paun, S.; Ion, A.D.; Bleotu, C.; Negoi, R.I.; Hostiuc, S. The epithelial to mesenchymal transition in pancreatic cancer: A systematic review. Pancreatology 2015, 5, 217–225. [Google Scholar] [CrossRef]
- Xie, D.; Xie, K. Pancreatic cancer stromal biology and therapy. Genes Dis. 2015, 2, 133–143. [Google Scholar] [CrossRef]
- Karayiannakis, A.J.; Syrigos, K.N.; Polychronidis, A.; Simopoulos, C. Expression patterns of alpha-, beta- and gamma-catenin in pancreatic cancer: Correlation with E-cadherin expression, pathological features and prognosis. Anticancer Res. 2001, 21, 4127–4134. [Google Scholar]
- De Wever, O.; Derycke, L.; Hendrix, A.; De Meerleer, G.; Godeau, F.; Depypere, H.; Bracke, M. Soluble cadherins as cancer biomarkers. Clin. Exp. Metastasis 2007, 24, 685–697. [Google Scholar] [CrossRef]
- Van Roy, F.; Berx, G. The cell–cell adhesion molecule E-cadherin. Cell. Mol. Life Sci. 2008, 65, 3756–3788. [Google Scholar] [CrossRef]
- Grabowska, M.M.; Day, M.L. Soluble E-cadherin: More than a symptom of disease. Front. Biosci. 2012, 17, 1948–1964. [Google Scholar] [CrossRef]
- Marambaud, P.; Shioi, J.; Serban, G.; Georgakopoulos, A.; Sarner, S.; Nagy, V.; Baki, L.; Wen, P.; Efthimiopoulos, S.; Shao, Z.; et al. A presenilin-1/gamma-secretase cleavage releases the Ecadherin intracellular domain and regulates disassembly of adherens junctions. EMBO J. 2002, 21, 1948–1956. [Google Scholar] [CrossRef]
- David, J.M.; Rajasekaran, A.K. Dishonorable Discharge: The Oncogenic Roles of Cleaved E-Cadherin Fragments. Cancer Res. 2012, 72, 1–7. [Google Scholar] [CrossRef]
- Lynch, C.C.; Vargo-Gogola, T.; Matrisian, L.M.; Fingletonet, B. Cleavage of E-Cadherin by Matrix Metalloproteinase-7 Promotes Cellular Proliferation in Nontransformed Cell Lines via Activation of RhoA. J. Oncol. 2010, 2010, 530745. [Google Scholar] [CrossRef]
- Ferber, E.C.; Kajita, M.; Wadlow, A.; Tobiansky, L.; Niessen, C.; Ariga, H.; Daniel, J.; Fujita, Y. A role for the cleaved cytoplasmic domain of e-cadherin in the nucleus. J. Biol. Chem. 2008, 283, 12691–12700. [Google Scholar] [CrossRef]
- Steinhusen, U.; Weiske, J.; Badock, V.; Tauber, R.; Bommert, K.; Huber, O. Cleavage and shedding of E-cadherin after induction of apoptosis. J. Biol. Chem. 2001, 276, 4972–4980. [Google Scholar] [CrossRef]
- Maretzky, T.; Reiss, K.; Ludwig, A.; Buchholz, J.; Scholz, F.; Proksch, E.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 mediates E-cadherin shedding and regulates epithelial cell-cell adhesion, migration, and beta-catenin translocation. Proc. Natl. Acad. Sci. USA 2005, 102, 9182–9187. [Google Scholar] [CrossRef]
- Ito, K.; Okamoto, I.; Araki, N.; Kawano, Y.; Nakao, M.; Fujiyama, S.; Tomita, K.; Mimori, T.; Saya, H. Calcium influx triggers the sequential proteolysis of extracellular and cytoplasmic domains of E-cadherin, leading to loss of beta-catenin from cell-cell contacts. Oncogene 1999, 18, 7080–7090. [Google Scholar] [CrossRef]
- Gil, O.D.; Lee, C.; Ariztia, E.V.; Wang, F.Q.; Smith, P.J.; Hope, J.M.; Fishman, D.A. Lysophosphatidic acid (LPA) promotes E-cadherin ectodomain shedding and OVCA429 cell invasion in an uPA-dependent manner. Gynecol. Oncol. 2008, 108, 361–369. [Google Scholar] [CrossRef]
- Noe, V.; Willems, J.; Vandekerckhove, J.; Roy, F.V.; Bruyneel, E.; Mareel, M. Inhibition of adhesion and induction of epithelial cell invasion by HAV-containing Ecadherin-specific peptides. J. Cell. Sci. 1999, 112, 127–135. [Google Scholar]
- Ryniers, F.; Stove, C.; Goethals, M.; Brackenier, L.; Noe, V.; Bracke, M.; Vandekerckhove, J.; Mareel, M.; Bruyneel, E. Plasmin produces an E-cadherin fragment that stimulates cancer cell invasion. Biol. Chem. 2002, 383, 159–165. [Google Scholar] [CrossRef]
- Nawrocki-Raby, B.; Gilles, C.; Polette, M.; Bruyneel, E.; Laronze, J.Y.; Bonnet, N.; Foidart, J.M.; Mareel, M.; Birembaut, P. Upregulation of MMPs by soluble E-cadherin in human lung tumor cells. Int. J. Cancer 2003, 105, 790–795. [Google Scholar] [CrossRef]
- Johnson, S.K.; Ramani, V.C.; Hennings, L.; Haun, R.S. Kallikrein 7 enhances pancreatic cancer cell invasion by shedding E-cadherin. Cancer 2007, 109, 1811–1820. [Google Scholar] [CrossRef]
- Gagliano, N.; Celesti, G.; Tacchini, L.; Pluchino, S.; Sforza, C.; Rasile, M.; Valerio, V.; Laghi, L.; Conte, V.; Procacci, P. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma: Characterization in a 3D-cell culture model. World J. Gastroenterol. 2016, 22, 4466–4483. [Google Scholar] [CrossRef]
- Christiansen, J.J.; Rajasekaran, A.K. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006, 66, 8319–8326. [Google Scholar] [CrossRef]
- Putzke, A.P.; Ventura, A.P.; Bailey, A.M.; Akture, C.; Opoku-Ansah, J.; Çelikta¸s, M.; Hwang, M.S.; Darling, D.S.; Coleman, I.M.; Nelson, P.S. Metastatic progression of prostate cancer and e-cadherin: Regulation by Zeb1 and Src family kinases. Am. J. Pathol. 2011, 179, 400–410. [Google Scholar] [CrossRef]
- Reddy, P.; Liu, L.; Ren, C.; Lindgren, P.; Boman, K.; Shen, Y.; Lundin, E.; Ottander, U.; Rytinki, M.; Liu, K. Formation of E-cadherin-mediated cell-cell adhesion activates AKT and mitogen activated protein kinase via phosphatidylinositol 3 kinase and ligand-independent activation of epidermal growth factor receptor in ovarian cancer cells. Mol. Endocrinol. 2005, 19, 2564–2578. [Google Scholar] [CrossRef] [PubMed]
- Lewis-Tuffin, L.J.; Rodriguez, F.; Giannini, C.; Scheithauer, B.; Necela, B.M.; Sarkaria, J.N.; Anastasiadis, P.Z. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PLoS ONE 2010, 5, e13665. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.M.; Li, A.; Olino, K.; Wolfgang, C.L.; Herman, J.M.; Schulick, R.D.; Iacobuzio-Donahue, C.; Hruban, R.H.; Goggins, M. Loss of E-cadherin expression and outcome among patients with resectable pancreatic adenocarcinomas. Mod. Pathol. 2011, 24, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Winter, J.M.; Ting, A.H.; Vilardell, F.; Gallmeier, E.; Baylin, S.B.; Hruban, R.H.; Kern, S.E.; Iacobuzio-Donahue, C.A. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin. Cancer Res. 2008, 14, 412–418. [Google Scholar] [CrossRef]
- Li, Y.J.; Ji, X.R. Relationship between expression of E-cadherin-catenin complex and clinicopathologic characteristics of pancreatic cancer. World J. Gastroenterol. 2003, 9, 368–372. [Google Scholar] [CrossRef]
- Iacobuzio-Donahue, C.A.; Fu, B.; Yachida, S.; Luo, M.; Abe, H.; Henderson, C.M.; Vilardell, F.; Wang, Z.; Keller, J.W.; Banerjee, P.; et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J. Clin. Oncol. 2009, 27, 1806–1813. [Google Scholar] [CrossRef]
- Al-Aynati, M.M.; Radulovich, N.; Riddell, R.H.; Tsao, M.S. Epithelial-cadherin and beta-catenin expression changes in pancreatic intraepithelial neoplasia. Clin. Cancer. Res. 2004, 10, 1235–1240. [Google Scholar] [CrossRef]
- Liu, Y.; Brand, R.E.; Turzhitsky, V.; Kim, Y.L.; Roy, H.K.; Hasabou, N.; Sturgis, C.; Shah, D.; Hall, C.; Backman, V. Optical markers in duodenal mucosa predict the presence of pancreatic cancer. Clin. Cancer Res. 2007, 13, 4392–4399. [Google Scholar] [CrossRef]
- Cates, J.M.; Byrd, R.H.; Fohn, L.E.; Tatsas, A.D.; Washington, M.K.; Black, C.C. Epithelial-mesenchymal transition markers in pancreatic ductal adenocarcinoma. Pancreas 2009, 38, e1–e6. [Google Scholar] [CrossRef]
- Menke, A.; Philippi, C.; Vogelmann, R.; Seidel, B.; Lutz, M.P.; Adler, G.; Wedlich, D. Down-regulation of E-cadherin gene expression by collagen type I and type III in pancreatic cancer cell lines. Cancer Res. 2001, 61, 3508–3517. [Google Scholar] [PubMed]
- Funel, N.; Costa, F.; Pettinari, L.; Taddeo, A.; Sala, A.; Chiriva-Internati, M.; Cobos, E.; Colombo, G.; Milzani, A.; Campani, D.; et al. Ukrain affects pancreas cancer cell phenotype in vitro by targeting MMP-9 and intra-/extracellular SPARC expression. Pancreatology 2010, 10, 545–552. [Google Scholar] [CrossRef] [PubMed]
- Bissell, M.J.; Radisky, D. Putting tumours in context. Nat. Rev. Cancer 2001, 1, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Kunz-Schugart, L.; Knuechel, R. Tumor-associated fibroblasts (Part I): Active stromal participants in tumor development and progression? Histol. Histopathol. 2002, 17, 599–621. [Google Scholar]
- Eritja, N.; Dolcet, X.; Matias-Guiu, X. Three-dimensional epithelial cultures: A tool to model cancer development and progression. Histol. Histopathol. 2013, 28, 1245–1256. [Google Scholar] [PubMed]
- Hirschhaeuser, F.; Menne, H.; Dittfeld, C.; West, J.; Mueller-Klieser, W.; Kunz-Schughart, L.A. Multicellular tumor spheroids: An underestimated tool is catching up again. J. Biotechnol. 2010, 148, 3–15. [Google Scholar] [CrossRef]
- Zeeberg, K.; Cardone, R.A.; Greco, M.R.; Saccomano, M.; Nøhr-Nielsen, A.; Alves, F.; Pedersen, S.F.; Reshkin, S.J. Assessment of different 3D culture systems to study tumor phenotype and chemosensitivity in pancreatic ductal adenocarcinoma. Int. J. Oncol. 2016, 49, 243–252. [Google Scholar] [CrossRef][Green Version]
- Bissell, M.J. Architecture is the Message: The role of extracellular matrix and 3-D structure in tissue-specific gene expression and breast cancer. Pezcoller Found. J. 2007, 16, 2–17. [Google Scholar]
- Inman, J.L.; Bissell, M.J. Apical polarity in three-dimensional culture systems: Where to now? J. Biol. 2010, 9, 2. [Google Scholar] [CrossRef]
- Coleman, S.J.; Watt, J.; Arumugam, P.; Solaini, L.; Carapuca, E.; Ghallab, M.; Grose, R.P.; Kocher, H.M. Pancreatic cancer organotypics: High throughput, preclinical models for pharmacological agent evaluation. World J. Gastroenterol. 2014, 20, 8471–8481. [Google Scholar] [CrossRef]
- Dufau, I.; Frongia, C.; Sicard, F.; Dedieu, L.; Cordelier, P.; Ausseil, F.; Ducommun, B.; Valette, A. Multicellular tumor spheroid model to evaluate spatio-temporal dynamics effect of chemotherapeutics: Application to the gemcitabine/CHK1 inhibitor combination in pancreatic cancer. BMC Cancer 2012, 12, 15. [Google Scholar] [CrossRef] [PubMed]
- Boj, S.F.; Hwang, C.I.; Baker, L.A.; Engle, D.D.; Tuveson, D.A.; Clevers, H. Model organoids provide new research opportunities for ductal pancreatic cancer. Mol. Cell. Oncol. 2015, 3, e1014757. [Google Scholar] [CrossRef] [PubMed]
- Park, C.C.; Bissell, M.J.; Barcellos-Hoff, M.H. The influence of the microenvironment on the malignant phenotype. Mol. Med. Today 2000, 6, 324–329. [Google Scholar] [CrossRef]
- Chu, G.C.; Kimmelman, A.C.; Hezel, A.F.; DePinho, R.A. Stromal biology of pancreatic cancer. J. Cell. Biochem. 2007, 101, 887–907. [Google Scholar] [CrossRef] [PubMed]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef]
- Grzesiak, J.J.; Bouvet, M. The alpha2beta1 integrin mediates the malignant phenotype on type I collagen in pancreatic cancer cell lines. Br. J. Cancer 2006, 94, 1311–1319. [Google Scholar] [CrossRef]
- Miyamoto, H.; Murakami, T.; Tsuchida, K.; Sugino, H.; Miyake, H.; Tashiro, S. Tumor-stroma interaction of human pancreatic cancer: Acquired resistance to anticancer drugs and proliferation regulation is dependent on extracellular matrix proteins. Pancreas 2004, 28, 38–44. [Google Scholar] [CrossRef]
- Procacci, P.; Moscheni, C.; Sartori, P.; Sommariva, M.; Gagliano, N. Tumor-Stroma Cross-Talk in Human Pancreatic Ductal Adenocarcinoma: A Focus on the Effect of the Extracellular Matrix on Tumor Cell Phenotype and Invasive Potential. Cells 2018, 7, 158. [Google Scholar] [CrossRef]
- Shintani, Y.; Fukumoto, Y.; Chaika, N.; Svoboda, R.; Wheelock, M.J.; Johnson, K.R. Collagen I-mediated up-regulation of N-cadherin requires cooperative signals from integrins and discoidin domain receptor 1. J. Cell. Biol. 2008, 180, 1277–1289. [Google Scholar] [CrossRef]
- Shintani, Y.; Maeda, M.; Chaika, N.; Johnson, K.R.; Wheelock, M.J. Collagen I promotes epithelial-to-mesenchymal transition in lung cancer cells via transforming growth factor-beta signaling. Am. J. Respir. Cell. Mol. Biol. 2008, 38, 95–104. [Google Scholar] [CrossRef]
- Micalizzi, D.S.; Farabaugh, S.M.; Ford, H.L. Epithelial-mesenchymal transition in cancer: Parallels between normal development and tumor progression. J. Mammary Gland. Biol. Neoplasia 2010, 15, 117–134. [Google Scholar] [CrossRef] [PubMed]
- Aiello, N.M.; Maddipati, R.; Norgard, R.J.; Balli, D.; Li, J.; Yuan, S.; Yamazoe, T.; Black, T.; Sahmoud, A.; Furth, E.E.; et al. EMT subtype influences epithelial plasticity and mode of cell migration. Dev. Cell 2018, 45, 681–695. [Google Scholar] [CrossRef] [PubMed]
- George, J.T.; Jolly, M.K.; Xu, S.; Somarelli, J.A.; Levine, H. Survival outcomes in cancer patients predicted by a partial EMT gene expression scoring metric. Cancer Res. 2017, 77, 6415–6428. [Google Scholar] [CrossRef] [PubMed]
- Ruscetti, M.; Dadashian, E.L.; Guo, W.; Quach, B.; Mulholland, D.J.; Park, J.W.; Tran, L.M.; Kobayashi, N.; Bianchi–Frias, D.; Xing, Y.; et al. HDAC inhibition impedes epithelial–mesenchymal plasticity and suppresses metastatic, castration–resistant prostate cancer. Oncogene 2016, 35, 3781–3795. [Google Scholar] [CrossRef]
- Sampson, V.B.; David, J.M.; Puig, I.; Patil, P.U.; de Herreros, A.G.; Thomas, G.V.; Rajasekaran, A.K. Wilmsʹ tumor protein induces an epithelial–mesenchymal hybrid differentiation state in clear cell renal cell carcinoma. PLoS ONE 2014, 9, e102041. [Google Scholar] [CrossRef]
- Hendrix, M.J.; Seftor, E.A.; Seftor, R.E.; Trevor, K.T. Experimental co–expression of vimentin and keratin intermediate filaments in human breast cancer cells results in phenotypic interconversion and increased invasive behavior. Am. J. Pathol. 1997, 150, 483–495. [Google Scholar]
- Siret, C.; Dobric, A.; Martirosyan, A.; Terciolo, C.; Germain, S.; Bonier, R.; Dirami, T.; Dusetti, N.; Tomasini, R.; Rubis, M.; et al. Cadherin-1 and cadherin-3 cooperation determines the aggressiveness of pancreatic ductal adenocarcinoma. Br. J. Cancer 2018, 118, 546–557. [Google Scholar] [CrossRef]
- Taniuchi, K.; Nakagawa, H.; Hosokawa, M.; Nakamura, T.; Eguchi, H.; Ohigashi, H.; Ishikawa, O.; Katagiri, T.; Nakamura, Y. Overexpressed P-cadherin/CDH3 promotes motility of pancreatic cancer cells by interacting with p120ctn and activating rho-family GTPases. Cancer Res. 2005, 65, 3092–3099. [Google Scholar] [CrossRef]
- Sakamoto, K.; Imai, K.; Higashi, T.; Taki, K.; Nakagawa, S.; Okabe, H.; Nitta, H.; Hayashi, H.; Chikamoto, A.; Ishiko, T.; et al. Significance of P-cadherin overexpression and possible mechanism of its regulation in intrahepatic cholangiocarcinoma and pancreatic cancer. Cancer Sci. 2015, 106, 1153–1162. [Google Scholar] [CrossRef]
- Reichert, M.; Bakir, B.; Moreira, L.; Pitarresi, J.R.; Feldmann, K.; Simon, L.; Suzuki, K.; Maddipati, R.; Rhim, A.D.; Schlitter, A.M.; et al. Regulation of epithelial plasticity determines metastatic organotropism in pancreatic cancer. Dev. Cell. 2018, 45, 696–711. [Google Scholar] [CrossRef]
- Liao, T.-T.; Yang, M.-H. Hybrid Epithelial/Mesenchymal State in Cancer Metastasis: Clinical Significance and Regulatory Mechanisms. Cells 2020, 9, 623. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Hou, D.; Wei, Z.; Zheng, S.; Zhang, Y.; Li, J. Extracellular and intracellular microRNAs in pancreatic cancer: From early diagnosis to reducing chemoresistance. ExRNA 2019, 1, 17. [Google Scholar] [CrossRef]
- Li, D.; Li, X.; Cao, W.; Qi, Y.; Yang, X. Antagonism of microRNA-99a promotes cell invasion and down-regulates E-cadherin expression in pancreatic cancer cells by regulating mammalian target of rapamycin. Acta Histochem. 2014, 116, 723–729. [Google Scholar] [CrossRef]
- Zhang, J.Q.; Chen, S.; Gu, J.N.; Zhu, Y.; Zhan, Q.; Cheng, D.F.; Chen, H.; Deng, X.X.; Shen, B.Y.; Peng, C.H. MicroRNA-300 promotes apoptosis and inhibits proliferation, migration, invasion and epithelial-mesenchymal transition via the Wnt/β-catenin signaling pathway by targeting CUL4B in pancreatic cancer cells. J. Cell. Biochem. 2018, 119, 1027–1040. [Google Scholar] [CrossRef]
- Lu, W.; Kang, Y. Epithelial-Mesenchymal Plasticity in Cancer Progression and Metastasis. Dev. Cell 2019, 49, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Cannon, A.; Thompson, C.; Hall, B.R.; Jain, M.; Kumar, S.; Batra, S.K. Desmoplasia in pancreatic ductal adenocarcinoma: Insight into pathological function and therapeutic potential. Genes Cancer 2018, 9, 78–86. [Google Scholar] [PubMed]
- Ren, B.; Cui, M.; Yang, G.; Wang, H.; Feng, M.; You, L.; Zhao, Y. Tumor microenvironment participates in metastasis of pancreatic cancer. Mol. Cancer 2018, 17, 108. [Google Scholar] [CrossRef]
- Liu, X.; Xu, J.; Zhang, B.; Liu, J.; Liang, C.; Meng, Q.; Hua, J.; Yu, X.; Shi, S. The reciprocal regulation between host tissue and immune cells in pancreatic ductal adenocarcinoma: New insights and therapeutic implications. Mol. Cancer 2019, 18, 184. [Google Scholar] [CrossRef]
- Dongre, A.; Weinberg, R.A. New insights into the mechanisms of epithelial-mesenchymal transition and implications for cancer. Nat. Rev. Mol. Cell Biol. 2019, 20, 69–84. [Google Scholar] [CrossRef]
- Romeo, E.; Caserta, C.A.; Rumio, C.; Marcucci, F. The Vicious Cross-Talk between Tumor Cells with an EMT Phenotype and Cells of the Immune System. Cells 2019, 8, 460. [Google Scholar] [CrossRef]
- Meng, F.; Li, C.; Li, W.; Gao, Z.; Guo, K.; Song, S. Interaction between pancreatic cancer cells and tumor-associated macrophages promotes the invasion of pancreatic cancer cells and the differentiation and migration of macrophages. IUBMB Life 2014, 66, 835–846. [Google Scholar] [CrossRef] [PubMed]
- Kuwada, K.; Kagawa, S.; Yoshida, R.; Sakamoto, S.; Ito, A.; Watanabe, M.; Ieda, T.; Kuroda, S.; Kikuchi, S.; Tazawa, H.; et al. The epithelial-to-mesenchymal transition induced by tumor-associated macrophages confers chemoresistance in peritoneally disseminated pancreatic cancer. J. Exp. Clin. Cancer Res. 2018, 37, 307. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells; partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Grosse-Steffen, T.; Giese, T.; Giese, N.; Longerich, T.; Schirmacher, P.; Hänsch, G.M.; Gaida, M.M. Epithelial-to-mesenchymal transition in pancreatic ductal adenocarcinoma and pancreatic tumor cell lines: The role of neutrophils and neutrophil-derived elastase. Clin. Dev. Immunol. 2012, 2012, 720768. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.G.; Tang, A.; Sharrow, S.O.; Udey, M.C. Murine dendritic epidermal T cells express the homophilic adhesion molecule E-cadherin. Epithelial Cell Biol. 1994, 3, 149–155. [Google Scholar]
- Munro, S.B.; Duclos, A.J.; Jackson, A.R.; Baines, M.G.; Blaschuk, O.W. Characterization of cadherins expressed by murine thymocytes. Cell. Immunol. 1996, 169, 309–312. [Google Scholar] [CrossRef]
- Müller, K.M.; Luedecker, C.J.; Udey, M.C.; Farr, A.G. Involvement of E-cadherin in thymus organogenesis and thymocyte maturation. Immunity 1997, 6, 257–264. [Google Scholar] [CrossRef]
- Mezouar, S.; Omar Osman, I.; Melenotte, C.; Slimani, C.; Chartier, C.; Raoult, D.; Mege, J.L.; Devaux, C.A. High concentrations of serum soluble e-cadherin in patients with Q fever. Front. Cell. Infect. Microbiol. 2019, 9, 219. [Google Scholar] [CrossRef]
- Esch, T.R.; Jonsson, M.V.; Levanos, V.A.; Poveromo, J.D.; Sorkin, B.C. Leukocytes infiltrating the submandibular glands of NOD mice express E-cadherin. J. Autoimmun. 2000, 15, 387–393. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Malissen, B.; Mantovani, A.; De Baetselier, P.; Van Ginderachter, J.A. Regulation and function of the E-cadherin/catenin complex in cells of the monocyte-macrophage lineage and DCs. Blood 2012, 119, 1623–1633. [Google Scholar] [CrossRef]
- Siddiqui, K.R.; Laffont, S.; Powrie, F. E-cadherin marks a subset of inflammatory dendritic cells that promote T cell-mediated colitis. Immunity 2010, 32, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Hu, X.; Hu, Y.; Teng, K.; Zhang, K.; Zheng, Y.; Hong, X.; Yu, K.; Wang, Y.; Liu, L. Anti-CD40-induced inflammatory E-cadherin+ dendritic cells enhance T cell responses and antitumour immunity in murine Lewis lung carcinoma. J. Exp. Clin. Cancer Res. 2015, 34, 11. [Google Scholar] [CrossRef]
- Pierre, P.; Turley, S.J.; Gatti, E.; Hull, M.; Meltzer, J.; Mirza, A.; Inaba, K.; Steinman, R.M.; Mellman, I. Developmental regulation of MHC class II transport in mouse dendritic cells. Nature 1997, 388, 787–792. [Google Scholar] [CrossRef] [PubMed]
- Riedl, E.; Stöckl, J.; Majdic, O.; Scheinecker, C.; Knapp, W.; Strobl, H. Ligation of E-cadherin on in vitro-generated immature Langerhans-type dendritic cells inhibits their maturation. Blood 2000, 96, 4276–4284. [Google Scholar] [CrossRef]
- Rescigno, M.; Urbano, M.; Valzasina, B.; Francolini, M.; Rotta, G.; Bonasio, R.; Granucci, F.; Kraehenbuhl, J.P.; Ricciardi-Castagnoli, P. Dendritic cells express tight junction proteins and penetrate gut epithelial monolayers to sample bacteria. Nat. Immunol. 2001, 2, 361–367. [Google Scholar] [CrossRef]
- Jiang, A.; Bloom, O.; Ono, S.; Cui, W.; Unternaehrer, J.; Jiang, S.; Whitney, J.A.; Connolly, J.; Banchereau, J.; Mellman, I. Disruption of E-cadherin-mediated adhesion induces a functionally distinct pathway of dendritic cell maturation. Immunity 2007, 27, 610–624. [Google Scholar] [CrossRef]
- Yu, W.; Yang, L.; Li, T.; Zhang, Y. Cadherin Signaling in Cancer: Its Functions and Role as a Therapeutic Target. Front. Oncol. 2019, 9, 989. [Google Scholar] [CrossRef]
- Manicassamy, S.; Reizis, B.; Ravindran, R.; Nakaya, H.; Salazar-Gonzalez, R.M.; Wang, Y.C.; Pulendran, B. Activation of beta-catenin in dendritic cells regulates immunity versus tolerance in the intestine. Science 2010, 329, 849–853. [Google Scholar] [CrossRef]
- Padovan, E.; Terracciano, L.; Certa, U.; Jacobs, B.; Reschner, A.; Bolli, M.; Spagnoli, G.C.; Borden, E.C.; Heberer, M. Interferon stimulated gene 15 constitutively produced by melanoma cells induces e-cadherin expression on human dendritic cells. Cancer Res. 2002, 62, 3453–3458. [Google Scholar]
- Locati, M.; Curtale, G.; Mantovani, A. Diversity; Mechanisms; and Significance of Macrophage Plasticity. Annu. Rev. Pathol. 2020, 15, 123–147. [Google Scholar] [CrossRef]
- Rehli, M.; Sulzbacher, S.; Pape, S.; Ravasi, T.; Wells, C.A.; Heinz, S.; Söllner, L.; El Chartouni, C.; Krause, S.W.; Steingrimsson, E.; et al. Transcription factor Tfec contributes to the IL-4-inducible expression of a small group of genes in mouse macrophages including the granulocyte colony-stimulating factor receptor. J. Immunol. 2005, 174, 7111–7122. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Bogaert, P.; van Hengel, J.; Guérin, C.J.; Berx, G.; Movahedi, K.; Van den Bergh, R.; Pereira-Fernandes, A.; Geuns, J.M.; Pircher, H.; et al. Alternatively activated macrophages engage in homotypic and heterotypic interactions through IL-4 and polyamine-induced E-cadherin/catenin complexes. Blood 2009, 114, 4664–4674. [Google Scholar] [CrossRef]
- Ghassabeh, G.H.; De Baetselier, P.; Brys, L.; Noël, W.; Van Ginderachter, J.A.; Meerschaut, S.; Beschin, A.; Brombacher, F.; Raes, G. Identification of a common gene signature for type II cytokine-associated myeloid cells elicited in vivo in different pathologic conditions. Blood 2006, 108, 575–583. [Google Scholar] [CrossRef]
- Brooks, P.J.; Glogauer, M.; McCulloch, C.A. An Overview of the Derivation and Function of Multinucleated Giant Cells and Their Role in Pathologic Processes. Am. J. Pathol. 2019, 189, 1145–1158. [Google Scholar] [CrossRef]
- Moreno, J.L.; Mikhailenko, I.; Tondravi, M.M.; Keegan, A.D. IL-4 promotes the formation of multinucleated giant cells from macrophage precursors by a STAT6-dependent; homotypic mechanism: Contribution of E-cadherin. J. Leukoc. Biol. 2007, 82, 1542–1553. [Google Scholar] [CrossRef]
- Wanat, K.A.; Rosenbach, M.; Zoiber, A.F.; Zhang, P.J.; Schaffer, A. E-cadherin is expressed by mono- and multinucleated histiocytes in cutaneous sarcoidal and foreign body granulomas. Am. J. Dermatopathol. 2014, 36, 651–654. [Google Scholar] [CrossRef]
- Cronan, M.R.; Beerman, R.W.; Rosenberg, A.F.; Saelens, J.W.; Johnson, M.G.; Oehlers, S.H.; Sisk, D.M.; Jurcic Smith, K.L.; Medvitz, N.A.; Miller, S.E.; et al. Macrophage Epithelial Reprogramming Underlies Mycobacterial Granuloma Formation and Promotes Infection. Immunity 2016, 45, 861–876. [Google Scholar] [CrossRef]
- Van den Bossche, J.; Laoui, D.; Naessens, T.; Smits, H.H.; Hokke, C.H.; Stijlemans, B.; Grooten, J.; De Baetselier, P.; Van Ginderachter, J.A. E-cadherin expression in macrophages dampens their inflammatory responsiveness in vitro; but does not modulate M2-regulated pathologies in vivo. Sci. Rep. 2015, 5, 12599. [Google Scholar] [CrossRef]
- Rosshart, S.; Hofmann, M.; Schweier, O.; Pfaff, A.K.; Yoshimoto, K.; Takeuchi, T.; Molnar, E.; Schamel, W.W.; Pircher, H. Interaction of KLRG1 with E-cadherin: New functional and structural insights. Eur. J. Immunol. 2008, 38, 3354–3364. [Google Scholar] [CrossRef]
- Henson, S.M.; Akbar, A.N. KLRG1--more than a marker for T cell senescence. Age (Dordr) 2009, 31, 285–291. [Google Scholar] [CrossRef]
- Henson, S.M.; Franzese, O.; Macaulay, R.; Libri, V.; Azevedo, R.I.; Kiani-Alikhan, S.; Plunkett, F.J.; Masters, J.E.; Jackson, S.; Griffiths, S.J.; et al. KLRG1 signaling induces defective Akt (ser473) phosphorylation and proliferative dysfunction of highly differentiated CD8+ T cells. Blood 2009, 113, 6619–6628. [Google Scholar] [CrossRef]
- Nakamura, S.; Kuroki, K.; Ohki, I.; Sasaki, K.; Kajikawa, M.; Maruyama, T.; Ito, M.; Kameda, Y.; Ikura, M.; Yamamoto, K.; et al. Molecular basis for E-cadherin recognition by killer cell lectin-like receptor G1 (KLRG1). J. Biol. Chem. 2009, 284, 27327–27335. [Google Scholar] [CrossRef] [PubMed]
- Schwartzkopff, S.; Gründemann, C.; Schweier, O.; Rosshart, S.; Karjalainen, K.E.; Becker, K.F.; Pircher, H. Tumor-associated E-cadherin mutations affect binding to the killer cell lectin-like receptor G1 in humans. J. Immunol. 2007, 179, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Karecla, P.I.; Green, S.J.; Bowden, S.J.; Coadwell, J.; Kilshaw, P.J. Identification of a binding site for integrin alphaEbeta7 in the N-terminal domain of E-cadherin. J. Biol. Chem. 1996, 271, 30909–30915. [Google Scholar] [CrossRef] [PubMed]
- Hardenberg, J.B.; Braun, A.; Schön, M.P. A Yin and Yang in Epithelial Immunology: The Roles of the αE(CD103)β7 Integrin in T Cells. J. Investig. Dermatol. 2018, 138, 23–31. [Google Scholar] [CrossRef]
- Franciszkiewicz, K.; Le Floc’h, A.; Jalil, A.; Vigant, F.; Robert, T.; Vergnon, I.; Mackiewicz, A.; Benihoud, K.; Validire, P.; Chouaib, S.; et al. Intratumoral induction of CD103 triggers tumor-specific CTL function and CCR5-dependent T-cell retention. Cancer Res. 2009, 69, 6249–6255. [Google Scholar] [CrossRef] [PubMed]
- French, J.J.; Cresswell, J.; Wong, W.K.; Seymour, K.; Charnley, R.M.; Kirby, J.A. T cell adhesion and cytolysis of pancreatic cancer cells: A role for E-cadherin in immunotherapy? Br. J. Cancer 2002, 87, 1034–1041. [Google Scholar] [CrossRef]
- Lohneis, P.; Sinn, M.; Bischoff, S.; Jühling, A.; Pelzer, U.; Wislocka, L.; Bahra, M.; Sinn, B.V.; Denkert, C.; Oettle, H.; et al. Cytotoxic tumour-infiltrating T lymphocytes influence outcome in resected pancreatic ductal adenocarcinoma. Eur. J. Cancer 2017, 8, 290–301. [Google Scholar] [CrossRef]
- Hou, Y.C.; Chao, Y.J.; Hsieh, M.H.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Low CD8+ T Cell Infiltration and High PD-L1 Expression Are Associated with Level of CD44+/CD133+ Cancer Stem Cells and Predict an Unfavorable Prognosis in Pancreatic Cancer. Cancers (Basel) 2019, 11, 541. [Google Scholar] [CrossRef]
- Miksch, R.C.; Schoenberg, M.B.; Weniger, M.; Bösch, F.; Ormanns, S.; Mayer, B.; Werner, J.; Bazhin, A.V.; D’Haese, J.G. Prognostic Impact of Tumor-Infiltrating Lymphocytes and Neutrophils on Survival of Patients with Upfront Resection of Pancreatic Cancer. Cancers (Basel) 2019, 11, 39. [Google Scholar] [CrossRef]
- Fu, C.; Jiang, A. Dendritic Cells and CD8 T Cell Immunity in Tumor Microenvironment. Front. Immunol. 2018, 9, 3059. [Google Scholar] [CrossRef]
- Pfirschke, C.; Siwicki, M.; Liao, H.W.; Pittet, M.J. Tumor Microenvironment: No Effector T Cells without Dendritic Cells. Cancer Cell 2017, 31, 614–615. [Google Scholar] [CrossRef]
- Sichien, D.; Lambrecht, B.N.; Guilliams, M.; Scott, C.L. Development of conventional dendritic cells: From common bone marrow progenitors to multiple subsets in peripheral tissues. Mucosal Immunol. 2017, 10, 831–844. [Google Scholar] [CrossRef]
- Tang, A.; Amagai, M.; Granger, L.G.; Stanley, J.R.; Udey, M.C. Adhesion of epidermal Langerhans cells to keratinocytes mediated by E-cadherin. Nature 1993, 361, 82–85. [Google Scholar] [CrossRef]
- Jakob, T.; Udey, M.C. Regulation of E-cadherin-mediated adhesion in Langerhans cell-like dendritic cells by inflammatory mediators that mobilize Langerhans cells in vivo. J. Immunol. 1998, 160, 4067–4073. [Google Scholar]
- Mayumi, N.; Watanabe, E.; Norose, Y.; Watari, E.; Kawana, S.; Geijtenbeek, T.B.; Takahashi, H. E-cadherin interactions are required for Langerhans cell differentiation. Eur. J. Immunol. 2013, 43, 270–280. [Google Scholar] [CrossRef]
- Fiorino, C.; Harrison, R.E. E-cadherin is important for cell differentiation during osteoclastogenesis. Bone 2016, 86, 106–118. [Google Scholar] [CrossRef]
- Helming, L.; Gordon, S. Molecular mediators of macrophage fusion. Trends Cell Biol. 2009, 19, 514–522. [Google Scholar] [CrossRef]
- Clawson, G.A.; Matters, G.L.; Xin, P.; McGovern, C.; Wafula, E.; dePamphilis, C.; Meckley, M.; Wong, J.; Stewart, L.; D’Jamoos, C.; et al. “Stealth dissemination” of macrophage-tumor cell fusions cultured from blood of patients with pancreatic ductal adenocarcinoma. PLoS ONE 2017, 12, e0184451. [Google Scholar] [CrossRef]
- Pawelek, J.M.; Chakraborty, A.K. The cancer cell--leukocyte fusion theory of metastasis. Adv. Cancer Res. 2008, 101, 397–444. [Google Scholar]
- Kemeny, L.V.; Kurgyis, Z.; Buknicz, T.; Groma, G.; Jakab, A.; Zanker, K.; Dittmar, T.; Kemeny, L.; Nemeth, I.B. Melanoma Cells Can Adopt the Phenotype of Stromal Fibroblasts and Macrophages by Spontaneous Cell Fusion in Vitro. Int. J. Mol. Sci. 2016, 17, 826. [Google Scholar] [CrossRef]
- Clark, C.E.; Hingorani, S.R.; Mick, R.; Combs, C.; Tuveson, D.A.; Vonderheide, R.H. Dynamics of the immune reaction to pancreatic cancer from inception to invasion. Cancer Res. 2007, 67, 9518–9527. [Google Scholar] [CrossRef]
- Anderson, K.G.; Stromnes, I.M.; Greenberg, P.D. Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 2017, 31, 311–325. [Google Scholar] [CrossRef]
- Melero, I.; Rouzaut, A.; Motz, G.T.; Coukos, G. T-cell and NK-cell infiltration into solid tumors: A key limiting factor for efficacious cancer immunotherapy. Cancer Discov. 2014, 4, 522–526. [Google Scholar]
- Peranzoni, E.; Lemoine, J.; Vimeux, L.; Feuillet, V.; Barrin, S.; Kantari-Mimoun, C.; Bercovici, N.; Guérin, M.; Biton, J.; Ouakrim, H.; et al. Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. USA 2018, 115, E4041–E4050. [Google Scholar] [CrossRef]
- Beatty, G.L.; Winograd, R.; Evans, R.A.; Long, K.B.; Luque, S.L.; Lee, J.W.; Clendenin, C.; Gladney, W.L.; Knoblock, D.M.; Guirnalda, P.D.; et al. Exclusion of T Cells From Pancreatic Carcinomas in Mice Is Regulated by Ly6C(low) F4/80(+) Extratumoral Macrophages. Gastroenterology 2015, 149, 201–210. [Google Scholar] [CrossRef]
- Lankadasari, M.B.; Mukhopadhyay, P.; Mohammed, S.; Harikumar, K.B. TAMing pancreatic cancer: Combat with a double edged sword. Mol. Cancer 2019, 18, 48. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G. Mechanisms of motility in metastasizing cells. Mol. Cancer Res. 2010, 8, 629–642. [Google Scholar] [CrossRef]
- Wicki, A.; Lehembre, F.; Wick, N.; Hantusch, B.; Kerjaschki, D.; Christofori, G. Tumor invasion in the absence of epithelial-mesenchymal transition: Podoplanin-mediated remodeling of the actin cytoskeleton. Cancer Cell 2006, 9, 261–272. [Google Scholar] [CrossRef]
- Friedl, P.; Gilmour, D. Collective cell migration in morphogenesis, regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2009, 10, 445–457. [Google Scholar] [CrossRef]
- Yilmaz, M.; Christofori, G.; Lehembre, F. Distinct mechanisms of tumor invasion and metastasis. Trends Mol. Med. 2007, 13, 535–541. [Google Scholar] [CrossRef]
- Bronsert, P.; Enderle-Ammour, K.; Bader, M.; Timme, S.; Kuehs, M.; Csanadi, A.; Kayser, G.; Kohler, I.; Bausch, D.; Hoeppner, J.; et al. Cancer cell invasion and EMT marker expression - a three-dimensional study of the human cancer-host interface. J. Pathol. 2014, 234, 410–422. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sommariva, M.; Gagliano, N. E-Cadherin in Pancreatic Ductal Adenocarcinoma: A Multifaceted Actor during EMT. Cells 2020, 9, 1040. https://doi.org/10.3390/cells9041040
Sommariva M, Gagliano N. E-Cadherin in Pancreatic Ductal Adenocarcinoma: A Multifaceted Actor during EMT. Cells. 2020; 9(4):1040. https://doi.org/10.3390/cells9041040
Chicago/Turabian StyleSommariva, Michele, and Nicoletta Gagliano. 2020. "E-Cadherin in Pancreatic Ductal Adenocarcinoma: A Multifaceted Actor during EMT" Cells 9, no. 4: 1040. https://doi.org/10.3390/cells9041040
APA StyleSommariva, M., & Gagliano, N. (2020). E-Cadherin in Pancreatic Ductal Adenocarcinoma: A Multifaceted Actor during EMT. Cells, 9(4), 1040. https://doi.org/10.3390/cells9041040