IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells


 ,
, 

Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Analysis of Publicly Available Genomics and Transcriptomic Databases of Breast Cancer
2.2. Chemicals and Drugs
2.3. Cell Cultures
2.4. Focal Adhesion Kinase (FAK) CRISPR/Cas9 Genome Editing
2.5. Western Blotting Analysis
2.6. Immunoprecipitation Assay
2.7. RNA Extraction and Real-Time PCR
2.8. Plasmids, Luciferase and Transfections Assays
2.9. Immunofluorescence Microscopy
2.10. YAP Nuclear Staining
2.11. Cell Proliferation Assay
2.12. Colony Formation Assay
2.13. Spheroid Formation Assay
2.14. Statistical Analysis
3. Results
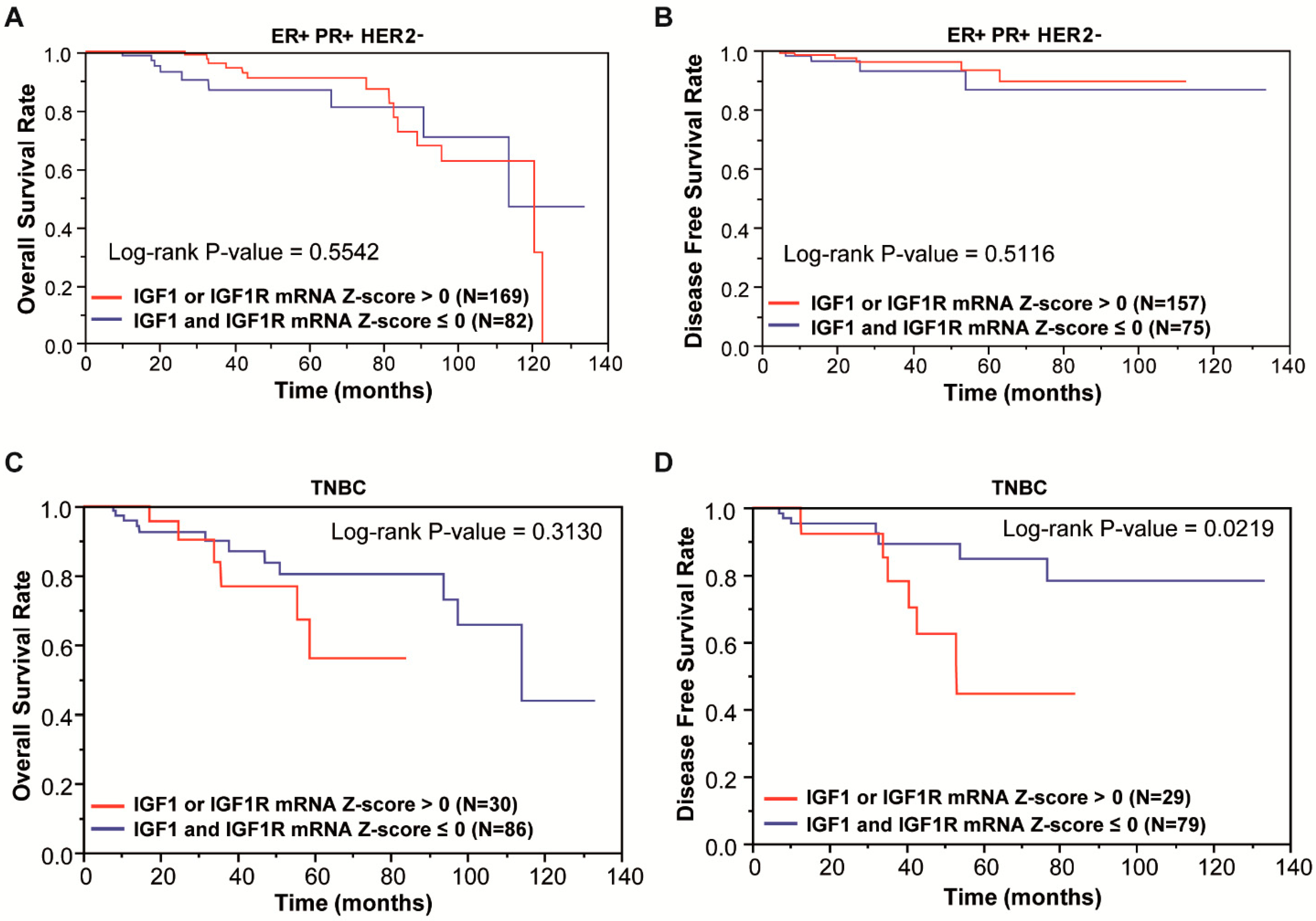
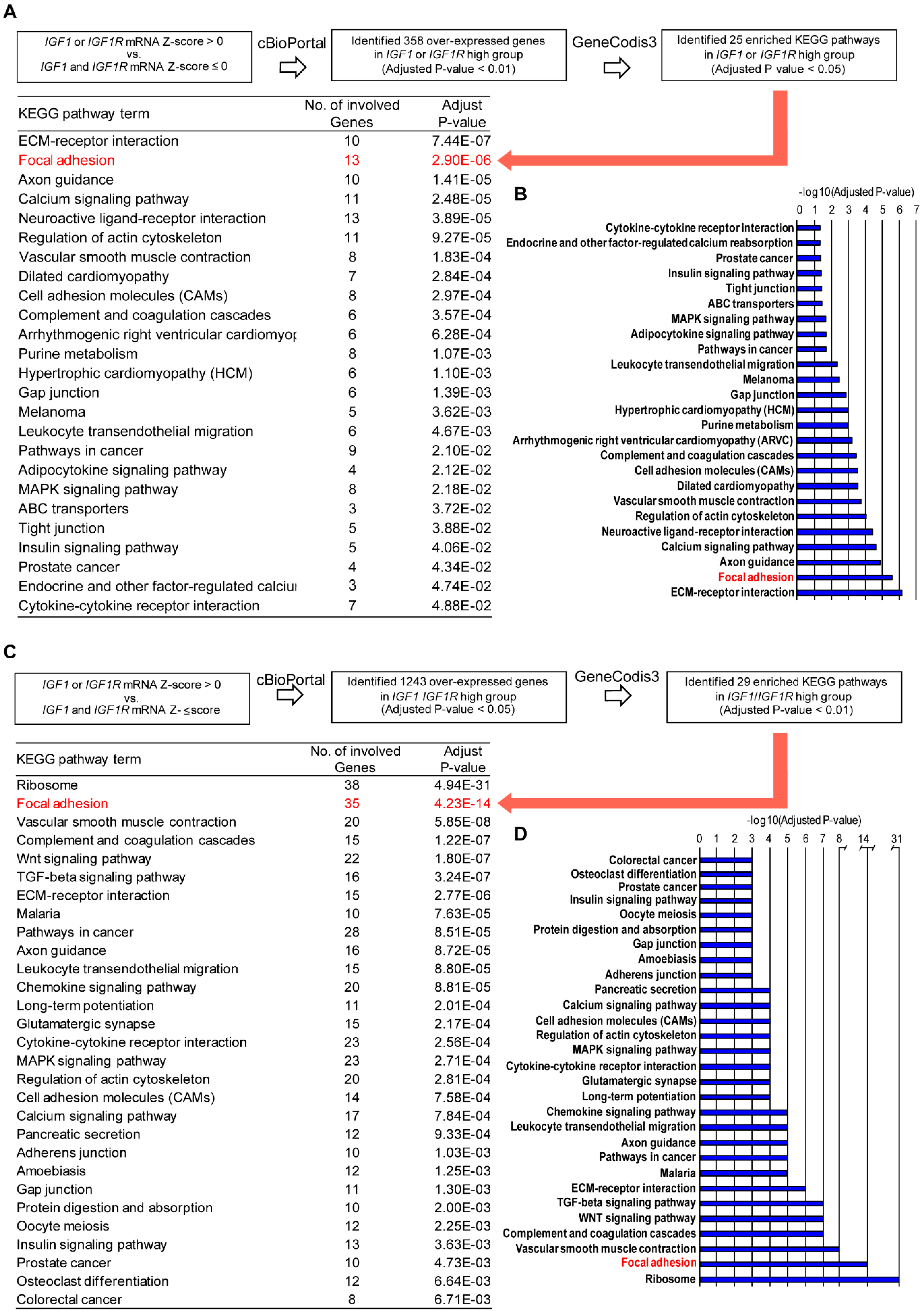
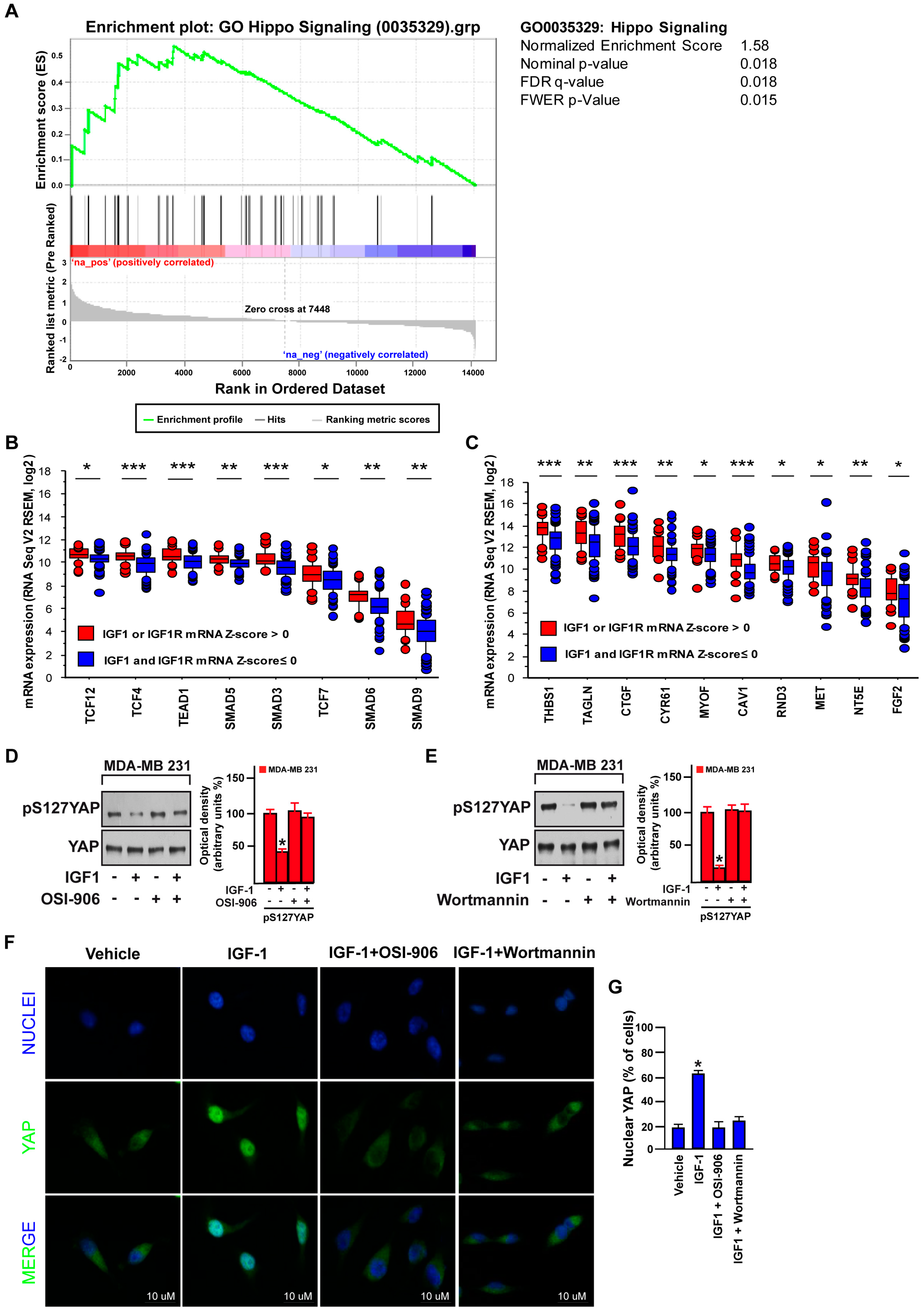
3.1. Focal Adhesion Is a Prominent Enriched KEGG Pathway Linked to the Expression of IGF-1/IGF-1R in TNBC
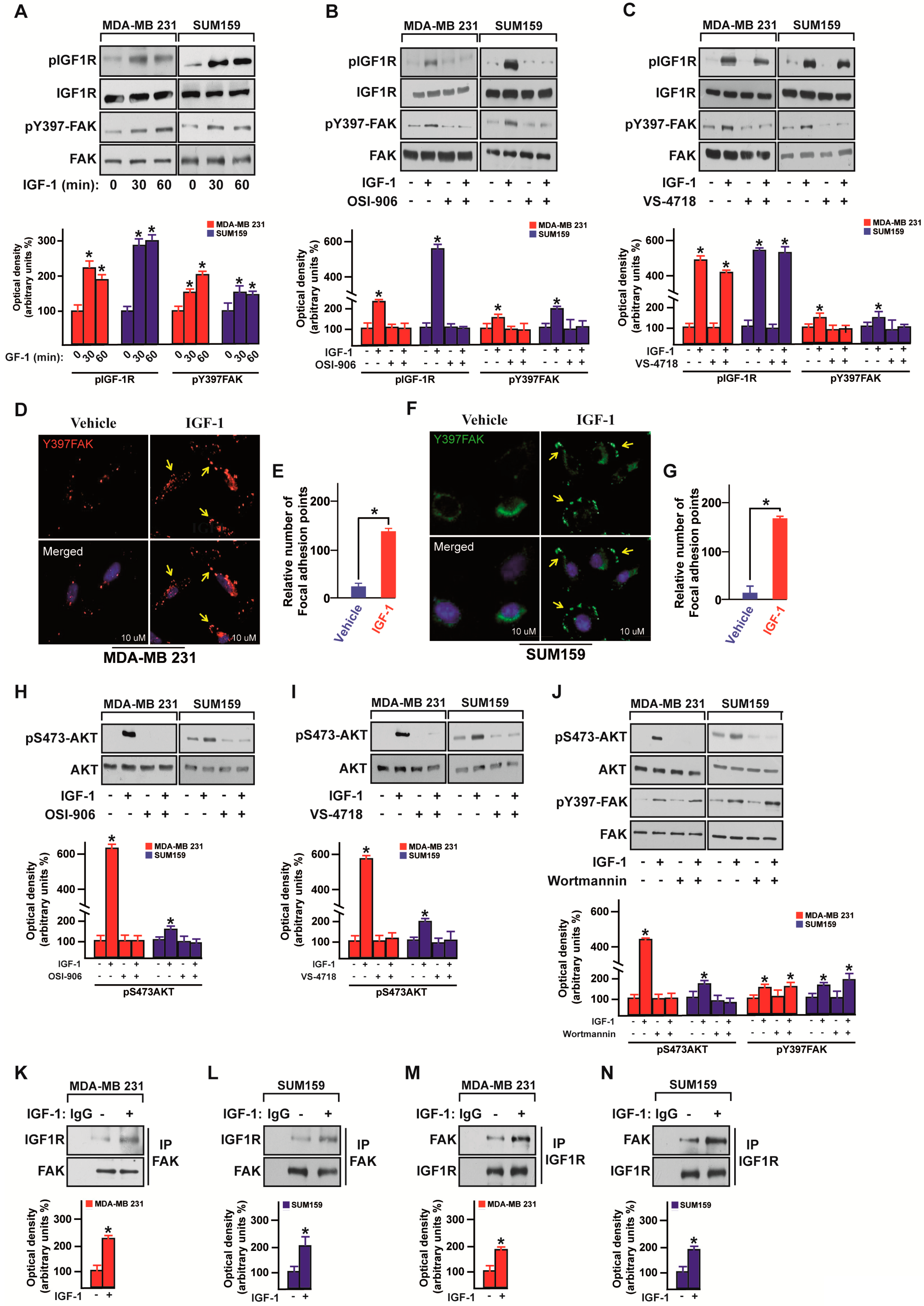
3.2. IGF-1/IGF-1R System Triggers FAK Activation
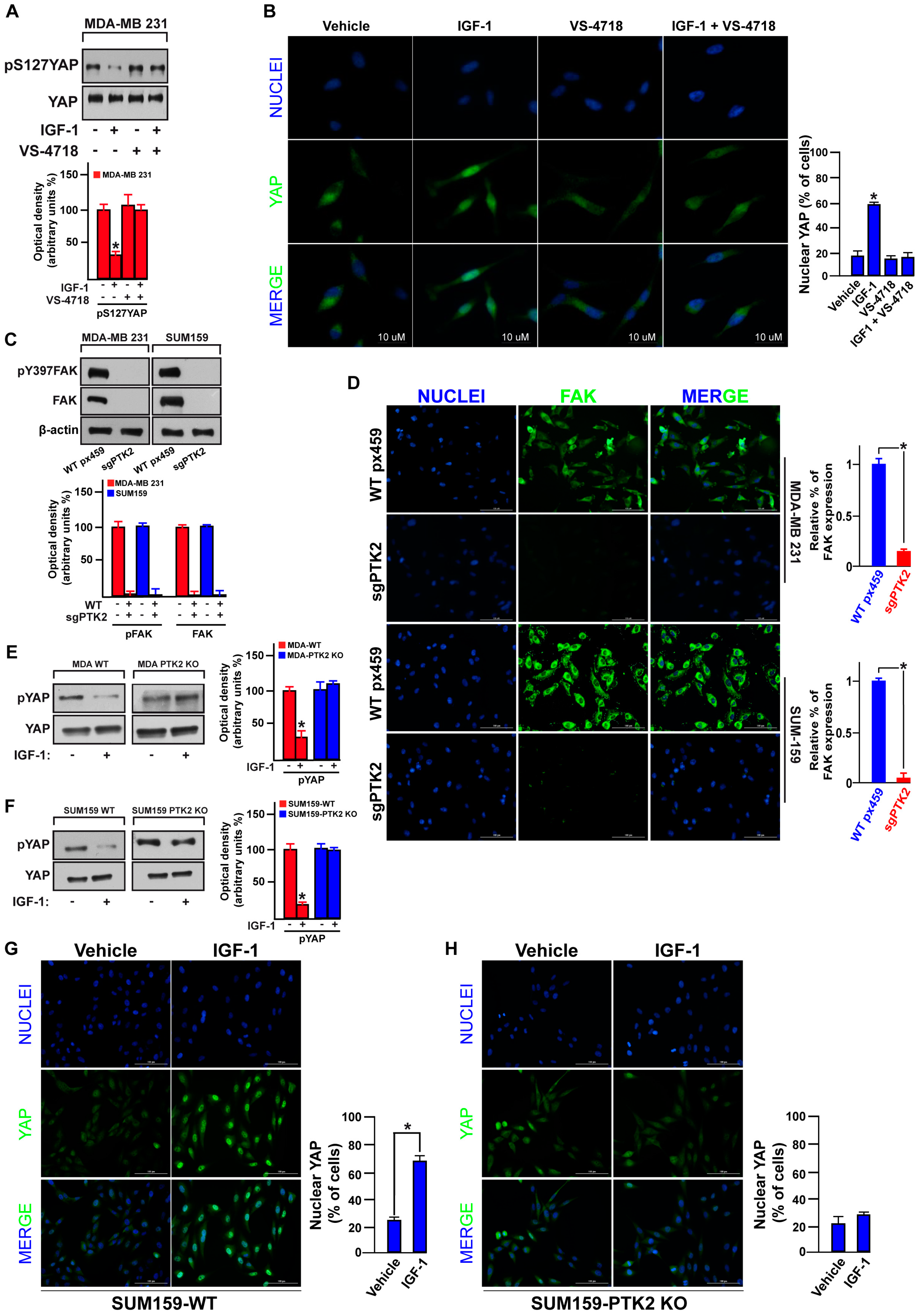
3.3. FAK Is Involved in the IGF-1/IGF-1R-Initiated YAP Activation
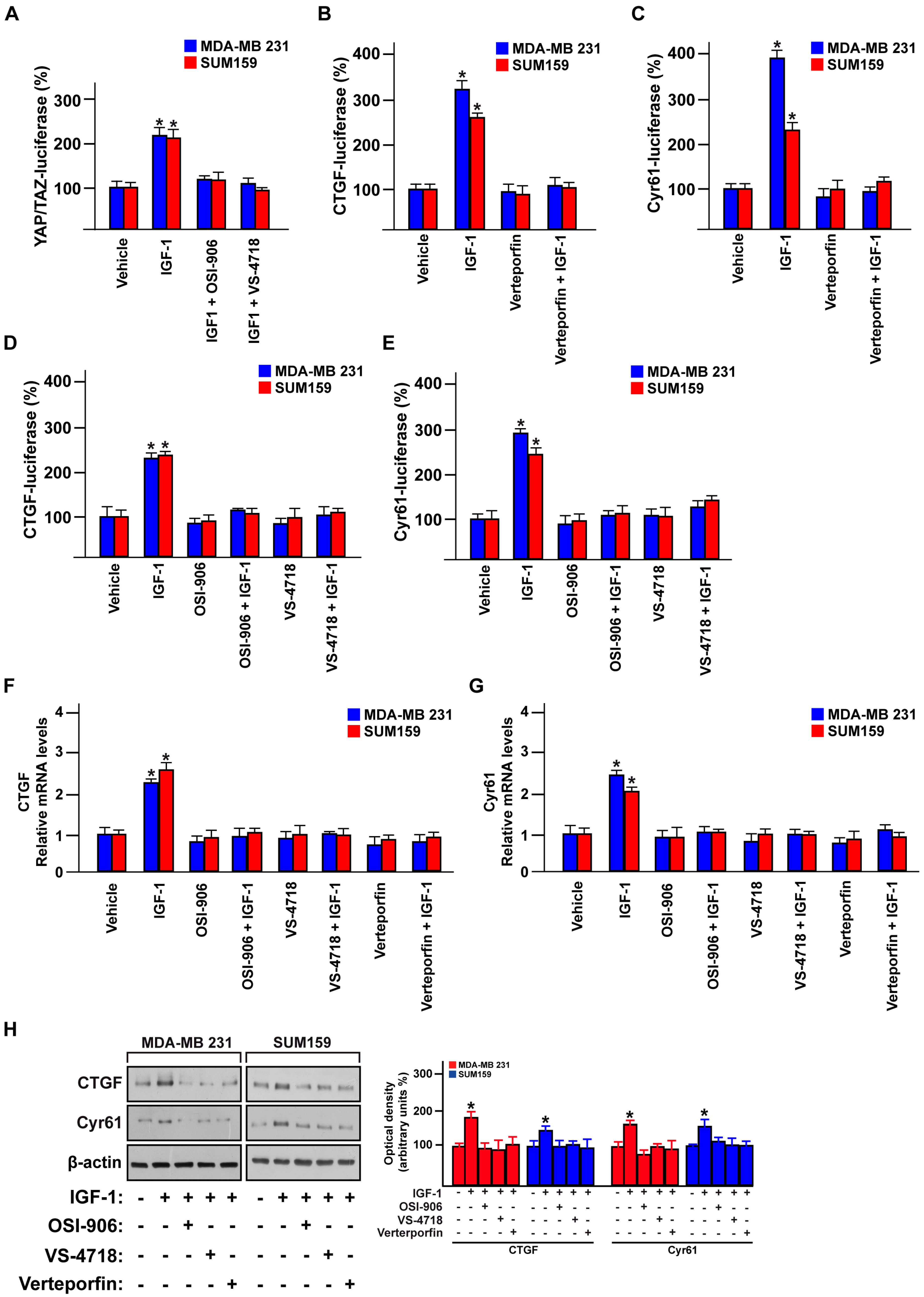
3.4. FAK Is Involved in the YAP-Dependent Transcriptional Activity Induced by IGF-1/IGF-1R
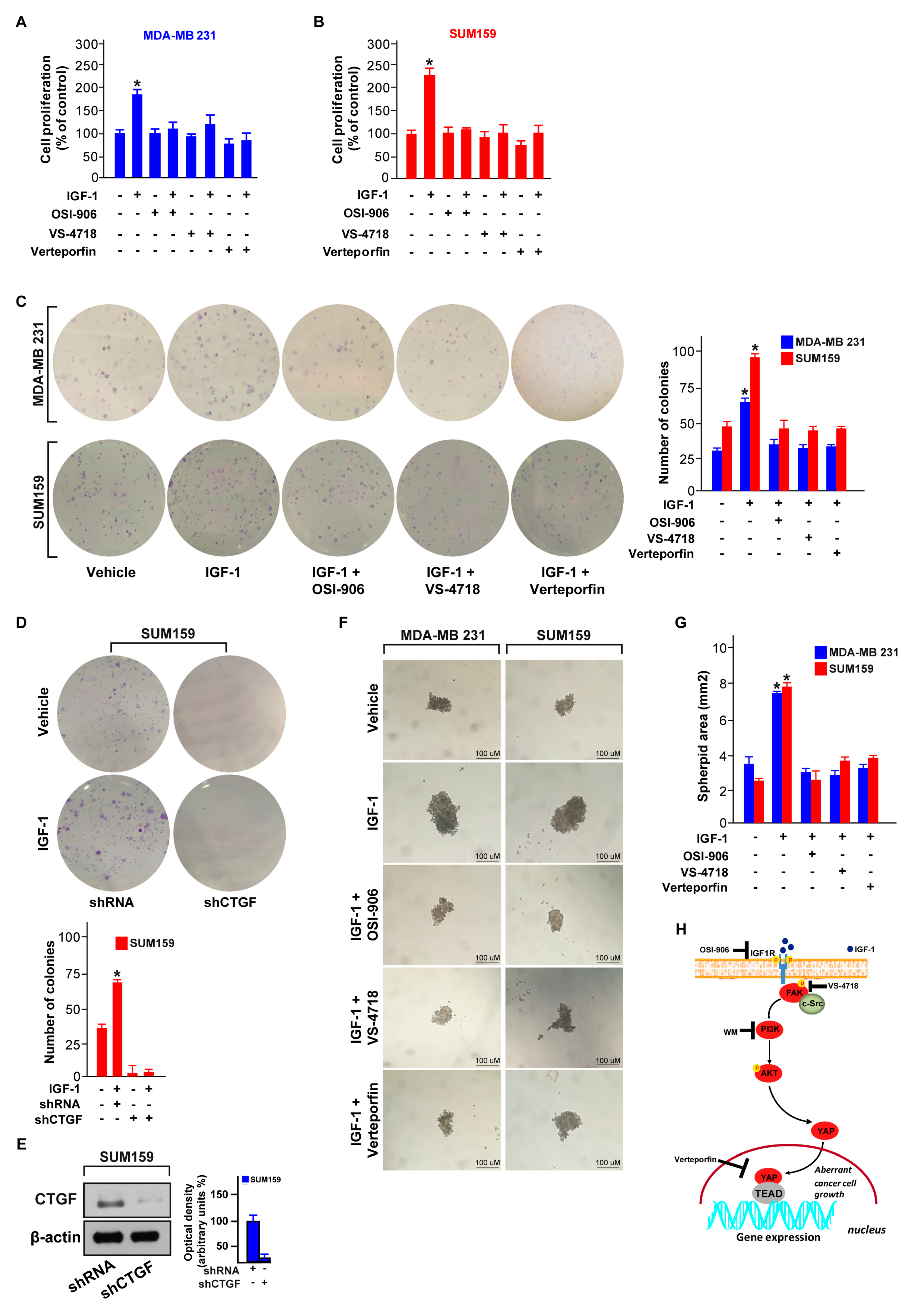
3.5. FAK and YAP Contribute to the Growth Responses Induced by IGF-1/IGF-1R
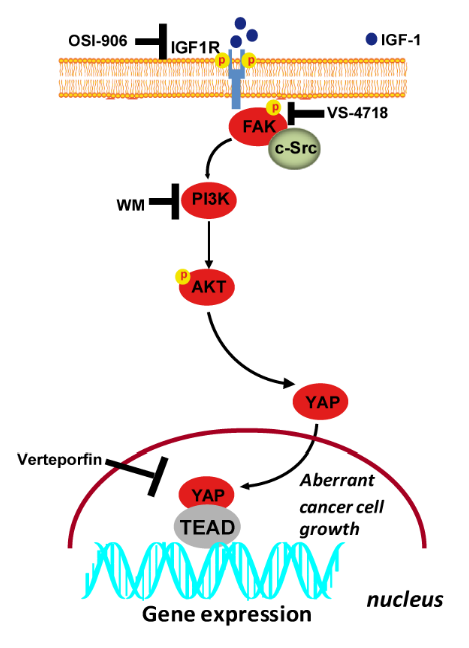
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| TNBC | triple-negative breast cancer |
| TCGA | The Cancer Genome Atlas |
| METABRIC | Molecular Taxonomy of Breast Cancer International Consortium |
| OS | overall survival |
| DFS | disease free survival |
| GSEA | gene set enrichment analysis |
| IGF-1 | insulin-like growth factor-1 |
| IGF-1R | insulin-like growth factor-1 receptor |
| IR | insulin receptor |
| PI3K | phosphatidylinositol 3-kinase |
| AKT | protein kinase B |
| FAK | focal adhesion kinase |
| YAP | yes-associated protein |
| TEAD | TEA domain family member |
| CRISPR | clustered regularly interspaced short palindromic repeats |
| sgRNA | single guide RNA |
| KO | knock out |
| WT | wild type |
| shRNA | short hairpin RNA |
| CTGF | connective tissue growth factor |
| Cyr61 | cellular communication network factor 1/cysteine-rich angiogenic inducer 61 |
References
- Tharmapalan, P.; Mahendralingam, M.; Berman, H.K.; Khokha, R. Mammary stem cells and progenitors: Targeting the roots of breast cancer for prevention. EMBO J. 2019, 38, 100852. [Google Scholar] [CrossRef] [PubMed]
- Walsh, E.W.; Keane, M.M.; Wink, D.A.; Callagy, G.; Glynn, S.A. Review of Triple Negative Breast Cancer and the Impact of Inducible Nitric Oxide Synthase on Tumor Biology and Patient Outcomes. Crit Rev. Oncog. 2016, 21, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Garrido-Castro, A.C.; Lin, N.U.; Polyak, K. Insights Into Molecular Classifications of Triple-Negative Breast Cancer: Improving Patient Selection for Treatment. Cancer Discov. 2019, 9, 176–198. [Google Scholar] [CrossRef] [PubMed]
- Nedeljkovic, M.; Damjanovic, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. [Google Scholar] [CrossRef]
- Pollak, M. Insulin and insulin-like growth factor signalling in neoplasia. Nat. Rev. Cancer 2009, 9, 224. [Google Scholar] [CrossRef]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef]
- Werner, H.; Lapkina-Gendler, L.; Achlaug, L.; Nagaraj, K.; Somri, L.; Yaron-Saminsky, D.; Pasmanik-Chor, M.; Sarfstein, R.; Laron, Z.; Yakar, S. Genome-Wide Profiling of Laron Syndrome Patients Identifies Novel Cancer Protection Pathways. Cells 2019, 8, 596. [Google Scholar] [CrossRef]
- Yang, Y.; Yee, D. Targeting insulin and insulin-like growth factor signaling in breast cancer. J. Mammary Gland Biol. Neoplasia 2012, 17, 251–261. [Google Scholar] [CrossRef]
- Crudden, C.; Song, D.; Cismas, S.; Trocmè, E.; Pasca, S.; Calin, G.A.; Girnita, A.; Girnita, L. Below the Surface: IGF-1R Therapeutic Targeting and Its Endocytic Journey. Cells 2019, 8, 1223. [Google Scholar] [CrossRef]
- Sarfstein, R.; Nagaraj, K.; LeRoith, D.; Werner, H. Differential Effects of Insulin and IGF1 Receptors on ERK and AKT Subcellular Distribution in Breast Cancer Cells. Cells 2019, 8, 1499. [Google Scholar] [CrossRef]
- Rosenzweig, S.A.; Atreya, H.S. Defining the pathway to insulin-like growth factor system targeting in cancer. Biochem. Pharmacol. 2010, 80, 1115–1124. [Google Scholar] [CrossRef] [PubMed]
- Mezi, S.; Todi, L.; Orsi, E.; Angeloni, A.; Mancini, P. Involvement of the Src-cortactin pathway in migration induced by IGF-1 and EGF in human breast cancer cells. Int. J. Oncol. 2012, 41, 2128–2138. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.A.; Campbell, C.I.; Gunther, E.J.; Chodosh, L.A.; Petrik, J.J.; Khokha, R.; Moorehead, R.A. Transgenic Overexpression of IGF-IR Disrupts Mammary Ductal Morphogenesis and Induces Tumor Formation. Oncogene 2007, 26, 1636–1644. [Google Scholar] [CrossRef] [PubMed]
- Law, J.H.; Habibi, G.; Hu, K.; Masoudi, H.; Wang, M.Y.; Stratford, A.L.; Park, E.; Gee, J.M.; Finlay, P.; Jones, H.E.; et al. Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res. 2008, 68, 10238–10246. [Google Scholar] [CrossRef]
- Endogenous Hormones and Breast Cancer Collaborative Group; Key, T.J.; Appleby, P.N.; Reeves, G.K.; Roddam, A.W. Insulin-like Growth Factor 1 (IGF1), IGF Binding Protein 3 (IGFBP3), and Breast Cancer Risk: Pooled Individual Data Analysis of 17 Prospective Studies. Lancet Oncol. 2010, 11, 530–542. [Google Scholar] [CrossRef]
- Hartog, H.; Horlings, H.M.; van der Vegt, B.; Kreike, B.; Ajouaou, A.; van de Vijver, M.J.; Marike Boezen, H.; de Bock, G.H.; van der Graaf, W.T.; Wesseling, J. Divergent effects of insulin-like growth factor-1 receptor expression on prognosis of estrogen receptor positive versus triple negative invasive ductal breast carcinoma. Breast Cancer Res. Treat. 2010, 129, 725–736. [Google Scholar] [CrossRef][Green Version]
- Litzenburger, B.C.; Creighton, C.J.; Tsimelzon, A.; Chan, B.T.; Hilsenbeck, S.G.; Wang, T.; Carboni, J.M.; Gottardis, M.M.; Huang, F.; Chang, J.C.; et al. High IGF-IR Activity in Triple-Negative Breast Cancer Cell Lines and Tumorgrafts Correlates With Sensitivity to anti-IGF-IR Therapy. Clin. Cancer Res. 2011, 17, 2314–2327. [Google Scholar] [CrossRef]
- Kleinschmidt, E.G.; Schlaepfer, D.D. Focal Adhesion Kinase Signaling in Unexpected Places. Curr. Opin. Cell Biol. 2017, 45, 24–30. [Google Scholar] [CrossRef]
- Sulzmaier, F.J.; Jean, C.; Schlaepfer, D.D. FAK in Cancer: Mechanistic Findings and Clinical Applications. Nat. Rev. Cancer 2014, 14, 598–610. [Google Scholar] [CrossRef]
- Frame, M.C.; Patel, H.; Serrels, B.; Lietha, D.; Eck, M.J. The FERM Domain: Organizing the Structure and Function of FAK. Nat. Rev. Mol. Cell Biol. 2010, 11, 802–814. [Google Scholar] [CrossRef]
- Tai, Y.; Chen, L.; Shen, T. Emerging Roles of Focal Adhesion Kinase in Cancer. Biomed. Res. Int. 2015, 2015, 690690. [Google Scholar] [CrossRef] [PubMed]
- Rigiracciolo, D.C.; Santolla, M.F.; Lappano, R.; Vivacqua, A.; Cirillo, F.; Galli, G.R.; Talia, M.; Muglia, L.; Pellegrino, M.; Nohata, N.; et al. Focal Adhesion Kinase (FAK) Activation by Estrogens Involves GPER in Triple-Negative Breast Cancer Cells. J. Exp. Clin. Cancer Res. 2019, 38, 58. [Google Scholar] [CrossRef] [PubMed]
- Shen, M.; Jiang, Y.; Wei, Y.; Ell, B.; Sheng, X.; Esposito, M.; Kang, J.; Hang, X.; Zheng, H.; Rowicki, M.; et al. Tinagl1 Suppresses Triple-Negative Breast Cancer Progression and Metastasis by Simultaneously Inhibiting Integrin/FAK and EGFR Signaling. Cancer Cell 2019, 35, 64–80.e7. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Chen, Q.; Gutkind, J.S. Oncotargeting G proteins: The Hippo in the room. Oncotarget 2014, 5, 10997–10999. [Google Scholar] [CrossRef]
- Pan, D. The Hippo Signaling Pathway in Development and Cancer. Dev. Cell 2010, 19, 491–505. [Google Scholar] [CrossRef]
- Huh, H.D.; Kim, D.H.; Jeong, H.; Park, H.W. Regulation of TEAD Transcription Factors in Cancer Biology. Cells 2019, 8, 600. [Google Scholar] [CrossRef]
- Moroishi, T.; Hansen, C.G.; Guan, K. The Emerging Roles of YAP and TAZ in Cancer. Nat. Rev. Cancer 2015, 15, 73–79. [Google Scholar] [CrossRef]
- Yu, F.; Zhao, B.; Guan, K. Hippo Pathway in Organ Size Control, Tissue Homeostasis, and Cancer. Cell 2015, 163, 811–828. [Google Scholar] [CrossRef]
- Wu, L.; Yang, X. Targeting the Hippo Pathway for Breast Cancer Therapy. Cancers 2018, 10, 422. [Google Scholar] [CrossRef]
- Andrade, D.; Mehta, M.; Griffith, J.; Panneerselvam, J.; Srivastava, A.; Kim, T.D.; Janknecht, R.; Herman, T.; Ramesh, R.; Munshi, A. YAP1 inhibition radiosensitizes triple negative breast cancer cells by targeting the DNA damage response and cell survival pathways. Oncotarget 2017, 8, 98495–98508. [Google Scholar] [CrossRef]
- Goto, Y.; Ando, T.; Izumi, H.; Feng, X.; Arang, N.; Gilardi, M.; Wang, Z.; Ando, K.; Gutkind, J.S. Muscarinic receptors promote castration-resistant growth of prostate cancer through a FAK-YAP signaling axis. Oncogene 2020. [Google Scholar] [CrossRef] [PubMed]
- Cancer Genome Atlas Network. Comprehensive Molecular Portraits of Human Breast Tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Pereira, B.; Chin, S.; Rueda, O.M.; Moen Vollan, H.; Provenzano, E.; Bardwell, H.A.; Pugh, M.; Jones, L.; Russell, R.; Sammut, S.; et al. The Somatic Mutation Profiles of 2,433 Breast Cancers Refines Their Genomic and Transcriptomic Landscapes. Nat. Commun. 2016, 7, 11479. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative Analysis of Complex Cancer Genomics and Clinical Profiles Using the cBioPortal. Sci Signal. 2013, 6, pl1. [Google Scholar] [CrossRef] [PubMed]
- Nogales-Cadenas, R.; Carmona-Saez, P.; Vazquez, M.; Vicente, C.; Yang, X.; Tirado, F.; Carazo, J.M.; Pascual-Montano, A. GeneCodis: Interpreting Gene Lists Through Enrichment Analysis and Integration of Diverse Biological Information. Nucleic Acids Res. 2009, 37, W317–W322. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New Perspectives on Genomes, Pathways, Diseases and Drugs. Nucleic Acids Res. 2017, 45, 353–361. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [PubMed]
- Lappano, R.; Rosano, C.; Santolla, M.F.; Pupo, M.; De Francesco, E.M.; De Marco, P.; Ponassi, M.; Spallarossa, A.; Ranise, A.; Maggiolini, M. Two novel GPER agonists induce gene expression changes and growth effects in cancer cells. Curr. Cancer Drug Targets 2012, 12, 531–542. [Google Scholar] [CrossRef]
- Feng, X.; Arang, N.; Rigiracciolo, D.C.; Lee, J.S.; Yeerna, H.; Wang, Z.; Lubrano, S.; Kishore, A.; Pachter, J.A.; König, G.M.; et al. A Platform of Synthetic Lethal Gene Interaction Networks Reveals That the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway Through FAK. Cancer Cell 2019, 35, 457–472. [Google Scholar] [CrossRef]
- Chaqour, B.; Yang, R.; Sha, Q. Mechanical stretch modulates the promoter activity of the profibrotic factor CCN2 through increased actin polymerization and NF-kappaB activation. J. Biol. Chem. 2006, 281, 20608–20622. [Google Scholar] [CrossRef]
- Zhao, Y.; Montminy, T.; Azad, T.; Lightbody, E.; Hao, Y.; SenGupta, S.; Asselin, E.; Nicol, C.; Yang, X. PI3K Positively Regulates YAP and TAZ in Mammary Tumorigenesis Through Multiple Signaling Pathways. Mol. Cancer Res. 2018, 16, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Pandey, D.P.; Lappano, R.; Albanito, L.; Madeo, A.; Maggiolini, M.; Picard, D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J. 2009, 28, 523–532. [Google Scholar] [CrossRef] [PubMed]
- Farabaugh, S.M.; Boone, D.N.; Lee, A.V. Role of IGF1R in Breast Cancer Subtypes, Stemness, and Lineage Differentiation. Front. Endocrinol. 2015, 6, 59. [Google Scholar] [CrossRef] [PubMed]
- Soljic, M.; Mrklic, I.; Tomic, S.; Omrcen, T.; Sutalo, N.; Bevanda, M.; Vrdoljak, E. Prognostic value of vitamin D receptor and insulin-like growth factor receptor 1 expression in triple-negative breast cancer. J. Clin. Pathol. 2017, 71, 34–39. [Google Scholar] [CrossRef]
- Davison, Z.; de Blacquière, G.E.; Westley, B.R.; May, F.E.B. Insulin-like Growth Factor-Dependent Proliferation and Survival of Triple-Negative Breast Cancer Cells: Implications for Therapy. Neoplasia 2011, 13, 504–515. [Google Scholar] [CrossRef]
- Creighton, C.J.; Casa, A.; Lazard, Z.; Huang, S.; Tsimelzon, A.; Hilsenbeck, S.G.; Osborne, C.K.; Lee, A.V. Insulin-like Growth factor-I Activates Gene Transcription Programs Strongly Associated With Poor Breast Cancer Prognosis. J. Clin. Oncol. 2008, 26, 4078–4085. [Google Scholar] [CrossRef]
- Yerushalmi, R.; Gelmon, K.A.; Leung, S.; Gao, D.; Cheang, M.; Pollak, M.; Turashvili, G.; Gilks, B.C.; Kennecke, H. Insulin-like Growth Factor Receptor (IGF-1R) in Breast Cancer Subtypes. Breast Cancer Res. Treat. 2012, 132, 131–142. [Google Scholar] [CrossRef]
- Zhai, J.; Lin, H.; Nie, Z.; Wu, J.; Cañete-Soler, R.; Schlaepfer, W.W.; Schlaepfer, D.D. Direct Interaction of Focal Adhesion Kinase With p190RhoGEF. J. Biol. Chem. 2003, 278, 24865–24873. [Google Scholar] [CrossRef]
- Sanabria-Figueroa, E.; Donnelly, S.M.; Foy, K.C.; Buss, M.C.; Castellino, R.C.; Paplomata, E.; Taliaferro-Smith, L.; Kaumaya, P.T.P.; Nahta, R. Insulin-like Growth factor-1 Receptor Signaling Increases the Invasive Potential of Human Epidermal Growth Factor Receptor 2-overexpressing Breast Cancer Cells via Src-focal Adhesion Kinase and Forkhead Box Protein M1. Mol. Pharmacol. 2015, 87, 150–161. [Google Scholar] [CrossRef]
- Taliaferro-Smith, L.; Oberlick, E.; Liu, T.; McGlothen, T.; Alcaide, T.; Tobin, R.; Donnelly, S.; Commander, R.; Kline, E.; Purnachandra Nagaraju, G.; et al. FAK Activation Is Required for IGF1R-mediated Regulation of EMT, Migration, and Invasion in Mesenchymal Triple Negative Breast Cancer Cells. Oncotarget 2015, 6, 4757–4772. [Google Scholar] [CrossRef]
- Hakuno, F.; Takahashi, S. IGF1 Receptor Signaling Pathways. J. Mol. Endocrinol. 2018, 61, 69–86. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Guan, K. The Hippo Pathway: Regulators and Regulations. Genes Dev. 2013, 27, 355–371. [Google Scholar] [CrossRef] [PubMed]
- Luo, J.; Yu, F. GPCR-Hippo Signaling in Cancer. Cells 2019, 8, 426. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Wang, S.; Wang, Z.; Feng, X.; Liu, P.; Lv, X.; Li, F.; Yu, F.; Sun, Y.; Yuan, H.; et al. Estrogen Regulates Hippo Signaling via GPER in Breast Cancer. J. Clin. Investig. 2015, 125, 2123–2135. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, G.; Ruggeri, N.; Zannini, A.; Ingallina, E.; Bertolio, R.; Marotta, C.; Neri, C.; Cappuzzello, E.; Forcato, M.; Rosato, A.; et al. Glucocorticoid Receptor Signalling Activates YAP in Breast Cancer. Nat. Commun. 2017, 8, 14073. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Kim, N.; Gumbiner, B.M. Regulation of Hippo Pathway by Mitogenic Growth Factors via Phosphoinositide 3-kinase and Phosphoinositide-Dependent kinase-1. Proc. Natl. Acad. Sci. USA 2013, 110, 2569–2574. [Google Scholar] [CrossRef]
- Zhu, H.; Wang, D.D.; Yuan, T.; Yan, F.J.; Zeng, C.M.; Dai, X.Y.; Chen, Z.B.; Chen, Y.; Zhou, T.; Fan, G.H.; et al. Multikinase Inhibitor CT-707 Targets Liver Cancer by Interrupting the Hypoxia-Activated IGF-1R-YAP Axis. Cancer Res. 2018, 78, 3995–4006. [Google Scholar] [CrossRef]
- Zhao, B.; Wei, X.; Li, W.; Udan, R.S.; Yang, Q.; Kim, J.; Xie, J.; Ikenoue, T.; Yu, J.; Li, L.; et al. Inactivation of YAP Oncoprotein by the Hippo Pathway Is Involved in Cell Contact Inhibition and Tissue Growth Control. Genes Dev. 2007, 21, 2747–2761. [Google Scholar] [CrossRef]
- Kim, N.; Gumbiner, B.M. Adhesion to Fibronectin Regulates Hippo Signaling via the FAK-Src-PI3K Pathway. J. Cell Biol. 2015, 210, 503–515. [Google Scholar] [CrossRef]
- Kim, M.; Kim, T.; Johnson, R.L.; Lim, D. Transcriptional Co-Repressor Function of the Hippo Pathway Transducers YAP and TAZ. Cell Rep. 2015, 11, 270–282. [Google Scholar] [CrossRef]
- Kim, M.K.; Jang, J.W.; Bae, S.C. DNA binding partners of YAP/TAZ. BMB Rep. 2018, 51, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Li, M.X.; Jin, L.T.; Wang, T.J.; Feng, Y.J.; Pan, C.P.; Zhao, D.M.; Shao, J. Identification of potential core genes in triple negative breast cancer using bioinformatics analysis. Onco Targets Ther. 2018, 11, 4105–4112. [Google Scholar] [CrossRef] [PubMed]
- Mehanna, J.; Haddad, F.G.; Eid, R.; Lambertini, M.; Kourie, H.R. Triple-negative breast cancer: Current perspective on the evolving therapeutic landscape. Int. J. Womens Health 2019, 11, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Lerma, E.; Peiro, G.; Ramón, T.; Fernandez, S.; Martinez, D.; Pons, C.; Muñoz, F.; Sabate, J.M.; Alonso, C.; Ojeda, B.; et al. Immunohistochemical Heterogeneity of Breast Carcinomas Negative for Estrogen Receptors, Progesterone Receptors and Her2/neu (Basal-Like Breast Carcinomas). Mod. Pathol. 2007, 20, 1200–1207. [Google Scholar] [CrossRef]
- Lee, B.Y.; Timpson, P.; Horvath, L.G.; Daly, R.J. FAK Signaling in Human Cancer as a Target for Therapeutics. Pharmacol. Ther. 2015, 146, 132–149. [Google Scholar] [CrossRef] [PubMed]
- Owens, L.V.; Xu, L.; Craven, R.J.; Dent, G.A.; Weiner, T.M.; Kornberg, L.; Liu, E.T.; Cance, W.G. Overexpression of the Focal Adhesion Kinase (p125FAK) in Invasive Human Tumors. Cancer Res. 1995, 55, 2752–2755. [Google Scholar]
- Pan, M.; Wu, C.; Kan, J.; Li, Q.; Chang, S.; Wu, C.; Li, C.; Yang, F.; Hou, M.; Yip, H.; et al. Impact of FAK Expression on the Cytotoxic Effects of CIK Therapy in Triple-Negative Breast Cancer. Cancers 2020, 12, 94. [Google Scholar] [CrossRef]
- Guan, J.L. Role of Focal Adhesion Kinase in Integrin Signaling. Int. J. Biochem. Cell Biol. 1997, 29, 1085–1096. [Google Scholar] [CrossRef]
- Kornberg, L.J. Focal Adhesion Kinase and Its Potential Involvement in Tumor Invasion and Metastasis. Head Neck 1998, 20, 745–752. [Google Scholar] [CrossRef]
- Andersson, S.; D’Arcy, P.; Larsson, O.; Sehat, B. Focal adhesion kinase (FAK) activates and stabilizes IGF-1 receptor. Biochem. Biophys. Res. Commun. 2009, 387, 36–41. [Google Scholar] [CrossRef]
- Watanabe, N.; Takaoka, M.; Sakurama, K.; Tomono, Y.; Hatakeyama, S.; Ohmori, O.; Motoki, T.; Shirakawa, Y.; Yamatsuji, T.; Haisa, M.; et al. Dual tyrosine kinase inhibitor for focal adhesion kinase and insulin-like growth factor-I receptor exhibits anticancer effect in esophageal adenocarcinoma in vitro and in vivo. Clin. Cancer Res. 2008, 14, 4631–4639. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.; Choi, S.; Mo, J. Role of the Hippo Pathway in Fibrosis and Cancer. Cells 2019, 8, 468. [Google Scholar] [CrossRef] [PubMed]
- Overholtzer, M.; Zhang, J.; Smolen, G.A.; Muir, B.; Li, W.; Sgroi, D.C.; Deng, C.; Brugge, J.S.; Haber, D.A. Transforming Properties of YAP, a Candidate Oncogene on the Chromosome 11q22 Amplicon. Proc. Natl. Acad. Sci. USA 2006, 103, 12405–12410. [Google Scholar] [CrossRef] [PubMed]
- Lamar, J.M.; Stern, P.; Liu, H.; Schindler, J.W.; Jiang, Z.; Hynes, R.O. The Hippo Pathway Target, YAP, Promotes Metastasis Through Its TEAD-interaction Domain. Proc. Natl. Acad. Sci. USA 2012, 109, 2441–2450. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Zhang, N.; Gray, R.S.; Li, H.; Ewald, A.J.; Zahnow, C.A.; Pan, D. A Temporal Requirement for Hippo Signaling in Mammary Gland Differentiation, Growth, and Tumorigenesis. Genes Dev. 2014, 28, 432–437. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.K.; Du, W.; Shelton, S.J.; Oldham, M.C.; DiPersio, C.M.; Klein, O.D. An FAK-YAP-mTOR Signaling Axis Regulates Stem Cell-Based Tissue Renewal in Mice. Cell Stem Cell 2017, 21, 91–106. [Google Scholar] [CrossRef]
- Lachowski, D.; Cortes, E.; Robinson, B.; Rice, A.; Rombouts, K.; Del Río Hernández, A.E. FAK Controls the Mechanical Activation of YAP, a Transcriptional Regulator Required for Durotaxis. FASEB J. 2018, 32, 1099–1107. [Google Scholar] [CrossRef]
- Zhang, H.; Kathawala, R.J.; Wang, Y.J.; Zhang, Y.K.; Patel, A.; Shukla, S.; Robey, R.W.; Talete, T.T.; Ashby, C.R.; Ambudkar, S.V.; et al. Linsitinib (OSI-906) antagonizes ATP-binding cassette subfamily G member 2 and subfamily C member 10-mediated drug resistance. Int. J. Biochem. Cell Biol. 2014, 51, 11–119. [Google Scholar] [CrossRef]
- Ji, N.; Yang, Y.; Cai, C.Y.; Lei, Z.N.; Wang, J.Q.; Gupta, P.; Teng, Q.X.; Chen, Z.S.; Kong, D.; Yang, D.-H. VS-4718 antagonizes multidrug resistance in ABCB1 and ABCG2-overexpressing cancer cells by inhibiting the efflux function of ABC transporters. Front. Pharmacol. 2018, 9, 1236. [Google Scholar] [CrossRef]
- Gavini, J.; Dommann, N.; Jakob, M.O.; Keogh, A.; Bouchez, L.C.; Karkampouna, S.; Kruithof-de Julio, M.; Medova, M.; Zimmer, Y.; Schàfli, M.; et al. Verteporfin-induced lysosomal compartment dysregulation potentiates the effect of sorafenib in hepatocellular carcinoma. Cell Death Dis. 2019, 10, 749. [Google Scholar] [CrossRef]
- Buck, E.; Gokhale, P.C.; Koujak, S.; Brown, E.; Eyzaguirre, A.; Tao, N.; Rosenfeld-Franklin, M.; Lerner, L.; Chiu, M.I.; Wild, R.; et al. Compensatory insulin receptor (IR) activation on inhibition of insulin-like growth factor-1 receptor (IGF-1R): Rationale for cotargeting IGF-1R and IR in cancer. Mol. Cancer Ther. 2010, 9, 2652–2664. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rigiracciolo, D.C.; Nohata, N.; Lappano, R.; Cirillo, F.; Talia, M.; Scordamaglia, D.; Gutkind, J.S.; Maggiolini, M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells 2020, 9, 1010. https://doi.org/10.3390/cells9041010
Rigiracciolo DC, Nohata N, Lappano R, Cirillo F, Talia M, Scordamaglia D, Gutkind JS, Maggiolini M. IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells. 2020; 9(4):1010. https://doi.org/10.3390/cells9041010
Chicago/Turabian StyleRigiracciolo, Damiano Cosimo, Nijiro Nohata, Rosamaria Lappano, Francesca Cirillo, Marianna Talia, Domenica Scordamaglia, J. Silvio Gutkind, and Marcello Maggiolini. 2020. "IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells" Cells 9, no. 4: 1010. https://doi.org/10.3390/cells9041010
APA StyleRigiracciolo, D. C., Nohata, N., Lappano, R., Cirillo, F., Talia, M., Scordamaglia, D., Gutkind, J. S., & Maggiolini, M. (2020). IGF-1/IGF-1R/FAK/YAP Transduction Signaling Prompts Growth Effects in Triple-Negative Breast Cancer (TNBC) Cells. Cells, 9(4), 1010. https://doi.org/10.3390/cells9041010