The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases

Abstract
1. Introduction
2. The Role of Prph2 in Photoreceptor Outer Segment Morphogenesis
3. Insight from Mouse Models into the Pathophysiology of Prph2 Mutations
3.1. Transgenic Mouse Models of Prph2 Mutations
3.1.1. Prph2R172W
3.1.2. Prph2C214S
3.1.3. Prph2P216L
3.1.4. Prph2L185P
3.2. Prph2 Knockin Mouse Models
3.2.1. Prph2307/+ and Prph2307/307
3.2.2. Prph2C213Y/+ and Prph2C213Y/C213Y
3.2.3. Prph2Y141C/+ and Prph2Y141C/Y141C
3.2.4. Prph2K153Δ/+ and Prph2K153Δ/K153Δ
3.2.5. Prph2N229S/+ and Prph2N229S/N229
4. Gene Therapy of PRPH2 Mutations
5. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Hemler, M.E. Tetraspanin functions and associated microdomains. Nat. Rev. Mol. Cell Biol. 2005, 6, 801–811. [Google Scholar] [CrossRef]
- Levy, S.; Shoham, T. The tetraspanin web modulates immune-signalling complexes. Nat. Rev. Immunol. 2005, 5, 136–148. [Google Scholar] [CrossRef] [PubMed]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef]
- Van Deventer, S.J.; Dunlock, V.E.; van Spriel, A.B. Molecular interactions shaping the tetraspanin web. Biochem. Soc. Trans. 2017, 45, 741–750. [Google Scholar] [CrossRef] [PubMed]
- Boucheix, C.; Rubinstein, E. Tetraspanins. Cell. Mol. Life Sci. Cmls 2001, 58, 1189–1205. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Tetraspanin proteins mediate cellular penetration, invasion, and fusion events and define a novel type of membrane microdomain. Annu. Rev. Cell Dev. Biol. 2003, 19, 397–422. [Google Scholar] [CrossRef]
- Kitadokoro, K.; Bordo, D.; Galli, G.; Petracca, R.; Falugi, F.; Abrignani, S.; Grandi, G.; Bolognesi, M. CD81 extracellular domain 3D structure: Insight into the tetraspanin superfamily structural motifs. Embo. J. 2001, 20, 12–18. [Google Scholar] [CrossRef]
- Murru, L.; Moretto, E.; Martano, G.; Passafaro, M. Tetraspanins shape the synapse. Mol. Cell. Neurosci. 2018, 91, 76–81. [Google Scholar] [CrossRef]
- Stipp, C.S.; Kolesnikova, T.V.; Hemler, M.E. Functional domains in tetraspanin proteins. Trends Biochem. Sci. 2003, 28, 106–112. [Google Scholar] [CrossRef]
- DeSalle, R.; Mares, R.; Garcia-España, A. Evolution of cysteine patterns in the large extracellular loop of tetraspanins from animals, fungi, plants and single-celled eukaryotes. Mol. Phylogenetics Evol. 2010, 56, 486–491. [Google Scholar] [CrossRef]
- Yang, X.; Kovalenko, O.V.; Tang, W.; Claas, C.; Stipp, C.S.; Hemler, M.E. Palmitoylation supports assembly and function of integrin-tetraspanin complexes. J. Cell Biol. 2004, 167, 1231–1240. [Google Scholar] [CrossRef]
- Skubitz, K.M.; Campbell, K.D.; Skubitz, A.P. CD63 associates with CD11/CD18 in large detergent-resistant complexes after translocation to the cell surface in human neutrophils. Febs Lett. 2000, 469, 52–56. [Google Scholar] [CrossRef]
- Claas, C.; Stipp, C.S.; Hemler, M.E. Evaluation of prototype transmembrane 4 superfamily protein complexes and their relation to lipid rafts. J. Biol. Chem. 2001, 276, 7974–7984. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Specific tetraspanin functions. J. Cell Biol. 2001, 155, 1103–1107. [Google Scholar] [CrossRef] [PubMed]
- Molday, R.S.; Hicks, D.; Molday, L. Peripherin. A rim-specific membrane protein of rod outer segment discs. Invest. Ophthalmol. Vis. Sci. 1987, 28, 50–61. [Google Scholar] [PubMed]
- Cheng, T.; Peachey, N.S.; Li, S.; Goto, Y.; Cao, Y.; Naash, M.I. The effect of peripherin/rds haploinsufficiency on rod and cone photoreceptors. J. Neurosci. Off. J. Soc. Neurosci. 1997, 17, 8118–8128. [Google Scholar] [CrossRef]
- Arikawa, K.; Molday, L.L.; Molday, R.S.; Williams, D.S. Localization of peripherin/rds in the disk membranes of cone and rod photoreceptors: Relationship to disk membrane morphogenesis and retinal degeneration. J. Cell Biol. 1992, 116, 659–667. [Google Scholar] [CrossRef] [PubMed]
- Wrigley, J.D.; Ahmed, T.; Nevett, C.L.; Findlay, J.B. Peripherin/rds influences membrane vesicle morphology. Implications for retinopathies. J. Biol. Chem. 2000, 275, 13191–13194. [Google Scholar] [CrossRef]
- Boesze-Battaglia, K. Fusion between retinal rod outer segment membranes and model membranes: Functional assays and role for peripherin/rds. Methods Enzymol. 2000, 316, 65–86. [Google Scholar]
- Conley, S.M.; Cai, X.; Naash, M.I. Nonviral ocular gene therapy: Assessment and future directions. Curr. Opin. Mol. Ther. 2008, 10, 456–463. [Google Scholar]
- Goldberg, A.F.; Molday, R.S. Subunit composition of the peripherin/rds-rom-1 disk rim complex from rod photoreceptors: Hydrodynamic evidence for a tetrameric quaternary structure. Biochemistry 1996, 35, 6144–6149. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Q.; Stricker, H.M.; Naash, M.I. Role of the second intradiscal loop of peripherin/rds in homo and hetero associations. Biochemistry 2005, 44, 4897–4904. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Conley, S.M.; Stuck, M.W.; Naash, M.I. Differences in RDS trafficking, assembly and function in cones versus rods: Insights from studies of C150S-RDS. Hum. Mol. Genet. 2010, 19, 4799–4812. [Google Scholar] [CrossRef] [PubMed]
- Bascom, R.A.; Manara, S.; Collins, L.; Molday, R.S.; Kalnins, V.I.; McInnes, R.R. Cloning of the cDNA for a novel photoreceptor membrane protein (rom-1) identifies a disk rim protein family implicated in human retinopathies. Neuron 1992, 8, 1171–1184. [Google Scholar] [CrossRef]
- Kevany, B.M.; Tsybovsky, Y.; Campuzano, I.D.; Schnier, P.D.; Engel, A.; Palczewski, K. Structural and functional analysis of the native peripherin-ROM1 complex isolated from photoreceptor cells. J. Biol. Chem. 2013, 288, 36272–36284. [Google Scholar] [CrossRef]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. PRPH2/RDS and ROM-1: Historical context, current views and future considerations. Prog Retin Eye Res. 2016, 52, 47–63. [Google Scholar] [CrossRef]
- Clarke, G.; Goldberg, A.F.; Vidgen, D.; Collins, L.; Ploder, L.; Schwarz, L.; Molday, L.L.; Rossant, J.; Szel, A.; Molday, R.S.; et al. Rom-1 is required for rod photoreceptor viability and the regulation of disk morphogenesis. Nat. Genet. 2000, 25, 67–73. [Google Scholar] [CrossRef]
- Chakraborty, D.; Ding, X.Q.; Conley, S.M.; Fliesler, S.J.; Naash, M.I. Differential requirements for retinal degeneration slow intermolecular disulfide-linked oligomerization in rods versus cones. Hum. Mol. Genet. 2009, 18, 797–808. [Google Scholar] [CrossRef]
- Boon, C.J.; den Hollander, A.I.; Hoyng, C.B.; Cremers, F.P.; Klevering, B.J.; Keunen, J.E. The spectrum of retinal dystrophies caused by mutations in the peripherin/RDS gene. Prog Retin Eye Res. 2008, 27, 213–235. [Google Scholar] [CrossRef]
- Francis, P.J.; Schultz, D.W.; Gregory, A.M.; Schain, M.B.; Barra, R.; Majewski, J.; Ott, J.; Acott, T.; Weleber, R.G.; Klein, M.L. Genetic and phenotypic heterogeneity in pattern dystrophy. Br. J. Ophthalmol. 2005, 89, 1115–1119. [Google Scholar] [CrossRef]
- Gocho, K.; Akeo, K.; Itoh, N.; Kameya, S.; Hayashi, T.; Katagiri, S.; Gekka, T.; Ohkuma, Y.; Tsuneoka, H.; Takahashi, H. High-Resolution Adaptive Optics Retinal Image Analysis at Early Stage Central Areolar Choroidal Dystrophy With PRPH2 Mutation. Ophthalmic Surg. Lasers Imaging Retin. 2016, 47, 1115–1126. [Google Scholar] [CrossRef] [PubMed]
- Jansen, H.G.; Sanyal, S. Development and degeneration of retina in rds mutant mice: Electron microscopy. J. Comp. Neurol. 1984, 224, 71–84. [Google Scholar] [CrossRef] [PubMed]
- Salinas, R.Y.; Pearring, J.N.; Ding, J.D.; Spencer, W.J.; Hao, Y.; Arshavsky, V.Y. Photoreceptor discs form through peripherin-dependent suppression of ciliary ectosome release. J. Cell Biol. 2017, 216, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Conley, S.M.; Stuck, M.W.; Watson, J.N.; Zulliger, R.; Burnett, J.L.; Naash, M.I. Prph2 initiates outer segment morphogenesis but maturation requires Prph2/Rom1 oligomerization. Hum. Mol. Genet. 2019, 28, 459–475. [Google Scholar] [CrossRef]
- May-Simera, H.; Nagel-Wolfrum, K.; Wolfrum, U. Cilia - The sensory antennae in the eye. Prog. Retin. Eye Res. 2017, 60, 144–180. [Google Scholar] [CrossRef]
- Sorokin, S. Centrioles and the formation of rudimentary cilia by fibroblasts and smooth muscle cells. J. Cell Biol. 1962, 15, 363–377. [Google Scholar] [CrossRef]
- Sedmak, T.; Wolfrum, U. Intraflagellar transport proteins in ciliogenesis of photoreceptor cells. Biol. Cell 2011, 103, 449–466. [Google Scholar] [CrossRef]
- Pearring, J.N.; Salinas, R.Y.; Baker, S.A.; Arshavsky, V.Y. Protein sorting, targeting and trafficking in photoreceptor cells. Prog. Retin. Eye Res. 2013, 36, 24–51. [Google Scholar] [CrossRef]
- Chakraborty, D.; Conley, S.M.; Al-Ubaidi, M.R.; Naash, M.I. Initiation of rod outer segment disc formation requires RDS. PLoS ONE 2014, 9, e98939. [Google Scholar] [CrossRef][Green Version]
- Wang, J.; Silva, M.; Haas, L.A.; Morsci, N.S.; Nguyen, K.C.; Hall, D.H.; Barr, M.M. C. elegans ciliated sensory neurons release extracellular vesicles that function in animal communication. Curr. Biol. 2014, 24, 519–525. [Google Scholar] [CrossRef]
- Cao, M.; Ning, J.; Hernandez-Lara, C.I.; Belzile, O.; Wang, Q.; Dutcher, S.K.; Liu, Y.; Snell, W.J. Uni-directional ciliary membrane protein trafficking by a cytoplasmic retrograde IFT motor and ciliary ectosome shedding. eLife 2015, 4, e05242. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Barr, M.M. Ciliary Extracellular Vesicles: Txt Msg Organelles. Cell. Mol. Neurobiol. 2016, 36, 449–457. [Google Scholar] [CrossRef] [PubMed]
- Nager, A.R.; Goldstein, J.S.; Herranz-Perez, V.; Portran, D.; Ye, F.; Garcia-Verdugo, J.M.; Nachury, M.V. An Actin Network Dispatches Ciliary GPCRs into Extracellular Vesicles to Modulate Signaling. Cell 2017, 168, 252–263.e214. [Google Scholar] [CrossRef] [PubMed]
- Phua, S.C.; Chiba, S.; Suzuki, M.; Su, E.; Roberson, E.C.; Pusapati, G.V.; Setou, M.; Rohatgi, R.; Reiter, J.F.; Ikegami, K.; et al. Dynamic Remodeling of Membrane Composition Drives Cell Cycle through Primary Cilia Excision. Cell 2017, 168, 264–279.e215. [Google Scholar] [CrossRef] [PubMed]
- Boesze-Battaglia, K.; Lamba, O.P.; Napoli, A.A., Jr.; Sinha, S.; Guo, Y.; Boesze-Battagliaa, K.; Stefano, F.P.; Milstein, M.L.; Kimler, V.A.; Ghatak, C.; et al. Fusion between retinal rod outer segment membranes and model membranes: A role for photoreceptor peripherin/rds Peripherin/rds fusogenic function correlates with subunit assembly An inducible amphipathic helix within the intrinsically disordered C terminus can participate in membrane curvature generation by peripherin-2/rds. Biochemistry 1998, 37, 9477–9487. [Google Scholar] [PubMed]
- Boesze-Battagliaa, K.; Stefano, F.P. Peripherin/rds fusogenic function correlates with subunit assembly. Exp. Eye Res. 2002, 75, 227–231. [Google Scholar] [CrossRef]
- Khattree, N.; Ritter, L.M.; Goldberg, A.F. Membrane curvature generation by a C-terminal amphipathic helix in peripherin-2/rds, a tetraspanin required for photoreceptor sensory cilium morphogenesis. J. Cell Sci. 2013, 126, 4659–4670. [Google Scholar] [CrossRef]
- Milstein, M.L.; Kimler, V.A.; Ghatak, C.; Ladokhin, A.S.; Goldberg, A.F.X. An inducible amphipathic helix within the intrinsically disordered C terminus can participate in membrane curvature generation by peripherin-2/rds. J. Biol. Chem. 2017, 292, 7850–7865. [Google Scholar] [CrossRef]
- Steinberg, R.H.; Fisher, S.K.; Anderson, D.H. Disc morphogenesis in vertebrate photoreceptors. J. Comp. Neurol. 1980, 190, 501–508. [Google Scholar] [CrossRef]
- Ding, J.D.; Salinas, R.Y.; Arshavsky, V.Y. Discs of mammalian rod photoreceptors form through the membrane evagination mechanism. J. Cell Biol. 2015, 211, 495–502. [Google Scholar] [CrossRef]
- Miraglia, S.; Godfrey, W.; Yin, A.H.; Atkins, K.; Warnke, R.; Holden, J.T.; Bray, R.A.; Waller, E.K.; Buck, D.W. A novel five-transmembrane hematopoietic stem cell antigen: Isolation, characterization, and molecular cloning. Blood 1997, 90, 5013–5021. [Google Scholar] [CrossRef] [PubMed]
- Weigmann, A.; Corbeil, D.; Hellwig, A.; Huttner, W.B. Prominin, a novel microvilli-specific polytopic membrane protein of the apical surface of epithelial cells, is targeted to plasmalemmal protrusions of non-epithelial cells. Proc. Natl. Acad. Sci. USA 1997, 94, 12425–12430. [Google Scholar] [CrossRef]
- Yin, A.H.; Miraglia, S.; Zanjani, E.D.; Almeida-Porada, G.; Ogawa, M.; Leary, A.G.; Olweus, J.; Kearney, J.; Buck, D.W. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood 1997, 90, 5002–5012. [Google Scholar] [CrossRef]
- Maw, M.A.; Corbeil, D.; Koch, J.; Hellwig, A.; Wilson-Wheeler, J.C.; Bridges, R.J.; Kumaramanickavel, G.; John, S.; Nancarrow, D.; Roper, K.; et al. A frameshift mutation in prominin (mouse)-like 1 causes human retinal degeneration. Hum. Mol. Genet. 2000, 9, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.; Anderson, D.W.; Papermaster, D.S. Prominin-1 localizes to the open rims of outer segment lamellae in Xenopus laevis rod and cone photoreceptors. Invest. Ophthalmol. Vis. Sci. 2012, 53, 361–373. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z.; Chen, Y.; Lillo, C.; Chien, J.; Yu, Z.; Michaelides, M.; Klein, M.; Howes, K.A.; Li, Y.; Kaminoh, Y.; et al. Mutant prominin 1 found in patients with macular degeneration disrupts photoreceptor disk morphogenesis in mice. J. Clin. Investig. 2008, 118, 2908–2916. [Google Scholar] [CrossRef]
- Zacchigna, S.; Oh, H.; Wilsch-Brauninger, M.; Missol-Kolka, E.; Jaszai, J.; Jansen, S.; Tanimoto, N.; Tonagel, F.; Seeliger, M.; Huttner, W.B.; et al. Loss of the cholesterol-binding protein prominin-1/CD133 causes disk dysmorphogenesis and photoreceptor degeneration. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 2297–2308. [Google Scholar] [CrossRef]
- Collison, F.T.; Fishman, G.A.; Nagasaki, T.; Zernant, J.; McAnany, J.J.; Park, J.C.; Allikmets, R. Characteristic Ocular Features in Cases of Autosomal Recessive PROM1 Cone-Rod Dystrophy. Investig. Ophthalmol. Vis. Sci. 2019, 60, 2347–2356. [Google Scholar] [CrossRef]
- Liang, J.; She, X.; Chen, J.; Zhai, Y.; Liu, Y.; Zheng, K.; Gong, Y.; Zhu, H.; Luo, X.; Sun, X. Identification of novel PROM1 mutations responsible for autosomal recessive maculopathy with rod-cone dystrophy. Graefe’s Arch. Clin. Exp. Ophthalmol. 2019, 257, 619–628. [Google Scholar] [CrossRef]
- Huttner, W.B.; Zimmerberg, J. Implications of lipid microdomains for membrane curvature, budding and fission. Curr. Opin. Cell Biol. 2001, 13, 478–484. [Google Scholar] [CrossRef]
- Iglic, A.; Hagerstrand, H.; Veranic, P.; Plemenitas, A.; Kralj-Iglic, V. Curvature-induced accumulation of anisotropic membrane components and raft formation in cylindrical membrane protrusions. J. Theor. Biol. 2006, 240, 368–373. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.I. Further studies on the question of the patency of saccules in outer segments of vertebrate photoreceptors. Vis. Res. 1970, 10, 445–453. [Google Scholar] [CrossRef]
- Anderson, D.H.; Fisher, S.K.; Steinberg, R.H. Mammalian cones: Disc shedding, phagocytosis, and renewal. Investig. Ophthalmol. Vis. Sci. 1978, 17, 117–133. [Google Scholar] [PubMed]
- Bunt, A.H. Fine structure and radioautography of rabbit photoreceptor cells. Investig. Ophthalmol. Vis. Sci. 1978, 17, 90–104. [Google Scholar]
- Carter-Dawson, L.D.; LaVail, M.M. Rods and cones in the mouse retina. I. Structural analysis using light and electron microscopy. J. Comp. Neurol. 1979, 188, 245–262. [Google Scholar] [CrossRef]
- Mears, A.J.; Kondo, M.; Swain, P.K.; Takada, Y.; Bush, R.A.; Saunders, T.L.; Sieving, P.A.; Swaroop, A. Nrl is required for rod photoreceptor development. Nat. Genet. 2001, 29, 447–452. [Google Scholar] [CrossRef]
- Daniele, L.L.; Lillo, C.; Lyubarsky, A.L.; Nikonov, S.S.; Philp, N.; Mears, A.J.; Swaroop, A.; Williams, D.S.; Pugh, E.N., Jr. Cone-like morphological, molecular, and electrophysiological features of the photoreceptors of the Nrl knockout mouse. Investig. Ophthalmol. Vis. Sci. 2005, 46, 2156–2167. [Google Scholar] [CrossRef]
- Conley, S.M.; Al-Ubaidi, M.R.; Han, Z.; Naash, M.I. Rim formation is not a prerequisite for distribution of cone photoreceptor outer segment proteins. FASEB J. 2014, 28, 3468–3479. [Google Scholar] [CrossRef]
- Farjo, R.; Skaggs, J.S.; Nagel, B.A.; Quiambao, A.B.; Nash, Z.A.; Fliesler, S.J.; Naash, M.I. Retention of function without normal disc morphogenesis occurs in cone but not rod photoreceptors. J. Cell Biol. 2006, 173, 59–68. [Google Scholar] [CrossRef]
- Moritz, O.L.; Molday, R.S. Molecular cloning, membrane topology, and localization of bovine rom-1 in rod and cone photoreceptor cells. Investig. Ophthalmol. Vis. Sci 1996, 37, 352–362. [Google Scholar]
- Conley, S.M.; Stuck, M.W.; Naash, M.I. Structural and functional relationships between photoreceptor tetraspanins and other superfamily members. Cell. Mol. Life Sci. 2012, 69, 1035–1047. [Google Scholar] [CrossRef] [PubMed]
- Loewen, C.J.; Molday, R.S. Disulfide-mediated oligomerization of Peripherin/Rds and Rom-1 in photoreceptor disk membranes. Implications for photoreceptor outer segment morphogenesis and degeneration. J. Biol. Chem. 2000, 275, 5370–5378. [Google Scholar] [CrossRef] [PubMed]
- Tian, G.; Ropelewski, P.; Nemet, I.; Lee, R.; Lodowski, K.H.; Imanishi, Y. An unconventional secretory pathway mediates the cilia targeting of peripherin/rds. J. Neurosci. Off. J. Soc. Neurosci. 2014, 34, 992–1006. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Ding, X.Q.; Fliesler, S.J.; Naash, M.I. Outer segment oligomerization of Rds: Evidence from mouse models and subcellular fractionation. Biochemistry 2008, 47, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. Retinal Degeneration Slow (RDS) Glycosylation Plays a Role in Cone Function and in the Regulation of RDS.ROM-1 Protein Complex Formation. J. Biol. Chem. 2015, 290, 27901–27913. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Strayve, D.G.; Makia, M.S.; Conley, S.M.; Kakahel, M.; Al-Ubaidi, M.R.; Naash, M.I. Novel molecular mechanisms for Prph2-associated pattern dystrophy. FASEB J. 2020, 34, 1211–1230. [Google Scholar] [CrossRef]
- Goldberg, A.F.; Loewen, C.J.; Molday, R.S. Cysteine residues of photoreceptor peripherin/rds: Role in subunit assembly and autosomal dominant retinitis pigmentosa. Biochemistry 1998, 37, 680–685. [Google Scholar] [CrossRef]
- Zulliger, R.; Conley, S.M.; Mwoyosvi, M.L.; Al-Ubaidi, M.R.; Naash, M.I. Oligomerization of Prph2 and Rom1 is essential for photoreceptor outer segment formation. Hum. Mol. Genet. 2018, 27, 3507–3518. [Google Scholar] [CrossRef]
- Chakraborty, D.; Conley, S.M.; Zulliger, R.; Naash, M.I. The K153Del PRPH2 mutation differentially impacts photoreceptor structure and function. Hum. Mol. Genet. 2016, 25, 3500–3514. [Google Scholar] [CrossRef]
- Weleber, R.G.; Carr, R.E.; Murphey, W.H.; Sheffield, V.C.; Stone, E.M. Phenotypic variation including retinitis pigmentosa, pattern dystrophy, and fundus flavimaculatus in a single family with a deletion of codon 153 or 154 of the peripherin/RDS gene. Arch. Ophthalmol. 1993, 111, 1531–1542. [Google Scholar] [CrossRef]
- Conley, S.M.; Stuck, M.W.; Burnett, J.L.; Chakraborty, D.; Azadi, S.; Fliesler, S.J.; Naash, M.I. Insights into the mechanisms of macular degeneration associated with the R172W mutation in RDS. Hum. Mol. Genet. 2014, 23, 3102–3114. [Google Scholar] [CrossRef] [PubMed]
- Stuck, M.W.; Conley, S.M.; Naash, M.I. The Y141C knockin mutation in RDS leads to complex phenotypes in the mouse. Hum. Mol. Genet. 2014, 23, 6260–6274. [Google Scholar] [CrossRef] [PubMed]
- Khani, S.C.; Karoukis, A.J.; Young, J.E.; Ambasudhan, R.; Burch, T.; Stockton, R.; Lewis, R.A.; Sullivan, L.S.; Daiger, S.P.; Reichel, E.; et al. Late-onset autosomal dominant macular dystrophy with choroidal neovascularization and nonexudative maculopathy associated with mutation in the RDS gene. Investig. Ophthalmol. Vis. Sci. 2003, 44, 3570–3577. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, W.; Lloyd, M.; Birch, D.G.; Bok, D.; Travis, G.H. Generation and analysis of transgenic mice expressing P216L-substituted rds/peripherin in rod photoreceptors. Investig. Ophthalmol. Vis. Sci. 1997, 38, 498–509. [Google Scholar]
- Stricker, H.M.; Ding, X.Q.; Quiambao, A.; Fliesler, S.J.; Naash, M.I. The Cys214→Ser mutation in peripherin/rds causes a loss-of-function phenotype in transgenic mice. Biochem. J. 2005, 388, 605–613. [Google Scholar] [CrossRef]
- Ding, X.Q.; Nour, M.; Ritter, L.M.; Goldberg, A.F.; Fliesler, S.J.; Naash, M.I. The R172W mutation in peripherin/rds causes a cone-rod dystrophy in transgenic mice. Hum. Mol. Genet. 2004, 13, 2075–2087. [Google Scholar] [CrossRef]
- Conley, S.; Nour, M.; Fliesler, S.J.; Naash, M.I. Late-onset cone photoreceptor degeneration induced by R172W mutation in Rds and partial rescue by gene supplementation. Investig. Ophthalmol. Vis. Sci. 2007, 48, 5397–5407. [Google Scholar] [CrossRef]
- Conley, S.M.; Stuck, M.W.; Watson, J.N.; Naash, M.I. Rom1 converts Y141C-Prph2-associated pattern dystrophy to retinitis pigmentosa. Hum. Mol. Genet. 2017, 26, 509–518. [Google Scholar] [CrossRef]
- Kedzierski, W.; Bok, D.; Travis, G.H. Transgenic analysis of rds/peripherin N-glycosylation: Effect on dimerization, interaction with rom1, and rescue of the rds null phenotype. J. Neurochem. 1999, 72, 430–438. [Google Scholar] [CrossRef]
- Poetsch, A.; Molday, L.L.; Molday, R.S. The cGMP-gated channel and related glutamic acid-rich proteins interact with peripherin-2 at the rim region of rod photoreceptor disc membranes. J. Biol. Chem. 2001, 276, 48009–48016. [Google Scholar] [CrossRef]
- Ritter, L.M.; Khattree, N.; Tam, B.; Moritz, O.L.; Schmitz, F.; Goldberg, A.F. In situ visualization of protein interactions in sensory neurons: Glutamic acid-rich proteins (GARPs) play differential roles for photoreceptor outer segment scaffolding. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 11231–11243. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Molday, L.L.; Molday, R.S.; Sarfare, S.S.; Woodruff, M.L.; Fain, G.L.; Kraft, T.W.; Pittler, S.J. Knockout of GARPs and the beta-subunit of the rod cGMP-gated channel disrupts disk morphogenesis and rod outer segment structural integrity. J. Cell Sci. 2009, 122, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Gilliam, J.C.; Chang, J.T.; Sandoval, I.M.; Zhang, Y.; Li, T.; Pittler, S.J.; Chiu, W.; Wensel, T.G. Three-dimensional architecture of the rod sensory cilium and its disruption in retinal neurodegeneration. Cell 2012, 151, 1029–1041. [Google Scholar] [CrossRef] [PubMed]
- Becirovic, E.; Nguyen, O.N.; Paparizos, C.; Butz, E.S.; Stern-Schneider, G.; Wolfrum, U.; Hauck, S.M.; Ueffing, M.; Wahl-Schott, C.; Michalakis, S.; et al. Peripherin-2 couples rhodopsin to the CNG channel in outer segments of rod photoreceptors. Hum. Mol. Genet. 2014, 23, 5989–5997. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, D.; Conley, S.M.; Pittler, S.J.; Naash, M.I. Role of RDS and Rhodopsin in Cngb1-Related Retinal Degeneration. Investig. Ophthalmol. Vis. Sci. 2016, 57, 787–797. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wroblewski, J.J.; Wells, J.A., 3rd; Eckstein, A.; Fitzke, F.; Jubb, C.; Keen, T.J.; Inglehearn, C.; Bhattacharya, S.; Arden, G.B.; Jay, M.; et al. Macular dystrophy associated with mutations at codon 172 in the human retinal degeneration slow gene. Ophthalmology 1994, 101, 12–22. [Google Scholar] [CrossRef]
- Piguet, B.; Heon, E.; Munier, F.L.; Grounauer, P.A.; Niemeyer, G.; Butler, N.; Schorderet, D.F.; Sheffield, V.C.; Stone, E.M. Full characterization of the maculopathy associated with an Arg-172-Trp mutation in the RDS/peripherin gene. Ophthalmic Genet. 1996, 17, 175–186. [Google Scholar] [CrossRef]
- Payne, A.M.; Downes, S.M.; Bessant, D.A.; Bird, A.C.; Bhattacharya, S.S. Founder effect, seen in the British population, of the 172 peripherin/RDS mutation-and further refinement of genetic positioning of the peripherin/RDS gene. Am. J. Hum. Genet. 1998, 62, 192–195. [Google Scholar] [CrossRef]
- Downes, S.M.; Fitzke, F.W.; Holder, G.E.; Payne, A.M.; Bessant, D.A.; Bhattacharya, S.S.; Bird, A.C. Clinical features of codon 172 RDS macular dystrophy: Similar phenotype in 12 families. Arch. Ophthalmol. 1999, 117, 1373–1383. [Google Scholar] [CrossRef]
- Saga, M.; Mashima, Y.; Akeo, K.; Oguchi, Y.; Kudoh, J.; Shimizu, N. A novel Cys-214-Ser mutation in the peripherin/RDS gene in a Japanese family with autosomal dominant retinitis pigmentosa. Hum. Genet. 1993, 92, 519–521. [Google Scholar] [CrossRef]
- Kajiwara, K.; Hahn, L.B.; Mukai, S.; Travis, G.H.; Berson, E.L.; Dryja, T.P. Mutations in the human retinal degeneration slow gene in autosomal dominant retinitis pigmentosa. Nature 1991, 354, 480–483. [Google Scholar] [CrossRef]
- Kajiwara, K.; Berson, E.L.; Dryja, T.P. Digenic retinitis pigmentosa due to mutations at the unlinked peripherin/RDS and ROM1 loci. Science 1994, 264, 1604–1608. [Google Scholar] [CrossRef] [PubMed]
- Dryja, T.P.; Hahn, L.B.; Kajiwara, K.; Berson, E.L. Dominant and digenic mutations in the peripherin/RDS and ROM1 genes in retinitis pigmentosa. Investig. Ophthalmol. Vis. Sci. 1997, 38, 1972–1982. [Google Scholar] [PubMed]
- Kedzierski, W.; Nusinowitz, S.; Birch, D.; Clarke, G.; McInnes, R.R.; Bok, D.; Travis, G.H. Deficiency of rds/peripherin causes photoreceptor death in mouse models of digenic and dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 2001, 98, 7718–7723. [Google Scholar] [CrossRef] [PubMed]
- Nour, M.; Ding, X.Q.; Stricker, H.; Fliesler, S.J.; Naash, M.I. Modulating expression of peripherin/rds in transgenic mice: Critical levels and the effect of overexpression. Investig. Ophthalmol. Vis. Sci. 2004, 45, 2514–2521. [Google Scholar] [CrossRef] [PubMed]
- Apfelstedt-Sylla, E.; Theischen, M.; Ruther, K.; Wedemann, H.; Gal, A.; Zrenner, E. Extensive intrafamilial and interfamilial phenotypic variation among patients with autosomal dominant retinal dystrophy and mutations in the human RDS/peripherin gene. Br. J. Ophthalmol. 1995, 79, 28–34. [Google Scholar] [CrossRef]
- McNally, N.; Kenna, P.F.; Rancourt, D.; Ahmed, T.; Stitt, A.; Colledge, W.H.; Lloyd, D.G.; Palfi, A.; O’Neill, B.; Humphries, M.M.; et al. Murine model of autosomal dominant retinitis pigmentosa generated by targeted deletion at codon 307 of the rds-peripherin gene. Hum. Mol. Genet. 2002, 11, 1005–1016. [Google Scholar] [CrossRef]
- Nichols, B.E.; Sheffield, V.C.; Vandenburgh, K.; Drack, A.V.; Kimura, A.E.; Stone, E.M. Butterfly-shaped pigment dystrophy of the fovea caused by a point mutation in codon 167 of the RDS gene. Nat. Genet. 1993, 3, 202–207. [Google Scholar] [CrossRef]
- Fossarello, M.; Bertini, C.; Galantuomo, M.S.; Cao, A.; Serra, A.; Pirastu, M. Deletion in the peripherin/RDS gene in two unrelated Sardinian families with autosomal dominant butterfly-shaped macular dystrophy. Arch. Ophthalmol. 1996, 114, 448–456. [Google Scholar] [CrossRef]
- Zhang, K.; Garibaldi, D.C.; Li, Y.; Green, W.R.; Zack, D.J. Butterfly-shaped pattern dystrophy: A genetic, clinical, and histopathological report. Arch. Ophthalmol. 2002, 120, 485–490. [Google Scholar] [CrossRef]
- Zulliger, R.; Conley, S.M.; Mwoyosvi, M.L.; Stuck, M.W.; Azadi, S.; Naash, M.I. SNAREs Interact with Retinal Degeneration Slow and Rod Outer Segment Membrane Protein-1 during Conventional and Unconventional Outer Segment Targeting. PLoS ONE 2015, 10, e0138508. [Google Scholar] [CrossRef] [PubMed]
- Travis, G.H.; Groshan, K.R.; Lloyd, M.; Bok, D. Complete rescue of photoreceptor dysplasia and degeneration in transgenic retinal degeneration slow (rds) mice. Neuron 1992, 9, 113–119. [Google Scholar] [CrossRef]
- Ali, R.R.; Sarra, G.M.; Stephens, C.; Alwis, M.D.; Bainbridge, J.W.; Munro, P.M.; Fauser, S.; Reichel, M.B.; Kinnon, C.; Hunt, D.M.; et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat. Genet. 2000, 25, 306–310. [Google Scholar] [CrossRef] [PubMed]
- Schlichtenbrede, F.C.; da Cruz, L.; Stephens, C.; Smith, A.J.; Georgiadis, A.; Thrasher, A.J.; Bainbridge, J.W.; Seeliger, M.W.; Ali, R.R. Long-term evaluation of retinal function in Prph2Rd2/Rd2 mice following AAV-mediated gene replacement therapy. J. Gene Med. 2003, 5, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.Q.; Quiambao, A.B.; Fitzgerald, J.B.; Cooper, M.J.; Conley, S.M.; Naash, M.I. Ocular delivery of compacted DNA-nanoparticles does not elicit toxicity in the mouse retina. PLoS ONE 2009, 4, e7410. [Google Scholar] [CrossRef]
- Cai, X.; Conley, S.M.; Nash, Z.; Fliesler, S.J.; Cooper, M.J.; Naash, M.I. Gene delivery to mitotic and postmitotic photoreceptors via compacted DNA nanoparticles results in improved phenotype in a mouse model of retinitis pigmentosa. FASEB J. 2010, 24, 1178–1191. [Google Scholar] [CrossRef]
- Han, Z.; Conley, S.M.; Makkia, R.S.; Cooper, M.J.; Naash, M.I. DNA nanoparticle-mediated ABCA4 delivery rescues Stargardt dystrophy in mice. J. Clin. Investig. 2012, 122, 3221–3226. [Google Scholar] [CrossRef]
- Han, Z.; Conley, S.M.; Makkia, R.; Guo, J.; Cooper, M.J.; Naash, M.I. Comparative analysis of DNA nanoparticles and AAVs for ocular gene delivery. PLoS ONE 2012, 7, e52189. [Google Scholar] [CrossRef]
- Han, Z.; Koirala, A.; Makkia, R.; Cooper, M.J.; Naash, M.I. Direct gene transfer with compacted DNA nanoparticles in retinal pigment epithelial cells: Expression, repeat delivery and lack of toxicity. Nanomedicine 2012, 7, 521–539. [Google Scholar] [CrossRef]
- Conley, S.M.; Naash, M.I. Gene therapy for PRPH2-associated ocular disease: Challenges and prospects. Cold Spring Harb. Perspect. Med. 2014, 4, a017376. [Google Scholar] [CrossRef][Green Version]
- Cai, X.; Nash, Z.; Conley, S.M.; Fliesler, S.J.; Cooper, M.J.; Naash, M.I. A partial structural and functional rescue of a retinitis pigmentosa model with compacted DNA nanoparticles. PLoS ONE 2009, 4, e5290. [Google Scholar] [CrossRef] [PubMed]
- Mao, H.; Gorbatyuk, M.S.; Rossmiller, B.; Hauswirth, W.W.; Lewin, A.S. Long-term rescue of retinal structure and function by rhodopsin RNA replacement with a single adeno-associated viral vector in P23H RHO transgenic mice. Hum. Gene Ther. 2012, 23, 356–366. [Google Scholar] [CrossRef] [PubMed]
- Cideciyan, A.V.; Sudharsan, R.; Dufour, V.L.; Massengill, M.T.; Iwabe, S.; Swider, M.; Lisi, B.; Sumaroka, A.; Marinho, L.F.; Appelbaum, T.; et al. Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proc. Natl. Acad. Sci. USA 2018, 115, E8547–E8556. [Google Scholar] [CrossRef] [PubMed]
- Palfi, A.; Ader, M.; Kiang, A.S.; Millington-Ward, S.; Clark, G.; O’Reilly, M.; McMahon, H.P.; Kenna, P.F.; Humphries, P.; Farrar, G.J. RNAi-based suppression and replacement of rds-peripherin in retinal organotypic culture. Hum. Mutat. 2006, 27, 260–268. [Google Scholar] [CrossRef]
- Petrs-Silva, H.; Yasumura, D.; Matthes, M.T.; LaVail, M.M.; Lewin, A.S.; Hauswirth, W.W. Suppression of rds expression by siRNA and gene replacement strategies for gene therapy using rAAV vector. Adv. Exp. Med. Biol. 2012, 723, 215–223. [Google Scholar]




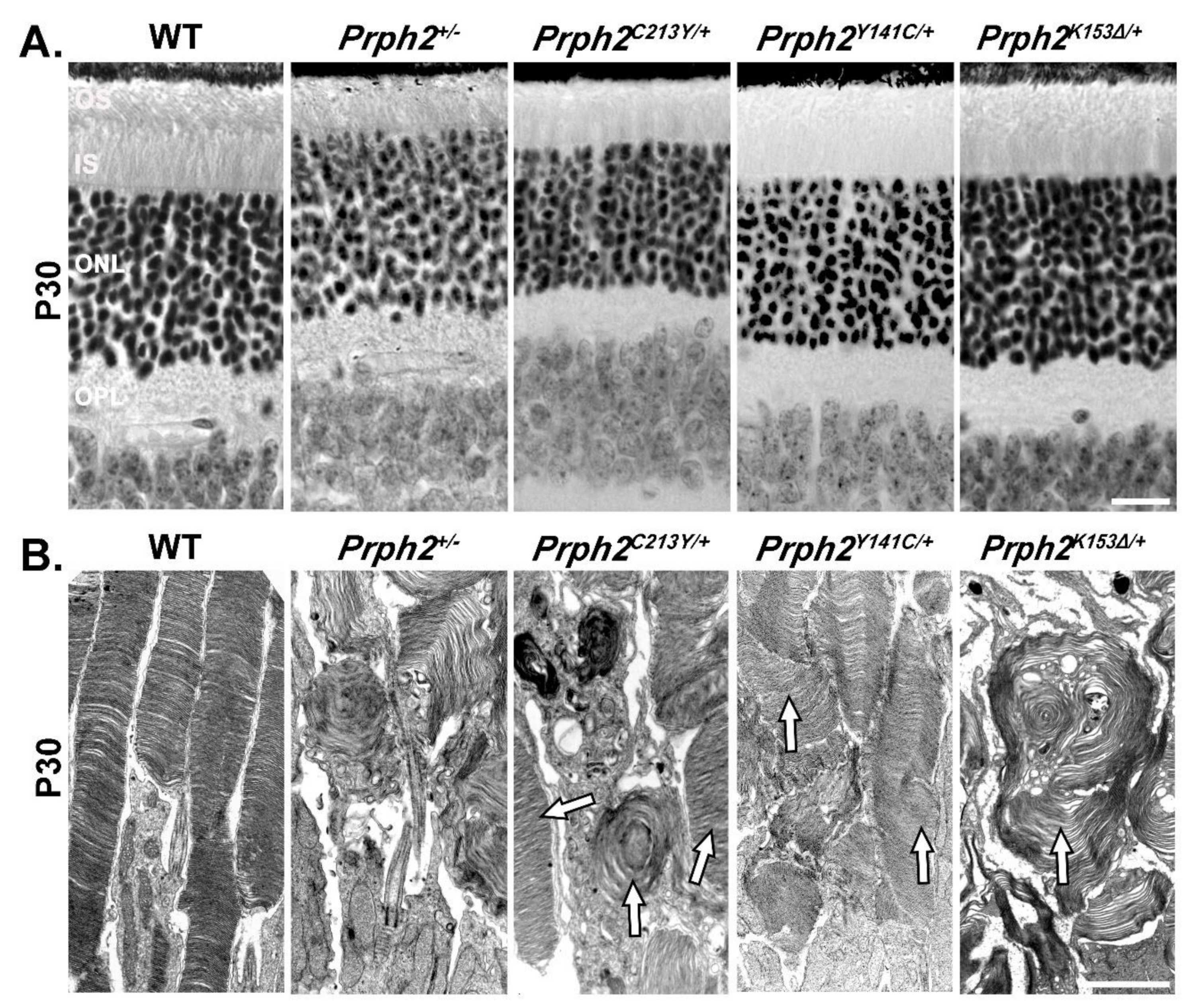{kind=link}
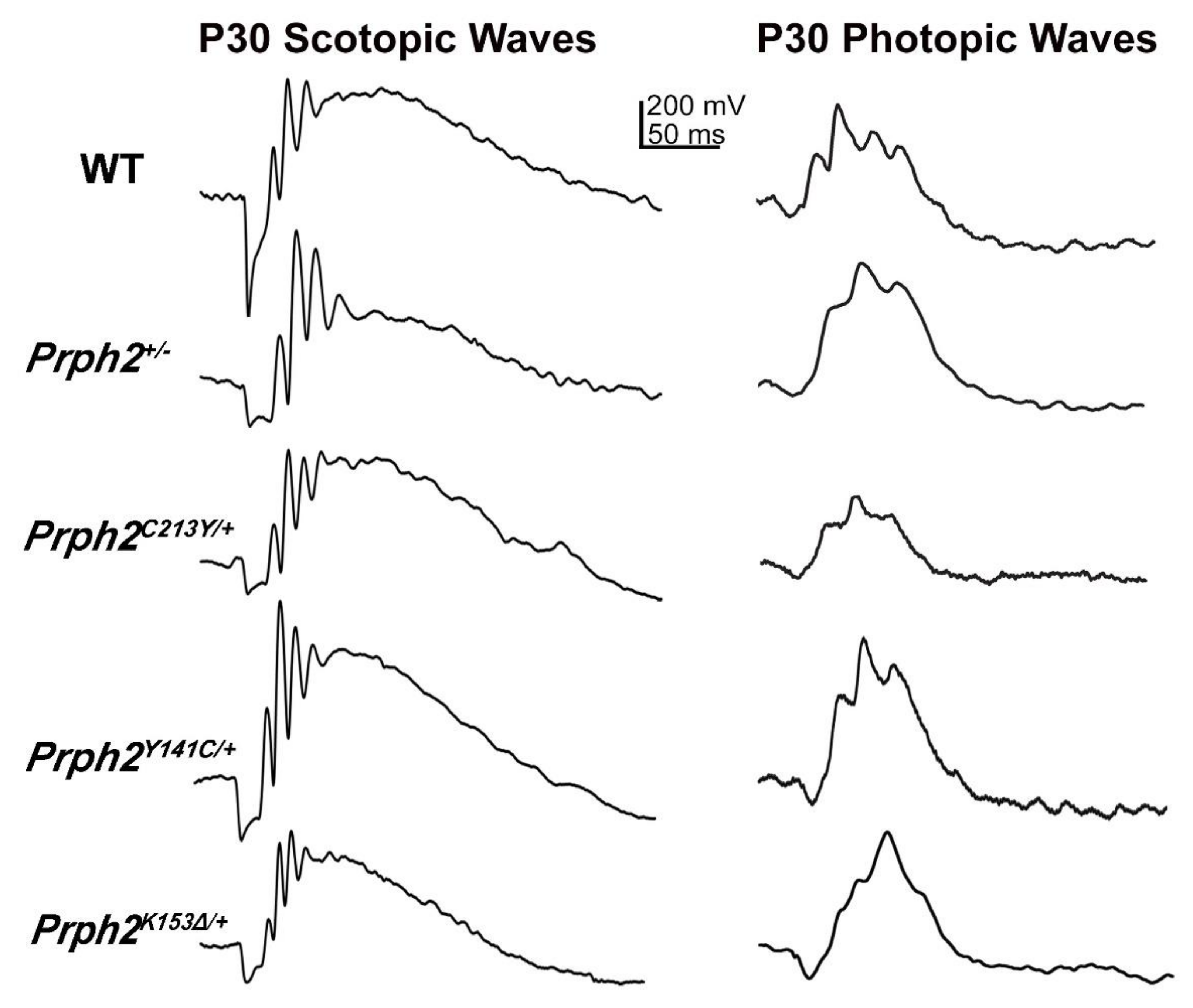{kind=link}
{kind=link}
| Genotype | Prph2+/− | Prph2−/− | Prph2K153∆/+ | Prph2K153∆/K153∆ | Prph2Y141C/+ | Prph2Y141C/Y141C |
|---|---|---|---|---|---|---|
| Mouse Rod Structure | Short OSs with whorl structures and ONL thinning at P180. | No OS structures. | Short OSs with whorl structures and ONL thinning at P180. | Almost no Oss with rare whorl structures and ONL thinning at P30, more severe at P180. | Short OS with longer discs and accumulation of vesicular structures, no ONL thinning at P30, ONL thinning at P180. | Small OS with flattened whorls and vesicular structures at P30, no OS present at P180, ONL thinning at P30, more severe at P180. |
| Mouse Cone Structure | Oss with whorl structures | Open OS with no lamella | Occasional whorl shaped OS, mostly open OS with no lamella | Occasional COS seen but mostly absent. | Abnormal and short COS structures. | Occasional COS seen but mostly absent. |
| Scotopic ERG | 57% and 33% reduction in a- and b-wave at P30. 74% and 48% reduction in a- and b-waves at P180, respectively. | 96% and 93% reduction in a- and b-wave at P30, respectively. | 63% and 49% reduction in a- and b-wave at P30. 83% and at 60% reduction in a- and b-wave at P180, respectively. | 90% and 89% reduction in a- and b-wave at P30, respectively. | 54% and 27% reduction in a- and b-wave at P30 50% and 25% reduction in a- and b-wave at P180, respectively. | 90% and 78% reduction in a- and b-wave at P30. 95% and 94% reduction in a- and b-wave at P180, respectively. |
| Photopic ERG | Photopic b-wave comparable to WT at P30. 35% reduction in photopic b-wave at P180. | 91% reduction in b-wave at P30. | 24% reduction in b-wave P30 and 50% at P180. | 64% reduction in b-wave at P30. | 10% reduction in b-wave at P30 and P180. | 64% reduction in b-wave at P30 and 90% at P180. |
| Complex formation | Prph2 complexes and distribution unchanged, 50% less Prph2 and Rom1. On NRL-/- background, higher order complexes decreased. | No Prph2 present, Rom1 still present but at lesser amount. | Prph2 complexes and distribution unchanged, while Rom1 shifted towards tetramers. On NRL−/− background, higher order complexes decreased, shift of Prph2 and Rom1 towards intermediate complexes. | No Prph2 dimers were formed, while Rom1 dimers were still formed. Prph2 interacted with Rom1. Prph2 and Rom1 restricted to tetramers. On NRL−/− background, no Prph2 dimers were formed, while Rom1 dimers were still formed. Prph2 did not interact with Rom1. Prph2 and Rom1 restricted to tetramers. | Prph2 occasionally found in abnormal high molecular weight aggregates. Abnormal aggregates were held together by intermolecular disulfide bonds. Rom1 also present in abnormal aggregates. Intermediate and higher order complexes formed but abnormal high molecular weight aggregates also present. | Prph2 almost exclusively found in abnormal high molecular weight aggregates. Abnormal aggregates were held together by intermolecular disulfide bonds. Rom1 also present in abnormal aggregates. Intermediate and higher order complexes reduced in favor of the abnormal high molecular weight aggregates. |
| Protein localization | Some rhodopsin detected in the IS and ONL. | Rhodopsin mislocalized to IS and ONL. | Small amount of rhodopsin and M-opsin mislocalized in the IS and ONL. | Huge amount of rhodopsin and Prph2 mislocalized in the IS and ONL. | NA | NA |
| Fundus | No abnormality | Flecking and splotches at P360 and older. | Flecking at P180 and no change at P365. | Severe flecking at P180 and big splotches at P365. | Flecking at P180. | Flecking at P180. |
| Rod defect in patients | NA | NA | RP | NA | Night blindness and RP reported in some patients. | NA |
| Cone defect in patients | NA | NA | Pattern dystrophy and fundus flavimaculs. | NA | Pattern dystrophy changed fundus in macula. | NA |
| Reference | [26,76,79] | [26,76,79] | [79] | [79] | [82] | [82] |
| Genotype | Prph2C213Y/+ | Prph2C213Y/C213Y | Prph2N229S/+ | Prph2N229S/N229S | Prph2C150S/+ | Prph2C150S/C150S | Prph2307/+ | Prph2307307 | |
|---|---|---|---|---|---|---|---|---|---|
| Rod structure mouse | Shortened OS, irregular structure, some disc structure better organized than in Prph2+/−. ONL thinning at P30. | Short OS formed with highly disorganized discs. Severe ONL thinning at P30. | Structure unaffected. | Modest ONL thinning at P180. | Modest elongation of discs, occasional formation of whorls. No ONL thinning. | Shortened OS, elongated discs curving into whorls. No ONL thinning. | ONL thinning starting at P60. Rod OS shortened, formation of whorls. | ONL thinning starting at P30 and rod OSs and absent. | |
| Cone structure mouse | Well organized lamella at P30 but slightly shorter. | Abnormally stacked lamella with whorl shaped structures. | Normal COS structures. | Occasional abnormal disc stacking at P180 | NA | Shortened OS, elongated discs curving into whorls. | NA | Cone OS are absent. | |
| Scotopic ERG | 60% and 47% reduction in a- and b-wave P30. 89% and 67% reduction in a- and b-wave at P180, respectively. | 96% and 94% reduction in a- and b-wave P30, respectively. | ERG a- and b-wave comparable to WT at P30 and P180. | ERG a- and b-wave comparable to WT at P30 and P180. | 50% and 30% reduction in a- and b-wave at P30, respectively. | 75% and 56% reduction in a- and b-wave P30, respectively. | 60% and 62% reduction in a- and b-wave at P180. 80% and 75% reduction in a- and b-wave at P300, respectively. | Scotopic ERG absent | |
| Photopic ERG | 33% and 46% reduction in b-wave at P30 and P180, respectively. | 79% reduction in b-wave at P30 ERG UV b-wave. | ERG b-wave comparable to WT at P30 and P180 | Normal at P30 but 22% reduction in b-wave at P180. | 29% reduction in b-wave at P30. | 64% reduction in b-wave at P30. | 60% and 80% reduction in b-wave at P180 and P300, respectively. | Photopic ERG absent | |
| Complex formation rods | Mutant Prph2 unable to interact with Rom1 in rods. Intermediate and higher order complex formation slightly impaired. Rom1 occasionally found in higher order complexes. | Mutant Prph2 unable to interact with Rom1 in rods. Protein levels of Rom1 decreased No intermediate or higher order complexes were formed Prph2 and Rom1 restricted to tetramers | Prph2 could not be glycosylated and formed normal Prph2/Rom1 complexes. | Prph2 could not be glycosylated. Amount of higher order complexes decreased, while the amount of intermediate complexes was increased. | Reduction in Prph2 protein levels, less pronounced than in Prph2+/−. Rom1 protein levels not affected. | Strong decrease in Prph2 and Rom1 protein levels. No intermediate or higher order complexes were formed. Prph2 and Rom1 restricted to tetramers. | |||
| Protein localization | Prph2 partially mislocalized to IS and ONL. Small amount of Rom1 mislocalized to IS. | Prph2 almost completely mislocalized to IS and ONL. Small amount of Rom1 mislocalized to IS. M- and S-Opsin as well as rhodopsin mislocalized to the IS and ONL. | None | None | None | None | NA | NA | |
| Fundus | Flecking at P180, persisted till P365. | Flecking at P180, more pronounced than in C213Y/+, replaced by splotches at P365. | None | None | NA | NA | NA | NA | |
| Rod defect patient | NA | NA | No patient model | NA | No patient model | NA | NA | NA | |
| Cone defect patient | Pattern dystrophy | NA | No patient model | NA | No patient model | NA | NA | NA | |
| Reference | [76] | [76] | [75] | [75] | [78] | [78] | [107] | [107] | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tebbe, L.; Kakakhel, M.; Makia, M.S.; Al-Ubaidi, M.R.; Naash, M.I. The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases. Cells 2020, 9, 784. https://doi.org/10.3390/cells9030784
Tebbe L, Kakakhel M, Makia MS, Al-Ubaidi MR, Naash MI. The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases. Cells. 2020; 9(3):784. https://doi.org/10.3390/cells9030784
Chicago/Turabian StyleTebbe, Lars, Mashal Kakakhel, Mustafa S. Makia, Muayyad R. Al-Ubaidi, and Muna I. Naash. 2020. "The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases" Cells 9, no. 3: 784. https://doi.org/10.3390/cells9030784
APA StyleTebbe, L., Kakakhel, M., Makia, M. S., Al-Ubaidi, M. R., & Naash, M. I. (2020). The Interplay between Peripherin 2 Complex Formation and Degenerative Retinal Diseases. Cells, 9(3), 784. https://doi.org/10.3390/cells9030784