Transcriptomic Insights into Mechanisms of Early Seed Maturation in the Garden Pea (Pisum sativum L.)

 ,
,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. RNA Extraction, Library Preparation, and Whole Transcriptome RNA Sequencing
2.3. RNA-Seq Read Processing and De Novo Transcriptome Assembly
2.4. Transcriptome Assembly Annotation
2.5. Incorporation of Open Access Transcriptomic Data into Analysis
2.6. Read Pseudoalignment and Estimation of Differential Expression
2.7. GO Over-Representation Tests
2.8. Sample Clustering
2.9. Term-Wise Transcript Clustering
2.10. Variant Calling and Single Nucleotide Polymorphism (SNP) Annotation
3. Results
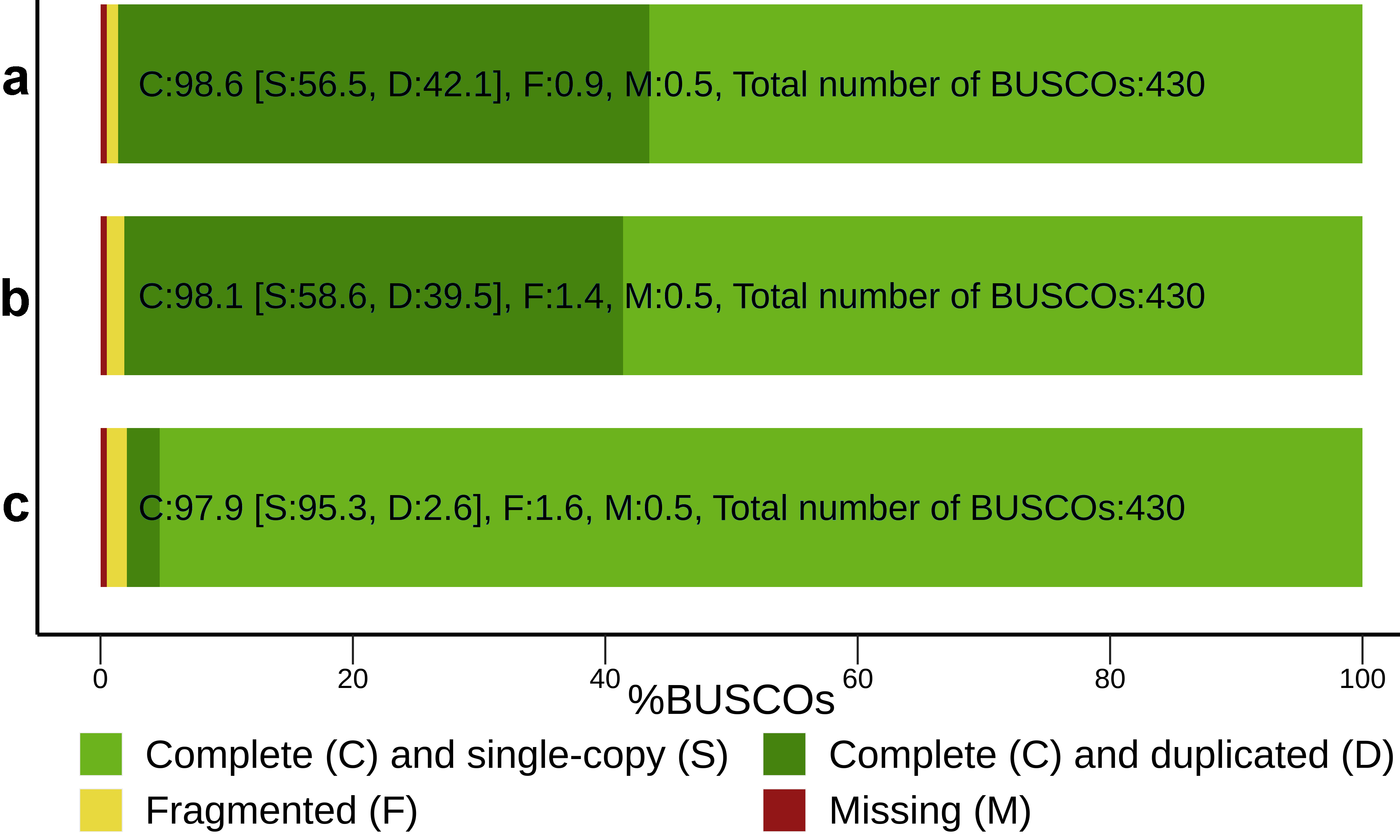
3.1. RNA-Seq De Novo Assembly and Annotation of Pea Seed Transcriptomes
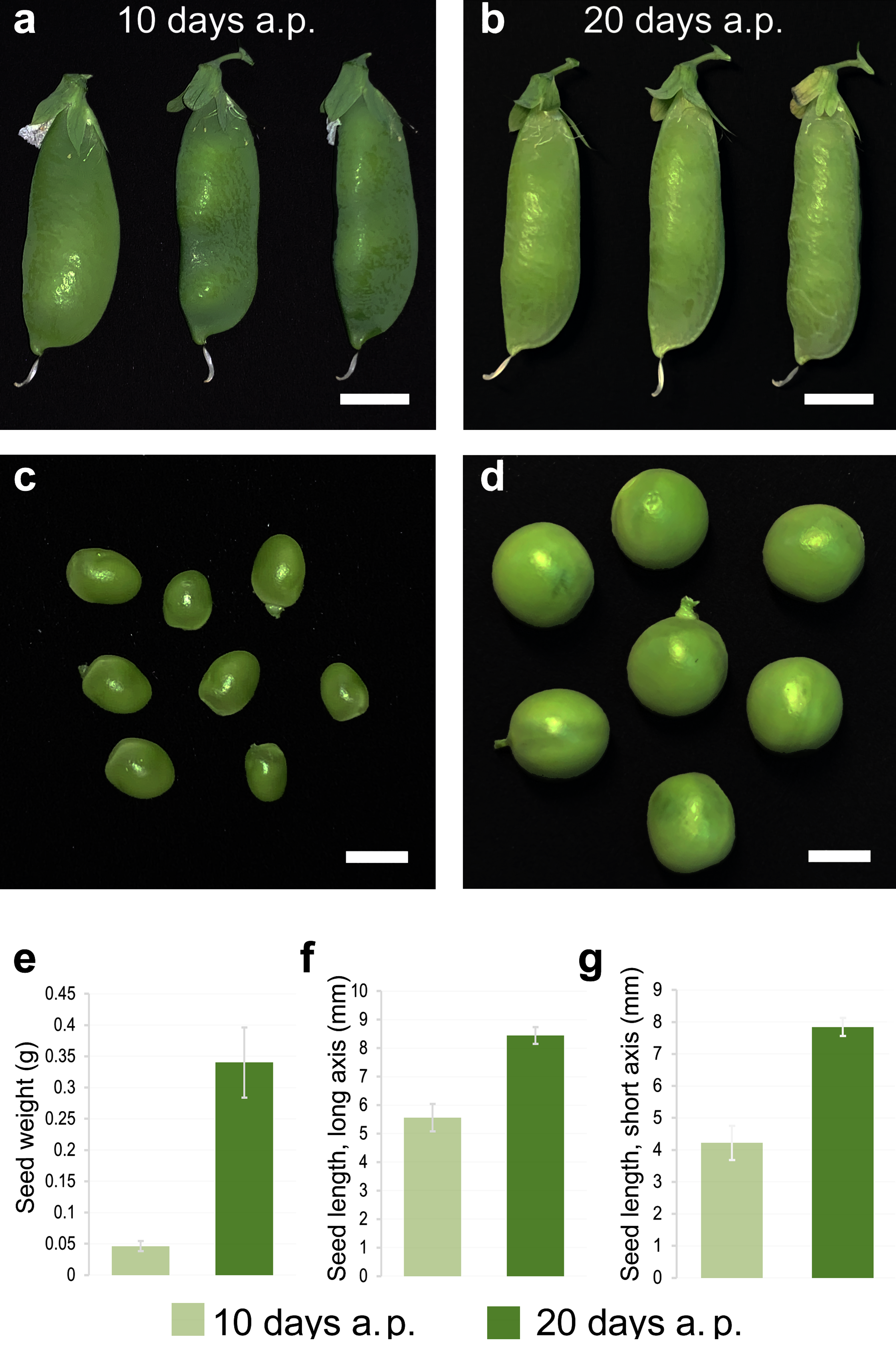
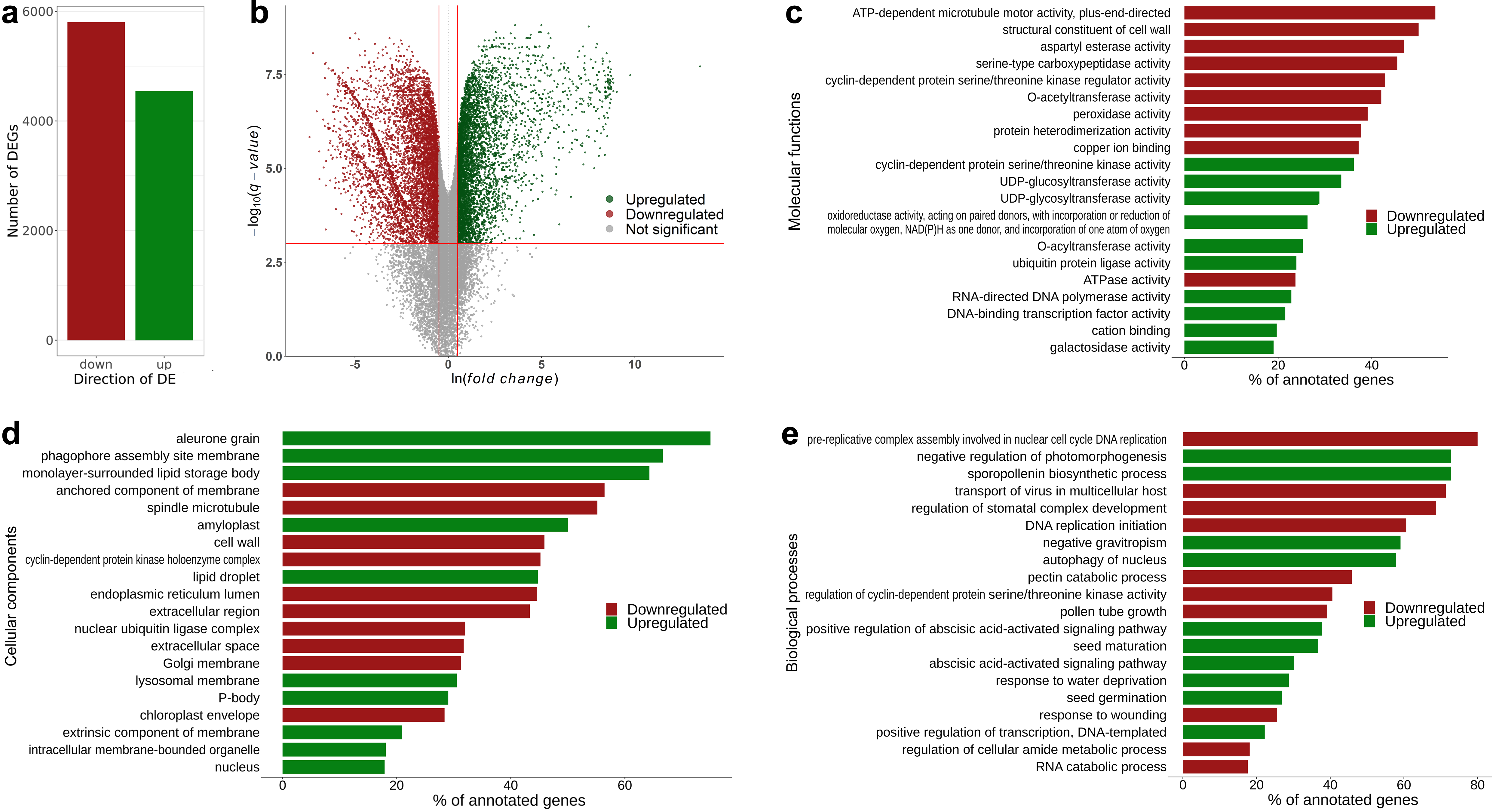
3.2. Transcriptomic Markers of Maturity during the Transition from Pre-Maturation (10 DAP) to Seed Filling (20 DAP)
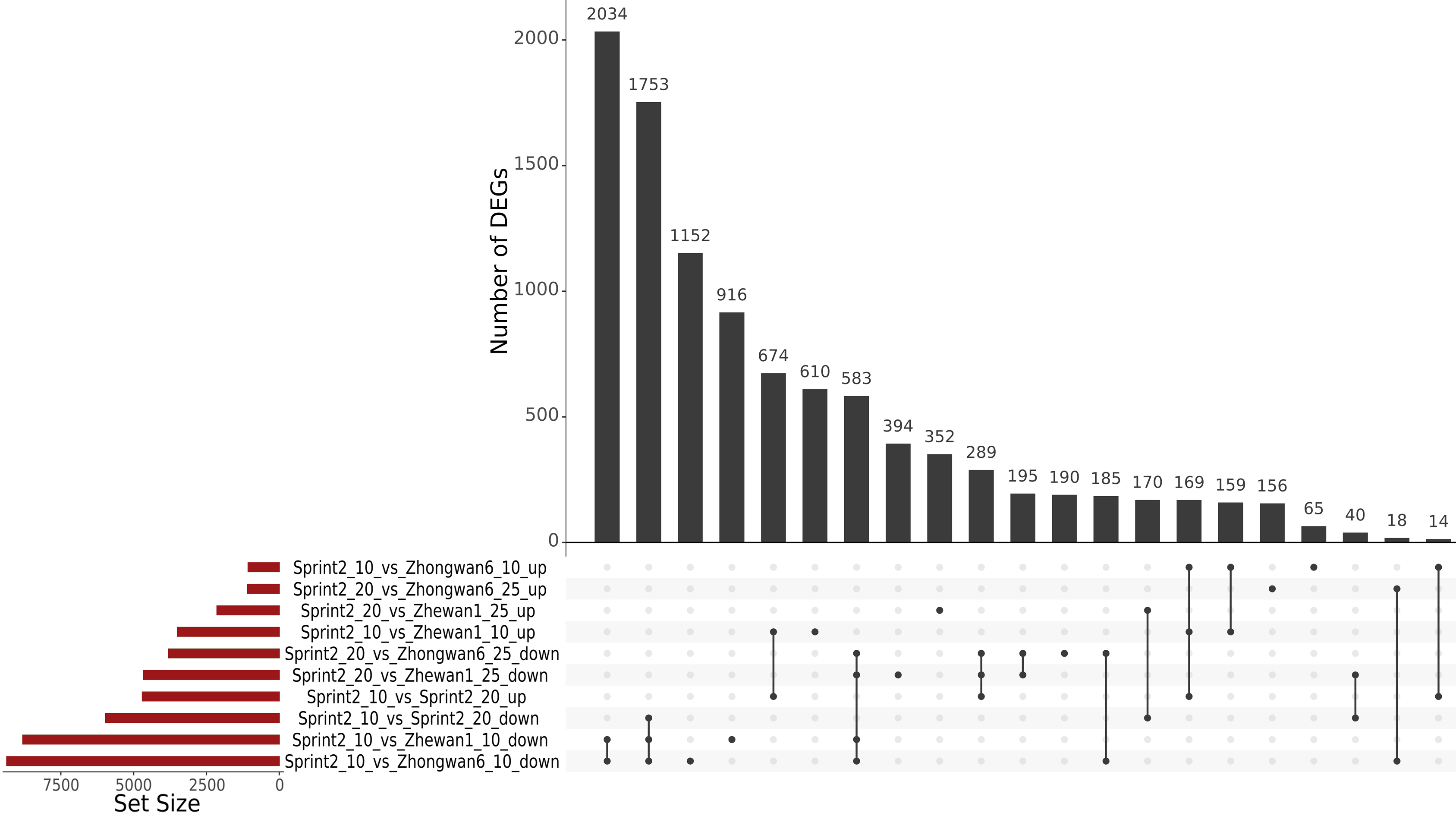
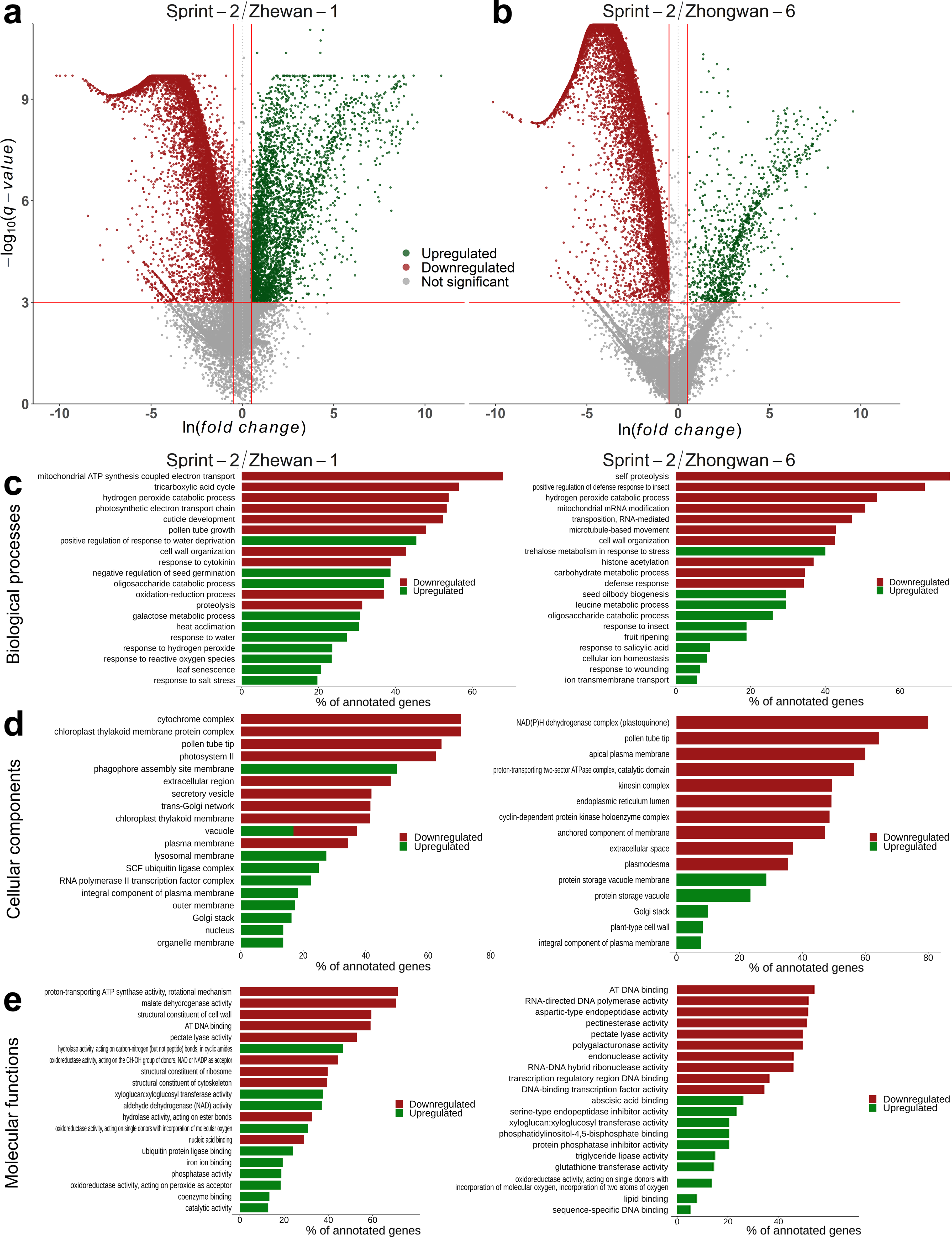
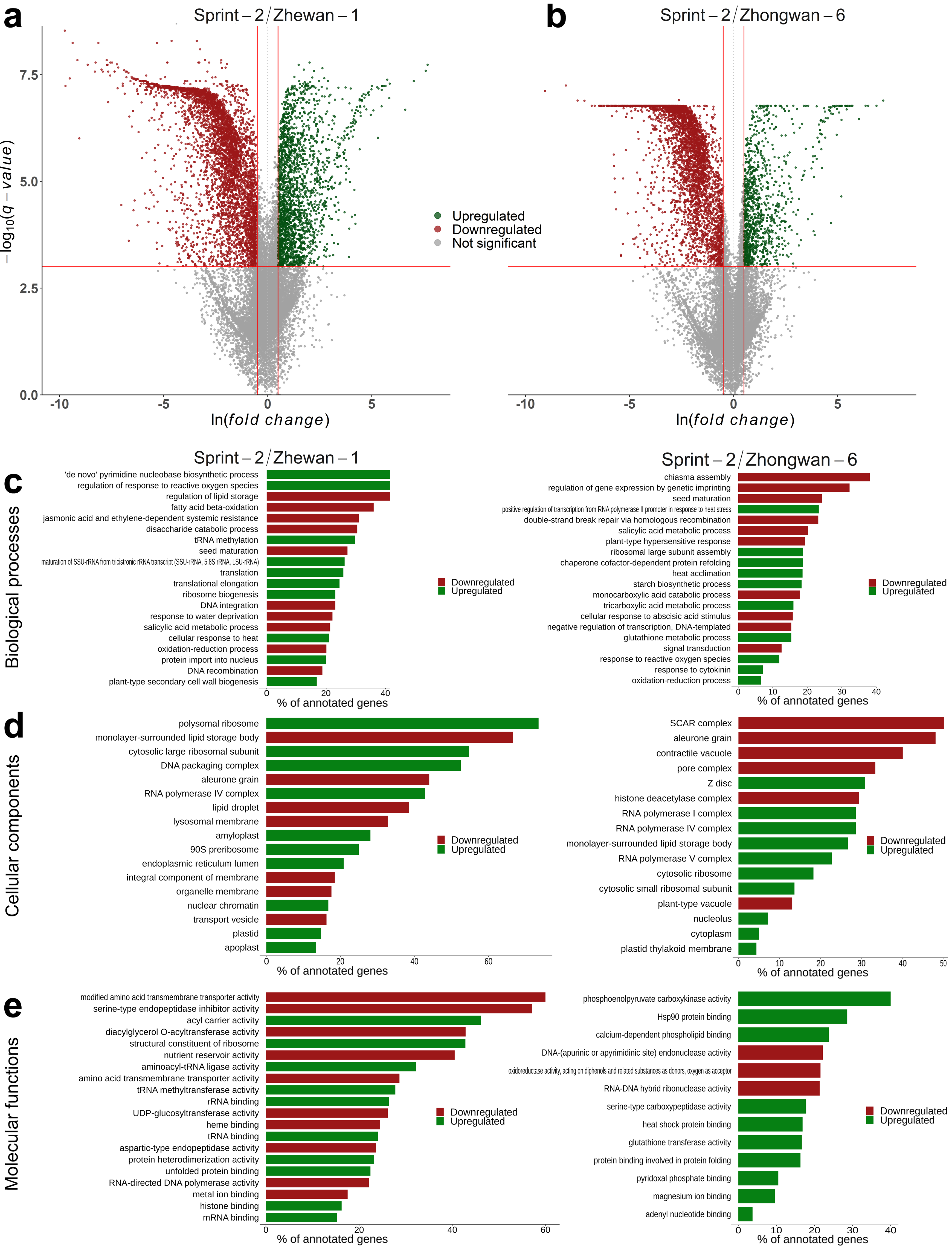
3.3. Sprint-2 Shows Significant Gene Expression Differences to Two Cultivars with Common Development Rates in Both Pre-Maturation and Maturation Stages
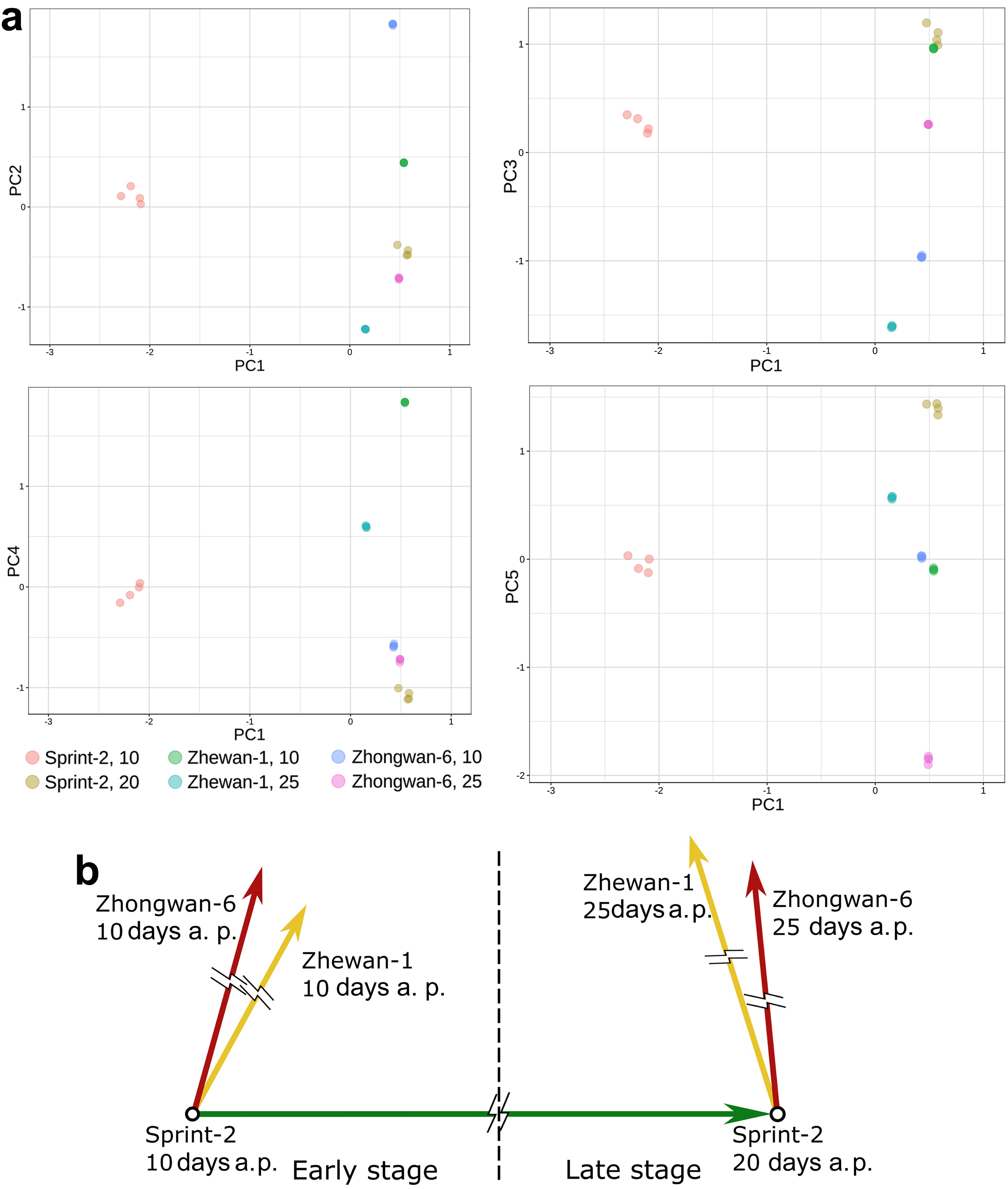
3.4. Evidence for the Non-Linear Maturation Timeline in Sprint-2
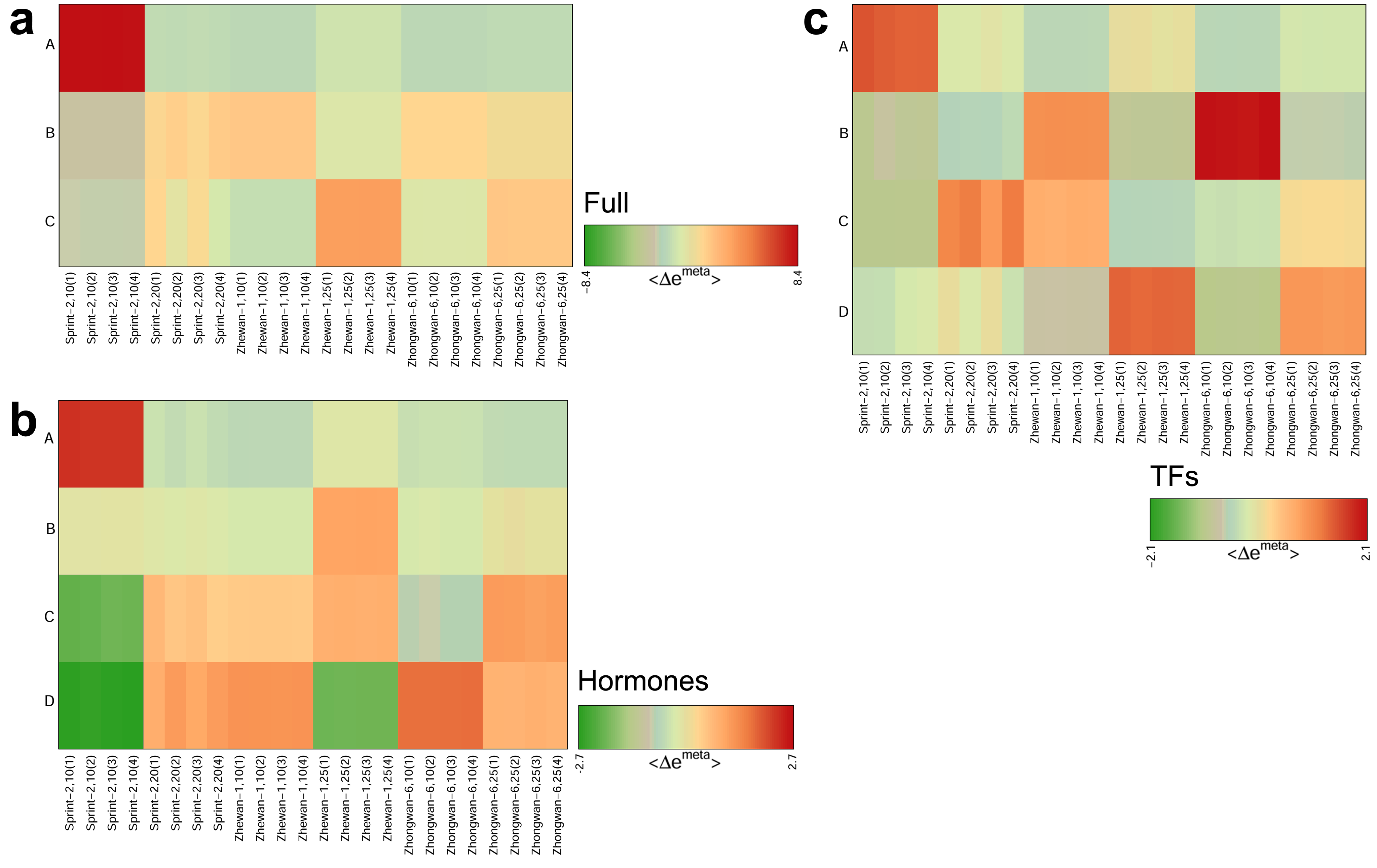
3.5. Hallmark Features of Altered Seed Maturation at a Pathway-Scale Resolution
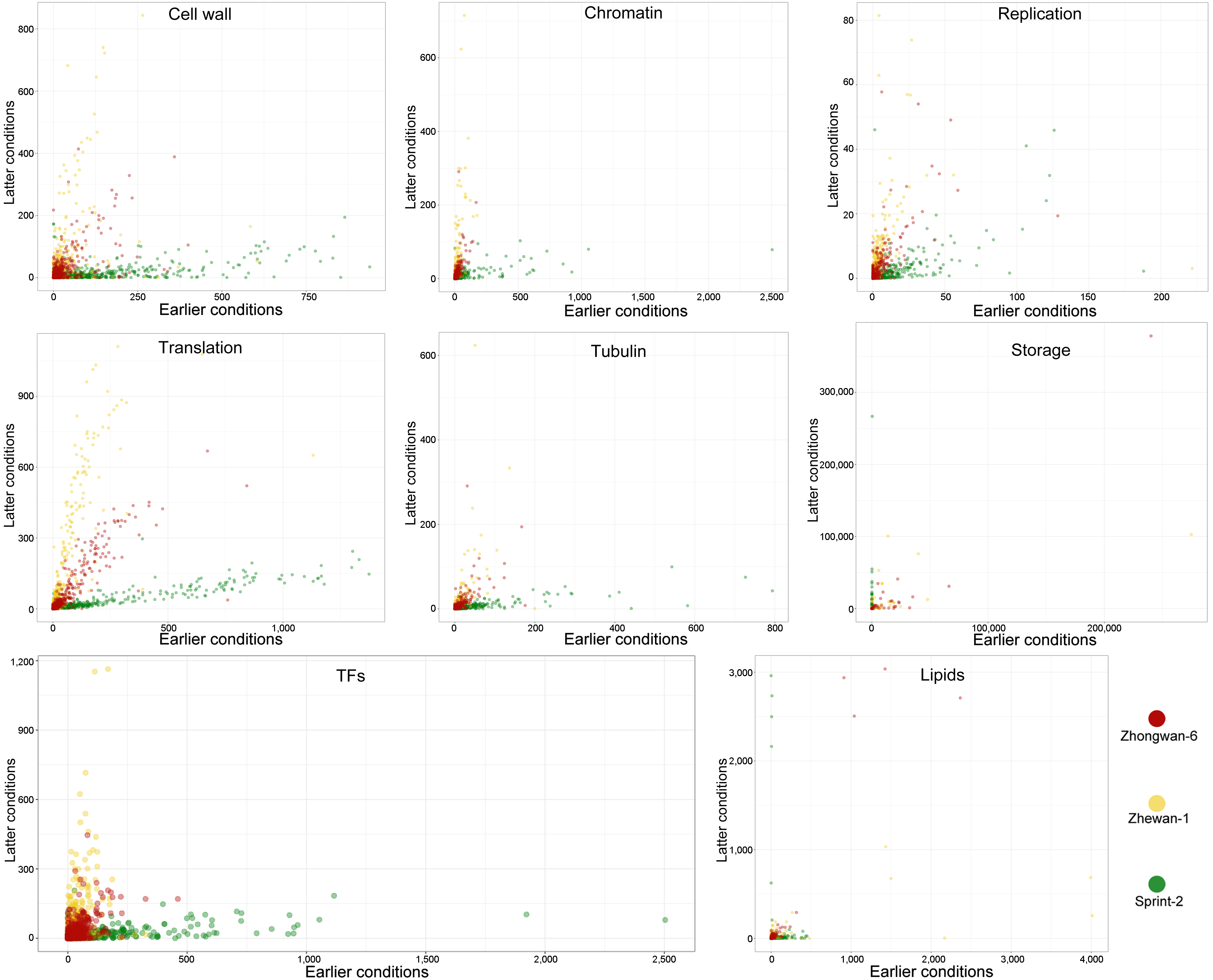
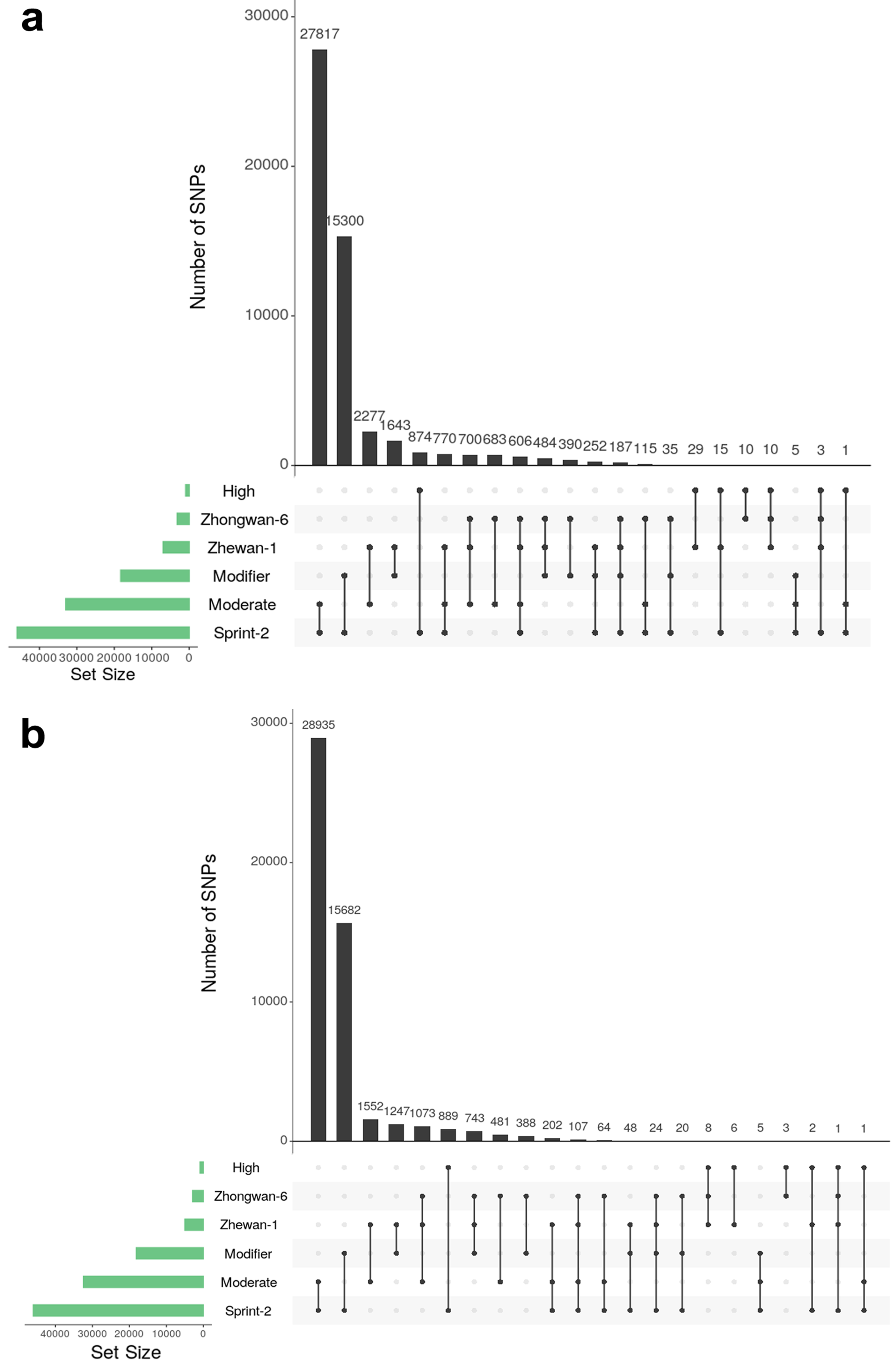
3.6. Single Nucleotide Polymorphism Sites Show a Non-Random Pattern of Distribution between Transcript Functional Groups
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bastianelli, D.; Grosjean, F.; Peyronnet, C.; Duparque, M.; Regnier, J.M. Feeding value of pea (Pisum sativum, L.) 1. Chemical composition of different categories of pea. Anim. Sci. 1998, 67, 609–619. [Google Scholar] [CrossRef]
- Kreplak, J.; Madoui, M.A.; Cápal, P.; Novák, P.; Labadie, K.; Aubert, G.; Bayer, P.E.; Gali, K.K.; Syme, R.A.; Main, D.; et al. A reference genome for pea provides insight into legume genome evolution. Nat. Genet. 2019, 51, 1411–1422. [Google Scholar] [CrossRef]
- Smykal, P.; Aubert, G.; Burstin, J.; Coyne, C.J.; Ellis, T.H.N.; Flavell, A.J.; Ford, R.; Hýbl, M.; Macas, J.; Neumann, P.; et al. Pea (Pisum sativum L.) in the Genomic Era. Agronomy 2012, 2, 74–115. [Google Scholar] [CrossRef]
- Weber, H.; Borisjuk, L.; Wobus, U. Molecular Physiology of Legume Seed Development. Annu. Rev. Plant Boil. 2005, 56, 253–279. [Google Scholar] [CrossRef] [PubMed]
- Caccere, R.; Teixeira, S.P.; Centeno, D.C.; Figueiredo-Ribeiro, R.D.C.L.; Braga, M. Metabolic and structural changes during early maturation of Inga vera seeds are consistent with the lack of a desiccation phase. J. Plant Physiol. 2013, 170, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Bayer, M.; Slane, D.; Jürgens, G. Early plant embryogenesis—Dark ages or dark matter? Curr. Opin. Plant Boil. 2017, 35, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Radoeva, T.; Vaddepalli, P.; Zhang, Z.; Weijers, D. Evolution, Initiation, and Diversity in Early Plant Embryogenesis. Dev. Cell 2019, 50, 533–543. [Google Scholar] [CrossRef]
- Nogueira, F.; Palmisano, G.; Soares, E.L.; Shah, M.; Soares, A.A.; Roepstorff, P.; Campos, F.; Domont, G.B. Proteomic profile of the nucellus of castor bean (Ricinus communis L.) seeds during development. J. Proteom. 2012, 75, 1933–1939. [Google Scholar] [CrossRef]
- Anderson, O.D. The Spectrum of Major Seed Storage Genes and Proteins in Oats (Avena sativa). PLoS ONE 2014, 9, e83569. [Google Scholar] [CrossRef]
- Guo, G.; Lv, D.; Yan, X.; Subburaj, S.; Ge, P.; Li, X.; Hu, Y.; Yan, Y.-M. Proteome characterization of developing grains in bread wheat cultivars (Triticum aestivum L.). BMC Plant Boil. 2012, 12, 147. [Google Scholar] [CrossRef]
- Wang, W.-Q.; Liu, S.-J.; Song, S.-Q.; Møller, I.M. Proteomics of seed development, desiccation tolerance, germination and vigor. Plant Physiol. Biochem. 2015, 86, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Devic, M.; Roscoe, T. Seed maturation: Simplification of control networks in plants. Plant Sci. 2016, 252, 335–346. [Google Scholar] [CrossRef] [PubMed]
- Parreira, J.; Balestrazzi, A.; Fevereiro, P.; Araújo, S. Maintaining Genome Integrity during Seed Development in Phaseolus vulgaris L.: Evidence from a Transcriptomic Profiling Study. Genes 2018, 9, 463. [Google Scholar] [CrossRef] [PubMed]
- Kurek, K.; Plitta-Michalak, B.; Ratajczak, E. Reactive Oxygen Species as Potential Drivers of the Seed Aging Process. Plants 2019, 8, 174. [Google Scholar] [CrossRef]
- Mertens, J.; Aliyu, H.; Cowan, D.A. LEA Proteins and the Evolution of the WHy Domain. Appl. Environ. Microbiol. 2018, 84, e00539-18. [Google Scholar] [CrossRef]
- Dintu, K.P.; Sibi, C.V.; Ravichandran, P.; Satheeshkumar, K. Vivipary in Ophiorrhiza mungos L.—A rare phenomenon in angiosperms. Plant Boil. 2014, 17, 294–295. [Google Scholar] [CrossRef]
- Mccarty, D.R.; Carson, C.B.; Stinard, P.S.; Robertson, D.S. Molecular Analysis of viviparous-1: An Abscisic Acid-Insensitive Mutant of Maize. Plant Cell 1989, 1, 523–532. [Google Scholar] [CrossRef]
- Agrawal, G.K. Screening of the Rice Viviparous Mutants Generated by Endogenous Retrotransposon Tos17 Insertion. Tagging of a Zeaxanthin Epoxidase Gene and a Novel OsTATC Gene. Plant Physiol. 2001, 125, 1248–1257. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, L.; Xu, X.; Qu, W.; Li, J.; Xu, X.; Wang, A. Seed development and viviparous germination in one accession of a tomato rin mutant. Breed. Sci. 2016, 66, 372–380. [Google Scholar] [CrossRef]
- Hong, L.; Su, W.; Zhang, Y.; Ye, C.; Shen, Y.; Li, Q.Q. Transcriptome profiling during mangrove viviparity in response to abscisic acid. Sci. Rep. 2018, 8, 770. [Google Scholar] [CrossRef]
- Farnsworth, E.J.; Farrant, J.M. Reductions in abscisic acid are linked with viviparous reproduction in mangroves. Am. J. Bot. 1998, 85, 760–769. [Google Scholar] [CrossRef] [PubMed]
- Copley, T.R.; Duceppe, M.-O.; O’Donoughue, L.S. Identification of novel loci associated with maturity and yield traits in early maturity soybean plant introduction lines. BMC Genom. 2018, 19, 167. [Google Scholar] [CrossRef] [PubMed]
- Tar’An, B.; Warkentin, T.; Somers, D.; Miranda, D.; Vandenberg, A.; Blade, S.; Bing, D. Identification of quantitative trait loci for grain yield, seed protein concentration and maturity in field pea (Pisum sativum L.). Euphytica 2004, 136, 297–306. [Google Scholar] [CrossRef]
- Rosenberg, L.A.; Rinne, R.W. Changes in Seed Constituents During Germination and Seedling Growth of Precociously Matured Soybean Seeds (Glycine max). Ann. Bot. 1987, 60, 705–712. [Google Scholar] [CrossRef]
- Rosenberg, L.A.; Rinne, R.W. Protein Synthesis during Natural and Precocious Soybean Seed (Glycine max [L.] Merr.) Maturation. Plant Physiol. 1988, 87, 474–478. [Google Scholar] [CrossRef]
- Pradhan, S.; Bandhiwal, N.; Shah, N.; Kant, C.; Gaur, R.; Bhatia, S. Global transcriptome analysis of developing chickpea (Cicer arietinum L.) seeds. Front. Plant Sci. 2014, 5, 698. [Google Scholar] [CrossRef]
- Jones, S.I.; Vodkin, L.O. Using RNA-Seq to Profile Soybean Seed Development from Fertilization to Maturity. PLoS ONE 2013, 8, e59270. [Google Scholar] [CrossRef]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.-L.; Shou, H. Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef]
- Gallardo, K.; Firnhaber, C.; Zuber, H.; Héricher, D.; Belghazi, M.; Henry, C.; Küster, H.; Thompson, R. A Combined Proteome and Transcriptome Analysis of DevelopingMedicago truncatulaSeeds. Mol. Cell. Proteom. 2007, 6, 2165–2179. [Google Scholar] [CrossRef]
- Terrasson, E.; Buitink, J.; Righetti, K.; Vu, B.L.; Pelletier, S.; Zinsmeister, J.; Lalanne, D.; Leprince, O. An emerging picture of the seed desiccome: Confirmed regulators and newcomers identified using transcriptome comparison. Front. Plant Sci. 2013, 4, 497. [Google Scholar] [CrossRef]
- Redekar, N.R.; Biyashev, R.M.; Jensen, R.V.; Helm, R.; Grabau, E.A.; Maroof, M.S. Genome-wide transcriptome analyses of developing seeds from low and normal phytic acid soybean lines. BMC Genom. 2015, 16, 1074. [Google Scholar] [CrossRef] [PubMed]
- Parreira, J.; Bouraada, J.; Fitzpatrick, M.; Silvestre, S.; Da Silva, A.B.; Da Silva, J.M.; Almeida, A.; Fevereiro, P.; Altelaar, M.; Araújo, S. Differential proteomics reveals the hallmarks of seed development in common bean (Phaseolus vulgaris L.). J. Proteom. 2016, 143, 188–198. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, G.; Xu, S.; Mao, W.; Hu, Q.; Gong, Y. Comparative Transcriptomic Analyses of Vegetable and Grain Pea (Pisum sativum L.) Seed Development. Front. Plant Sci. 2015, 6, 189. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Osuna, D.; Colville, L.; Lorenzo, O.; Graeber, K.; Küster, H.; Leubner-Metzger, G.; Kranner, I. Transcriptome-Wide Mapping of Pea Seed Ageing Reveals a Pivotal Role for Genes Related to Oxidative Stress and Programmed Cell Death. PLoS ONE 2013, 8, e78471. [Google Scholar] [CrossRef]
- Mamontova, T.; Afonin, A.; Ihling, C.; Soboleva, A.; Lukasheva, E.; Sulima, A.S.; Shtark, O.Y.; Akhtemova, G.A.; Povydysh, M.N.; Sinz, A.; et al. Profiling of Seed Proteome in Pea (Pisum sativum L.) Lines Characterized with High and Low Responsivity to Combined Inoculation with Nodule Bacteria and Arbuscular Mycorrhizal Fungi. Molecules 2019, 24, 1603. [Google Scholar] [CrossRef]
- Borisov, A.Y.; Rozov, S.M.; Tsyganov, V.E.; Morzhina, E.V.; Lebsky, V.K.; Tikhonovich, I.A. Sequential functioning of Sym-13 and Sym-31, two genes affecting symbiosome development in root nodules of pea (Pisum sativum L.). Mol. Genet. Genom. 1997, 254, 592–598. [Google Scholar] [CrossRef]
- Berdnikov, V.A.; Rozov, S.M.R.; Bogdanova, V.S. Construction of a series of laboratory pea lines. In Special Plant Genetics; Academy of Sciences of the Ukrainian SSR: Kiev, USSR, 1989; Volume 1, pp. 26–27. [Google Scholar]
- Knott, C.M. A key for stages of development of the pea (Pisum sativum). Ann. Appl. Boil. 1987, 111, 233–245. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap: A Fast, Accurate, Splice-Aware Aligner (No. LBNL-7065E). Available online: https://www.osti.gov/biblio/1241166 (accessed on 17 March 2014).
- Song, L.; Florea, L. Rcorrector: Efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 2015, 4, 48. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Boil. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef]
- Alves-Carvalho, S.; Aubert, G.; Carrère, S.; Cruaud, C.; Brochot, A.-L.; Jacquin, F.; Klein, A.; Martin, C.; Boucherot, K.; Kreplak, J.; et al. Full-lengthde novoassembly of RNA-seq data in pea (Pisum sativumL.) provides a gene expression atlas and gives insights into root nodulation in this species. Plant J. 2015, 84, 1–19. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 2018, 34, 3094–3100. [Google Scholar] [CrossRef] [PubMed]
- Bushmanova, E.; Suvorov, V.; Prjibelski, A.D.; Antipov, D.; Lapidus, A. rnaQUAST: A quality assessment tool for de novo transcriptome assemblies: Table 1. Bioinformatics 2016, 32, 2210–2212. [Google Scholar] [CrossRef]
- Bryant, D.M.; Johnson, K.; DiTommaso, T.; Tickle, T.; Couger, M.B.; Payzin-Dogru, D.; Lee, T.J.; Leigh, N.; Kuo, T.-H.; Davis, F.G.; et al. A Tissue-Mapped Axolotl De Novo Transcriptome Enables Identification of Limb Regeneration Factors. Cell Rep. 2017, 18, 762–776. [Google Scholar] [CrossRef]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef]
- Sonnhammer, E.L.; von Heijne, G.; Krogh, A. A hidden Markov model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1998, 6, 175–182. [Google Scholar]
- Nielsen, H. Predicting Secretory Proteins with SignalP. In Advanced Structural Safety Studies; Springer Science and Business Media LLC: Berlin/Heidelberg, Germany, 2017; Volume 1611, pp. 59–73. [Google Scholar]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2013, 37, 1250–1258. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Morishima, K. BlastKOALA and GhostKOALA: KEGG Tools for Functional Characterization of Genome and Metagenome Sequences. J. Mol. Boil. 2016, 428, 726–731. [Google Scholar] [CrossRef] [PubMed]
- Huerta-Cepas, J.; Szklarczyk, D.; Forslund, S.K.; Cook, H.V.; Heller, D.; Walter, M.C.; Rattei, T.; Mende, D.R.; Sunagawa, S.; Kuhn, M.; et al. eggNOG 4.5: A hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 2015, 44, D286–D293. [Google Scholar] [CrossRef] [PubMed]
- SRA Toolkit Development Team. SRA Toolkit, Version 2.9.6. Available online: https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?cmd=show&f=software&m=software&s=software (accessed on 7 May 2019).
- Seqtk. Available online: https://omictools.com/seqtk-tool (accessed on 7 May 2019).
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Pimentel, H.; Bray, N.L.; Puente, S.; Melsted, P.; Pachter, L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 2017, 14, 687–690. [Google Scholar] [CrossRef]
- topGO: Enrichment analysis for Gene Ontology. R Package, version 2.34.0. Available online: https://bioconductor.org/packages/release/bioc/html/topGO.html (accessed on 7 May 2019).
- Ginestet, C. ggplot2: Elegant Graphics for Data Analysis. J. R. Stat. Soc. Ser. A 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Neuwirth, E. RColorBrewer: ColorBrewer palettes. R Packag. version 1.1-2 2014. Available online: https://www.rdocumentation.org/packages/RColorBrewer/versions/1.1-2 (accessed on 21 December 2019).
- Vu, V.Q. ggbiplot: A ggplot2 based biplot. R package version 0.55. Available online: https://www.rdocumentation.org/packages/ggbiplot/versions/0.55 (accessed on 30 June 2019).
- Witten, D.M. Classification and clustering of sequencing data using a Poisson model. Ann. Appl. Stat. 2011, 5, 2493–2518. [Google Scholar] [CrossRef]
- Kolde, R. pheatmap: Pretty Heatmaps. R package, version 1.0.12. Available online: https://cran.r-project.org/web/packages/pheatmap/pheatmap.pdf (accessed on 4 January 2019).
- Wild, F. An open source LSA package for R. R Package version 0.73.1 2015. Available online: https://cran.r-project.org/web/packages/lsa/index.html (accessed on 15 July 2019).
- Löffler-Wirth, H.; Kalcher, M.; Binder, H. oposSOM: R-package for high-dimensional portraying of genome-wide expression landscapes on bioconductor: Fig. 1. Bioinform. 2015, 31, 3225–3227. [Google Scholar] [CrossRef]
- Cook, R.D. Detection of Influential Observation in Linear Regression. Technometrics 1977, 19, 15–18. [Google Scholar]
- The R Project for Statistical Computing. Available online: https://www.r-project.org/index.html (accessed on 22 March 2020).
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2012, 29, 15–21. [Google Scholar] [CrossRef]
- PicardTools. Available online: http://broadinstitute.github.io/picard (accessed on 22 March 2020).
- Van der Auwera, G.A.; Carneiro, M.O.; Hartl, C.; Poplin, R.; Del Angel, G.; Levy-Moonshine, A.; Jordan, T.; Shakir, K.; Roazen, D.; Thibault, J.; et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr. Protoc. Bioinform. 2013, 43, 11.10.1–11.10.33. [Google Scholar]
- Cingolani, P.; Platts, A.E.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Davidson, N.; Hawkins, A.D.K.; Oshlack, A. SuperTranscripts: A data driven reference for analysis and visualisation of transcriptomes. Genome Boil. 2017, 18, 148. [Google Scholar] [CrossRef] [PubMed]
- Conway, J.R.; Lex, A.; Gehlenborg, N. UpSetR: An R package for the visualization of intersecting sets and their properties. Bioinformatics 2017, 33, 2938–2940. [Google Scholar] [CrossRef] [PubMed]
- Nodine, M.D.; Bartel, B. MicroRNAs prevent precocious gene expression and enable pattern formation during plant embryogenesis. Genes Dev. 2010, 24, 2678–2692. [Google Scholar] [CrossRef]
- Willmann, M.R.; Mehalick, A.J.; Packer, R.L.; Jenik, P. MicroRNAs regulate the timing of embryo maturation in Arabidopsis. Plant Physiol. 2011, 155, 1871–1884. [Google Scholar] [CrossRef]
- Hajduch, M.; Casteel, J.E.; Hurrelmeyer, K.E.; Song, Z.; Agrawal, G.K.; Thelen, J.J. Proteomic Analysis of Seed Filling in Brassica napus. Developmental Characterization of Metabolic Isozymes Using High-Resolution Two-Dimensional Gel Electrophoresis1[W]. Plant Physiol. 2006, 141, 32–46. [Google Scholar] [CrossRef]
- Hajduch, M.; Hearne, L.B.; Miernyk, J.A.; Casteel, J.E.; Joshi, T.; Agrawal, G.K.; Song, Z.; Zhou, M.; Xu, N.; Thelen, J.J. Systems Analysis of Seed Filling in Arabidopsis: Using General Linear Modeling to Assess Concordance of Transcript and Protein Expression1[C][W][OA]. Plant Physiol. 2010, 152, 2078–2087. [Google Scholar] [CrossRef]
- Aghamirzaie, D.; Nabiyouni, M.; Fang, Y.; Klumas, C.; Heath, L.S.; Grene, R.; Collakova, E. Changes in RNA Splicing in Developing Soybean (Glycine max) Embryos. Biology 2013, 2, 1311–1337. [Google Scholar] [CrossRef]
- Srinivasan, A.; Jiménez-Gómez, J.M.; Fornara, F.; Soppe, W.J.J.; Brambilla, V. Alternative splicing enhances transcriptome complexity in desiccating seeds. J. Integr. Plant Boil. 2016, 58, 947–958. [Google Scholar] [CrossRef]
- Sato, Y.; Antonio, B.; Namiki, N.; Takehisa, H.; Minami, H.; Kamatsuki, K.; Sugimoto, K.; Shimizu, Y.; Hirochika, H.; Nagamura, Y. RiceXPro: A platform for monitoring gene expression in japonica rice grown under natural field conditions. Nucleic Acids Res. 2010, 39, D1141–D1148. [Google Scholar] [CrossRef] [PubMed]
- Damaris, R.N.; Lin, Z.; Yang, P.; He, D. The Rice Alpha-Amylase, Conserved Regulator of Seed Maturation and Germination. Int. J. Mol. Sci. 2019, 20, 450. [Google Scholar] [CrossRef] [PubMed]
- Rivin, C.J.; Grudt, T. Abscisic Acid and the Developmental Regulation of Embryo Storage Proteins in Maize. Plant Physiol. 1991, 95, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, S.S.; Miksche, J.P. DNA, RNA, protein and heterochromatin changes during embryo development and germination of soybean (Glycine max L.). J. Mol. Histol. 1983, 15, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Larkins, B.A.; Dilkes, B.P.; Dante, R.A.; Coelho, C.; Woo, Y.; Liu, Y. Investigating the hows and whys of DNA endoreduplication. J. Exp. Bot. 2001, 52, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Bensen, R.J.; Boyer, J.S.; Mullet, J.E. Water Deficit-Induced Changes in Abscisic Acid, Growth, Polysomes, and Translatable RNA in Soybean Hypocotyls. Plant Physiol. 1988, 88, 289–294. [Google Scholar] [CrossRef]
- Guo, J.; Wang, S.; Valerius, O.; Hall, H.; Zeng, Q.; Li, J.-F.; Weston, D.J.; Ellis, B.E.; Chen, J. Involvement of Arabidopsis RACK1 in protein translation and its regulation by abscisic acid. Plant Physiol. 2010, 155, 370–383. [Google Scholar] [CrossRef]
- Negi, P.; Rai, A.N.; Suprasanna, P. Moving through the Stressed Genome: Emerging Regulatory Roles for Transposons in Plant Stress Response. Front. Plant Sci. 2016, 7, 1448. [Google Scholar] [CrossRef]
- Dubin, M.; Scheid, O.M.; Becker, C. Transposons: A blessing curse. Curr. Opin. Plant Boil. 2018, 42, 23–29. [Google Scholar] [CrossRef]
- Do Kim, K.; El Baidouri, M.; Abernathy, B.; Iwata-Otsubo, A.; Chavarro, C.; Gonzales, M.; Libault, M.; Grimwood, J.; Jackson, S.A. A Comparative Epigenomic Analysis of Polyploidy-Derived Genes in Soybean and Common Bean. Plant Physiol. 2015, 168, 1433–1447. [Google Scholar] [CrossRef]
- An, Y.-Q.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic Changes of Genome-Wide DNA Methylation during Soybean Seed Development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.-Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.-F.; Chen, P.-Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L.; et al. Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. 2017, 114, E9730–E9739. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lang, Z.; Zhu, J. Dynamics and function of DNA methylation in plants. Nat. Rev. Mol. Cell Boil. 2018, 19, 489–506. [Google Scholar] [CrossRef] [PubMed]
- Zabala, G.; Vodkin, L.O. Methylation Affects Transposition and Splicing of a Large CACTA Transposon from a MYB Transcription Factor Regulating Anthocyanin Synthase Genes in Soybean Seed Coats. PLoS ONE 2014, 9, e111959. [Google Scholar] [CrossRef]
- Corem, S.; Faigenboim, A.; Jouffroy, O.; Maumus, F.; Arazi, T.; Bouché, N. Redistribution of CHH Methylation and Small Interfering RNAs across the Genome of Tomato ddm1 Mutants. Plant Cell 2018, 30, 1628–1644. [Google Scholar] [CrossRef]
- Ito, H.; Gaubert, H.; Bucher, E.; Mirouze, M.; Vaillant, I.; Paszkowski, J. An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 2011, 472, 115–119. [Google Scholar] [CrossRef]
- Ito, H.; Yoshida, T.; Tsukahara, S.; Kawabe, A. Evolution of the ONSEN retrotransposon family activated upon heat stress in Brassicaceae. Gene 2013, 518, 256–261. [Google Scholar] [CrossRef]
- Ito, H.; Kim, J.-M.; Matsunaga, W.; Saze, H.; Matsui, A.; Endo, T.A.; Harukawa, Y.; Takagi, H.; Yaegashi, H.; Masuta, Y.; et al. A Stress-Activated Transposon in Arabidopsis Induces Transgenerational Abscisic Acid Insensitivity. Sci. Rep. 2016, 6, 23181. [Google Scholar] [CrossRef]














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Assembly | Per Transcript Statistics | Longest Isoform Statistics | Percent of Reads Aligned | ||||
|---|---|---|---|---|---|---|---|
| N50 | Median Contig Length | Average Contig Length | N50 | Median Contig Length | Average Contig Length | ||
| Initial | 1970 | 781 | 1199.79 | 1677 | 461 | 901.07 | 98.83 |
| Final | 2034 | 1415 | 1636.26 | 2073 | 1454 | 1665.17 | 77.62 |
| Cultivar | Moderate | High | Modifier | |||
|---|---|---|---|---|---|---|
| Homozygous | Heterozygous | Homozygous | Heterozygous | Homozygous | Heterozygous | |
| Sprint-2 | NA * | 29,314 | NA * | 893 | NA * | 15,799 |
| Zhewan-1 | 2934 | 4353 | 17 | 57 | 2062 | 2566 |
| Zhongwan-6 | 1725 | 2104 | 12 | 23 | 1175 | 1096 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Malovichko, Y.V.; Shtark, O.Y.; Vasileva, E.N.; Nizhnikov, A.A.; Antonets, K.S. Transcriptomic Insights into Mechanisms of Early Seed Maturation in the Garden Pea (Pisum sativum L.). Cells 2020, 9, 779. https://doi.org/10.3390/cells9030779
Malovichko YV, Shtark OY, Vasileva EN, Nizhnikov AA, Antonets KS. Transcriptomic Insights into Mechanisms of Early Seed Maturation in the Garden Pea (Pisum sativum L.). Cells. 2020; 9(3):779. https://doi.org/10.3390/cells9030779
Chicago/Turabian StyleMalovichko, Yury V., Oksana Y. Shtark, Ekaterina N. Vasileva, Anton A. Nizhnikov, and Kirill S. Antonets. 2020. "Transcriptomic Insights into Mechanisms of Early Seed Maturation in the Garden Pea (Pisum sativum L.)" Cells 9, no. 3: 779. https://doi.org/10.3390/cells9030779
APA StyleMalovichko, Y. V., Shtark, O. Y., Vasileva, E. N., Nizhnikov, A. A., & Antonets, K. S. (2020). Transcriptomic Insights into Mechanisms of Early Seed Maturation in the Garden Pea (Pisum sativum L.). Cells, 9(3), 779. https://doi.org/10.3390/cells9030779