Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib+




 ,
, 
Abstract

1. Introduction
2. Materials and Methods
2.1. Cell Lines and Reagents
2.2. Immunoblotting
2.3. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
2.4. Cell Viability Assays
2.5. RNAi Transfection
2.6. Radiolabeled Glucose Uptake Assay
2.7. Gas Chromatography–Mass Spectrometry (GC–MS)
2.8. Metabolomics Data Analysis
2.9. Seahorse XFe96 Metabolic Assays
2.10. Analysis of Human Datasets
2.11. Statistical Analysis
3. Results
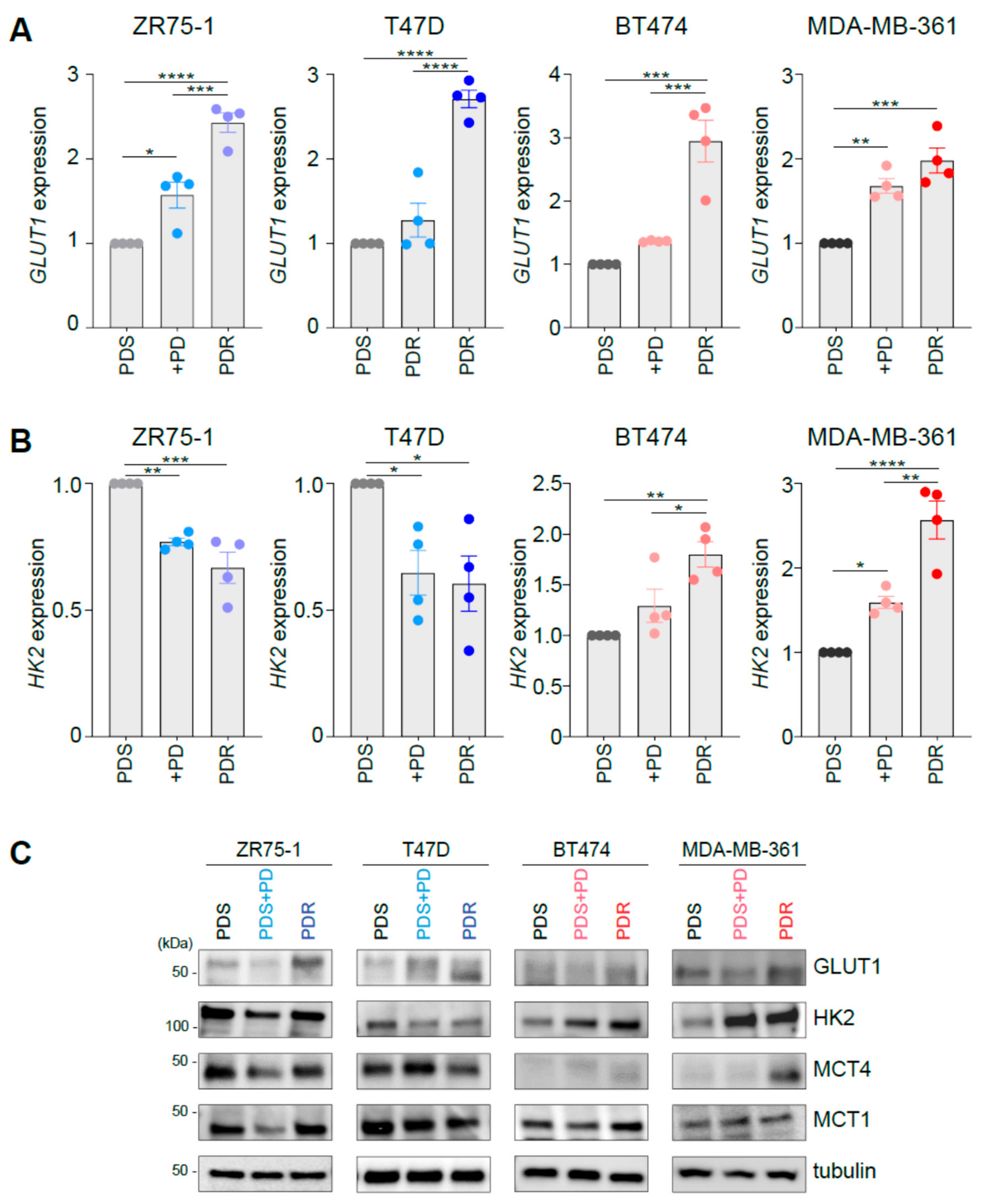
3.1. Palbociclib Impacts on the Expression of Key Players Involved in Glucose Catabolism
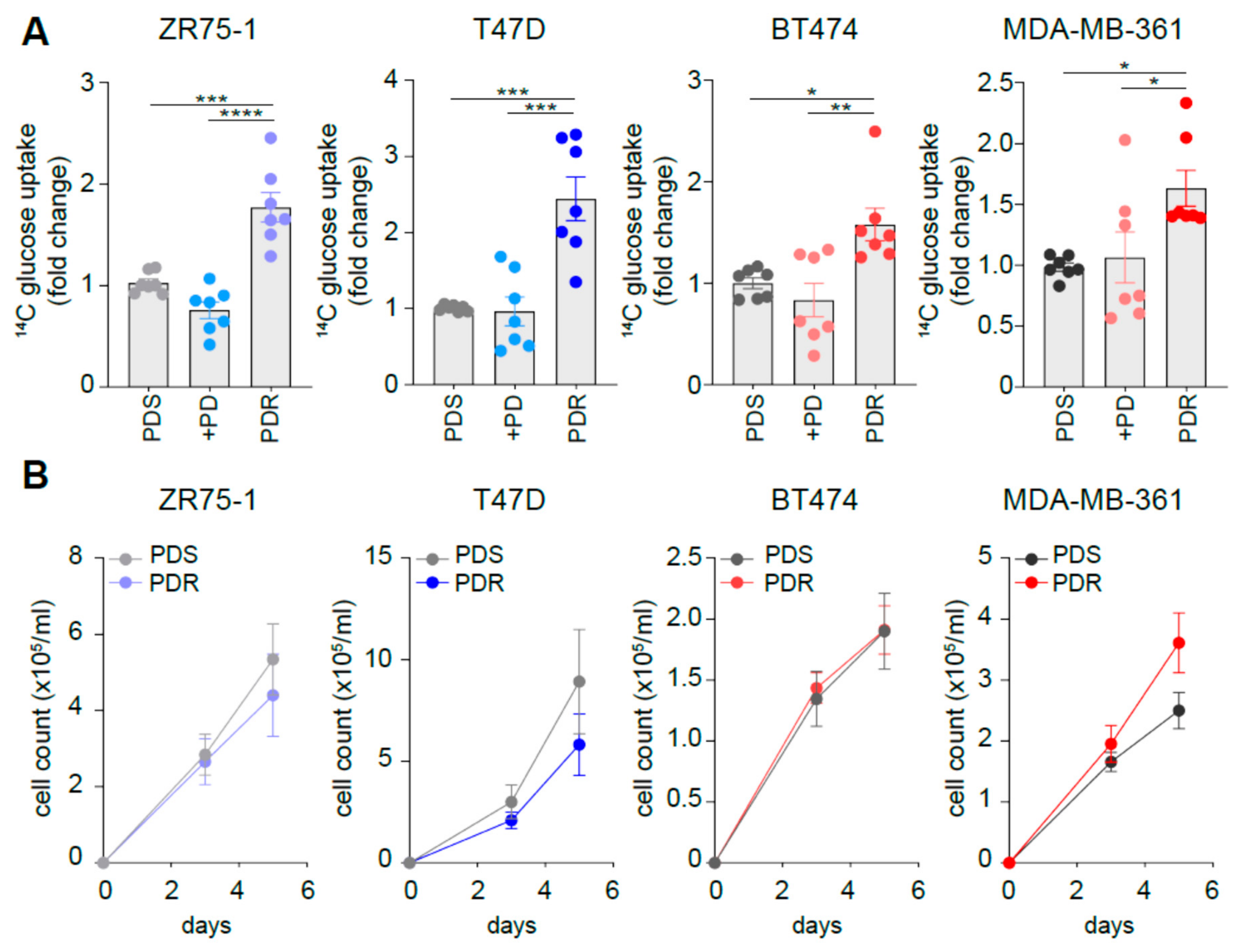
3.2. Resistant Cells Show Comparable Growth Rates When Compared to Sensitive Counterpart While Increasing Glucose Uptake
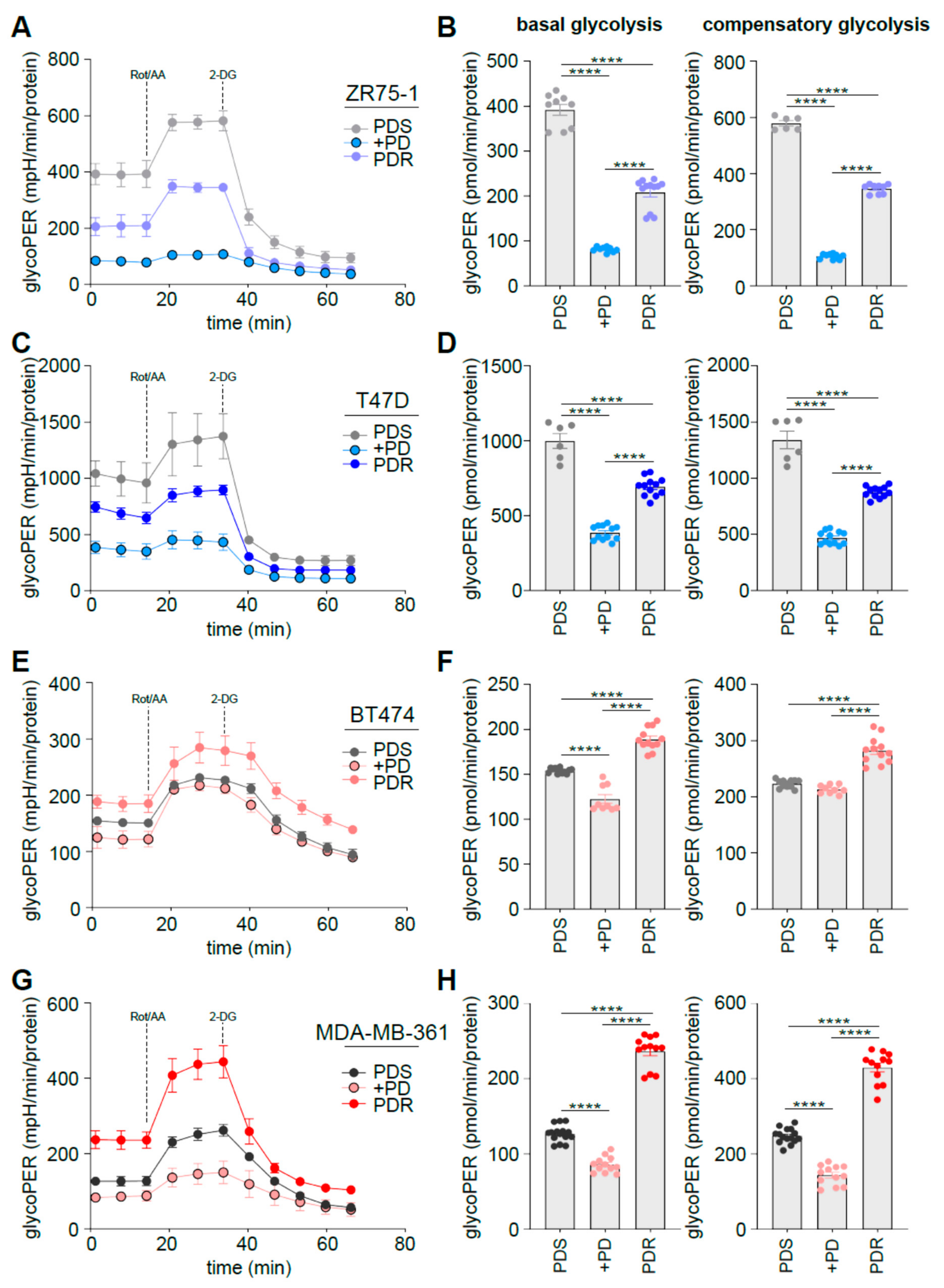
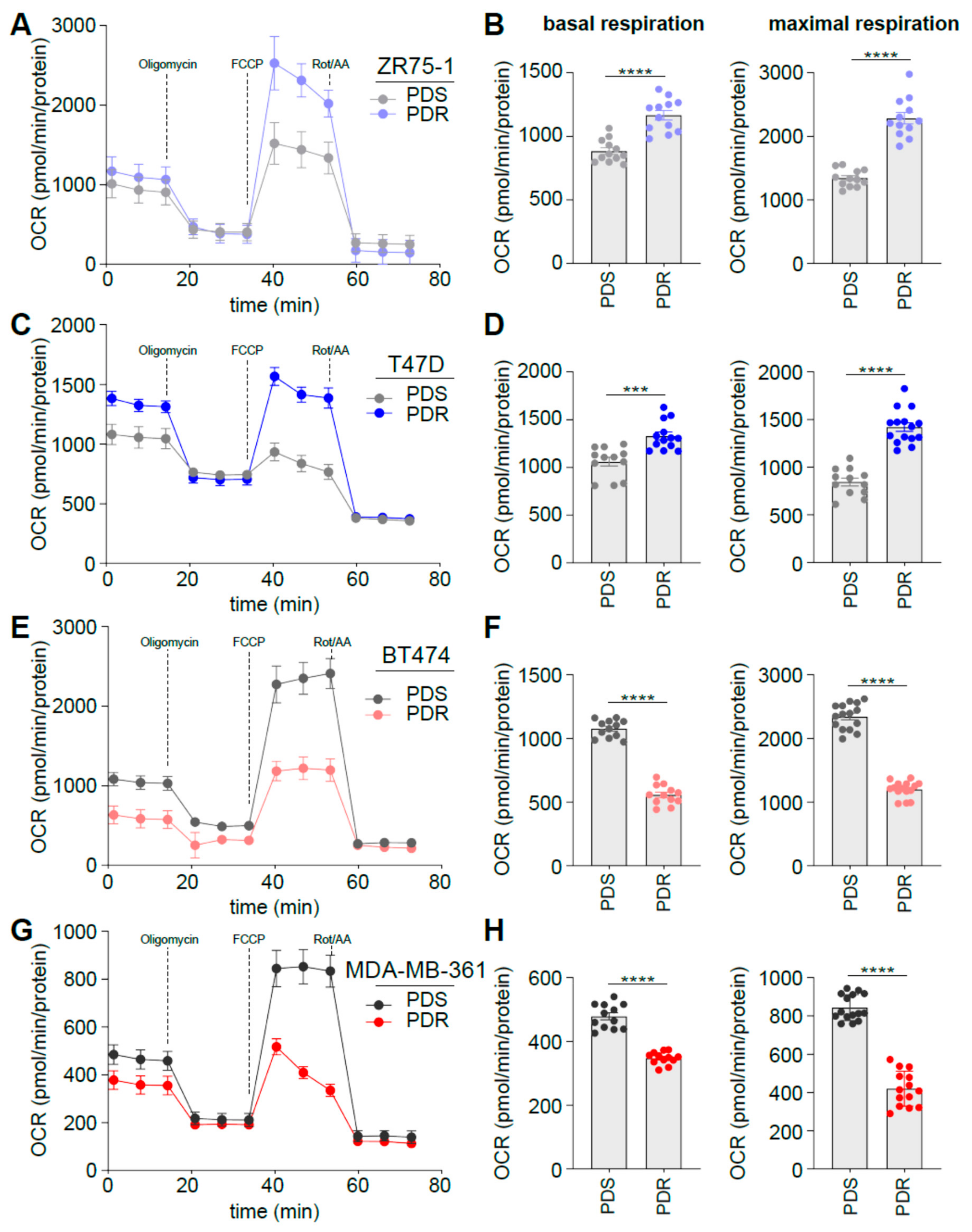
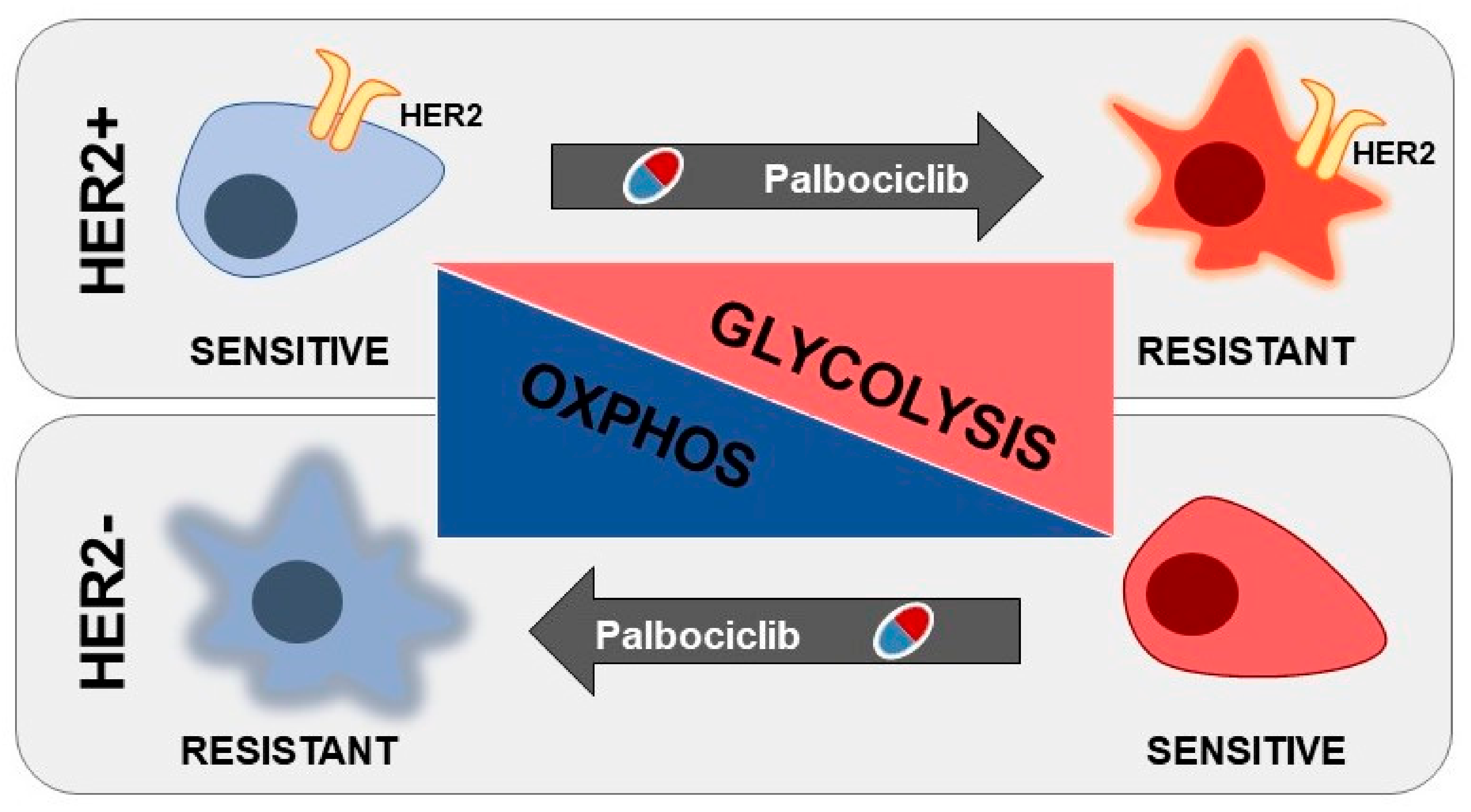
3.3. Human Epidermal Growth Factor Receptor 2 (HER2) Status Impacts on Glucose Catabolism and Reveals Different Glucose Dependency during Acute and Chronic Palbociclib Administration
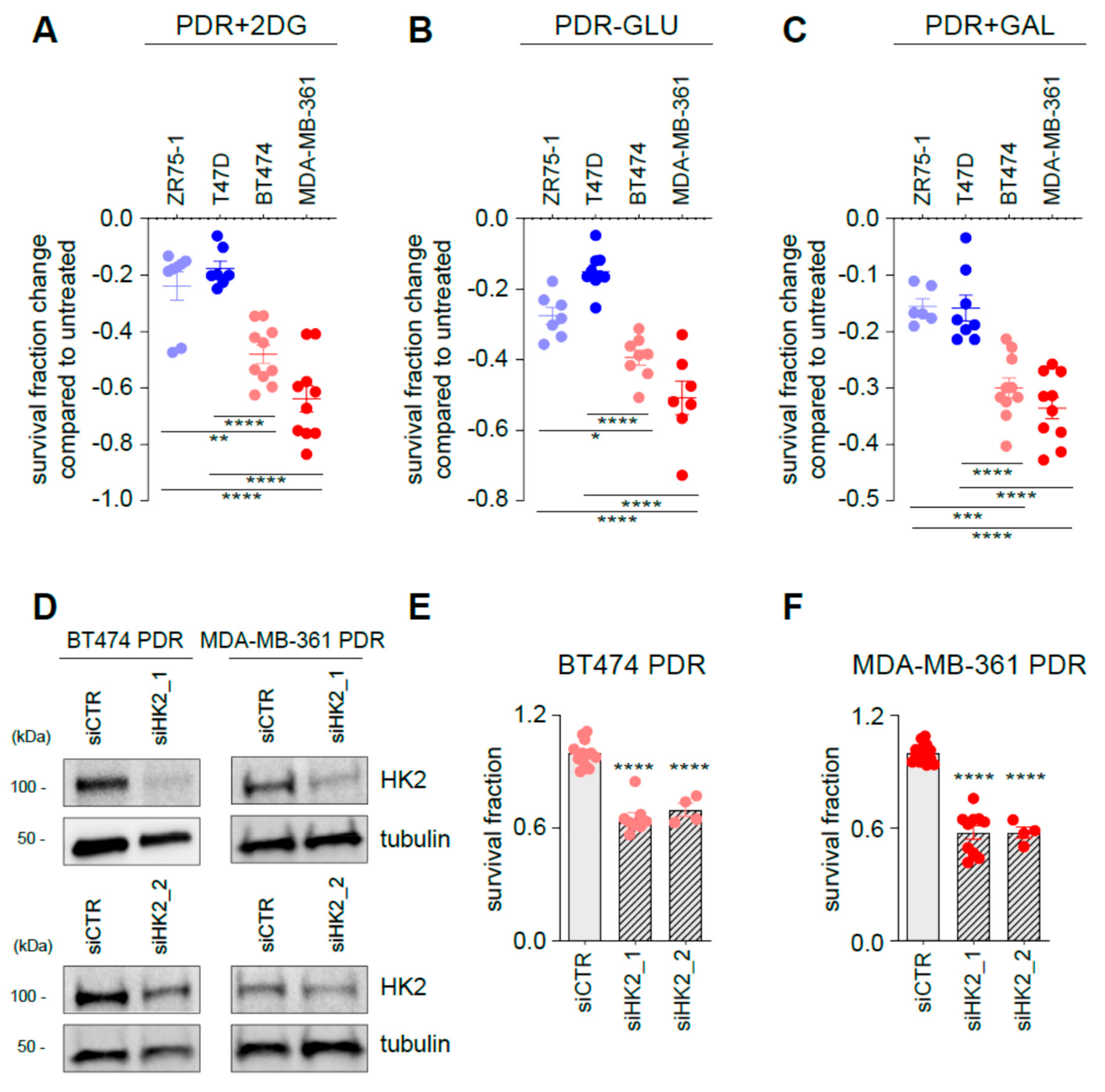
3.4. Targeting Glycolysis Resensitizes ER+/HER2+ Palbociclib-Resistant (PDR) Cells to Palbociclib
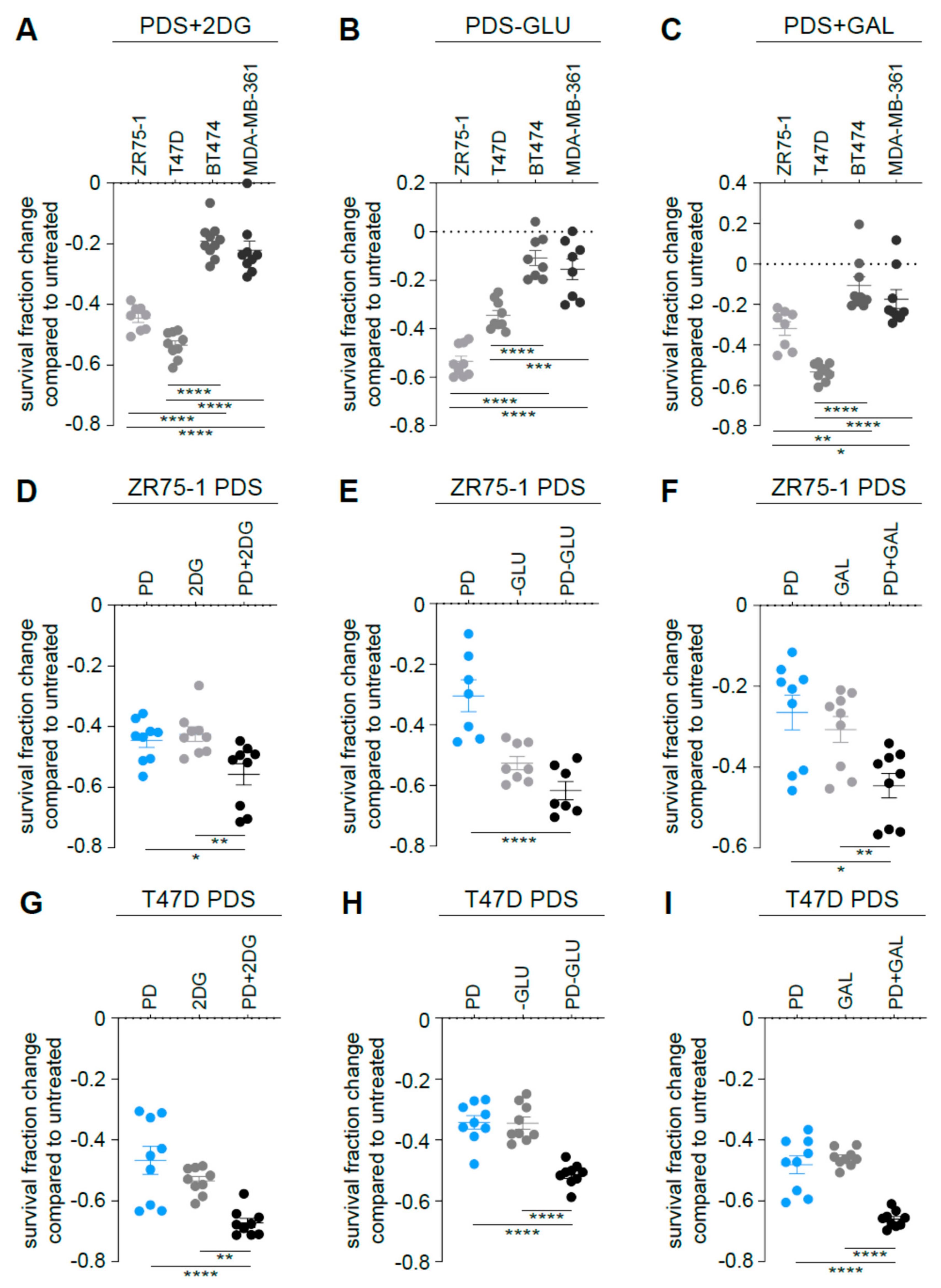
3.5. Targeting Glycolysis Potentiates Palbociclib Effects on ER+/HER2− Parental Cells
3.6. Metabolomic Profiling Identifies a Distinct Intracellular Metabolites Pattern between HER2+ and HER2− ER+ PDR Cells
3.7. Enhanced HK2 Expression Levels Identify a Subset of Poor Prognosis Tumors
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Finn, R.S.; Crown, J.P.; Lang, I.; Boer, K.; Bondarenko, I.M.; Kulyk, S.O.; Ettl, J.; Patel, R.; Pinter, T.; Schmidt, M.; et al. The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): A randomised phase 2 study. Lancet Oncol. 2015, 16, 25–35. [Google Scholar] [CrossRef]
- Turner, N.C.; Huang Bartlett, C.; Cristofanilli, M. Palbociclib in Hormone-Receptor-Positive Advanced Breast Cancer. N. Engl. J. Med. 2015, 373, 1672–1673. [Google Scholar] [CrossRef] [PubMed]
- Hortobagyi, G.N.; Stemmer, S.M.; Burris, H.A.; Yap, Y.S.; Sonke, G.S.; Paluch-Shimon, S.; Campone, M.; Blackwell, K.L.; André, F.; Winer, E.P.; et al. Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N. Engl. J. Med. 2018, 379, 2582. [Google Scholar] [CrossRef] [PubMed]
- Goetz, M.P.; Toi, M.; Campone, M.; Sohn, J.; Paluch-Shimon, S.; Huober, J.; Park, I.H.; Trédan, O.; Chen, S.C.; Manso, L.; et al. MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J. Clin. Oncol. 2017, 35, 3638–3646. [Google Scholar] [CrossRef] [PubMed]
- Sobhani, N.; D’Angelo, A.; Pittacolo, M.; Roviello, G.; Miccoli, A.; Corona, S.P.; Bernocchi, O.; Generali, D.; Otto, T. Updates on the CDK4/6 Inhibitory Strategy and Combinations in Breast Cancer. Cells 2019, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.S.; Martin, M.; Rugo, H.S.; Jones, S.E.; Im, S.A.; Gelmon, K.A.; Harbeck, N.; Lipatov, O.N.; Walshe, J.M.; Moulder, S.L.; et al. PALOMA-2: Primary results from a phase III trial of palbociclib (P) with letrozole (L) compared with letrozole alone in postmenopausal women with ER+/HER2– advanced breast cancer (ABC). J. Clin. Oncol. 2017, 34, 507. [Google Scholar] [CrossRef]
- Pandey, K.; An, H.J.; Kim, S.K.; Lee, S.A.; Kim, S.; Lim, S.M.; Kim, G.M.; Sohn, J.; Moon, Y.W. Molecular mechanisms of resistance to CDK4/6 inhibitors in breast cancer: A review. Int. J. Cancer 2019, 145, 1179–1188. [Google Scholar] [CrossRef]
- Guarducci, C.; Bonechi, M.; Boccalini, G.; Benelli, M.; Risi, E.; Di Leo, A.; Malorni, L.; Migliaccio, I. Mechanisms of Resistance to CDK4/6 Inhibitors in Breast Cancer and Potential Biomarkers of Response. Breast Care 2017, 12, 304–308. [Google Scholar] [CrossRef]
- Vijayaraghavan, S.; Karakas, C.; Doostan, I.; Chen, X.; Bui, T.; Yi, M.; Raghavendra, A.S.; Zhao, Y.; Bashour, S.I.; Ibrahim, N.K.; et al. CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat. Commun. 2017, 8, 15916. [Google Scholar] [CrossRef]
- Wang, H.; Nicolay, B.N.; Chick, J.M.; Gao, X.; Geng, Y.; Ren, H.; Gao, H.; Yang, G.; Williams, J.A.; Suski, J.M.; et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 2017, 546, 426–430. [Google Scholar] [CrossRef]
- Lopez-Mejia, I.C.; Lagarrigue, S.; Giralt, A.; Martinez-Carreres, L.; Zanou, N.; Denechaud, P.D.; Castillo-Armengol, J.; Chavey, C.; Orpinell, M.; Delacuisine, B.; et al. CDK4 Phosphorylates AMPKα2 to Inhibit Its Activity and Repress Fatty Acid Oxidation. Mol. Cell 2017, 68, 336–349. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Dominy, J.E.; Choi, Y.J.; Jurczak, M.; Tolliday, N.; Camporez, J.P.; Chim, H.; Lim, J.H.; Ruan, H.B.; Yang, X.; et al. Cyclin D1-Cdk4 controls glucose metabolism independently of cell cycle progression. Nature 2014, 510, 547–551. [Google Scholar] [CrossRef] [PubMed]
- Guarducci, C.; Bonechi, M.; Benelli, M.; Biagioni, C.; Boccalini, G.; Romagnoli, D.; Verardo, R.; Schiff, R.; Osborne, C.K.; De Angelis, C.; et al. Cyclin E1 and Rb modulation as common events at time of resistance to palbociclib in hormone receptor-positive breast cancer. NPJ Breast Cancer 2018, 4, 38. [Google Scholar] [CrossRef] [PubMed]
- Bacci, M.; Lorito, N.; Ippolito, L.; Ramazzotti, M.; Luti, S.; Romagnoli, S.; Parri, M.; Bianchini, F.; Cappellesso, F.; Virga, F.; et al. Reprogramming of Amino Acid Transporters to Support Aspartate and Glutamate Dependency Sustains Endocrine Resistance in Breast Cancer. Cell Rep. 2019, 28, 104–118. [Google Scholar] [CrossRef]
- Budczies, J.; Klauschen, F.; Sinn, B.V.; Győrffy, B.; Schmitt, W.D.; Darb-Esfahani, S.; Denkert, C. Cutoff Finder: A comprehensive and straightforward Web application enabling rapid biomarker cutoff optimization. PLoS ONE 2012, 7, e51862. [Google Scholar] [CrossRef]
- Lánczky, A.; Nagy, Á.; Bottai, G.; Munkácsy, G.; Szabó, A.; Santarpia, L.; Győrffy, B. miRpower: A web-tool to validate survival-associated miRNAs utilizing expression data from 2178 breast cancer patients. Breast Cancer Res. Treat. 2016, 160, 439–446. [Google Scholar] [CrossRef]
- Franco, J.; Balaji, U.; Freinkman, E.; Witkiewicz, A.K.; Knudsen, E.S. Metabolic Reprogramming of Pancreatic Cancer Mediated by CDK4/6 Inhibition Elicits Unique Vulnerabilities. Cell Rep. 2016, 14, 979–990. [Google Scholar] [CrossRef]
- Adekola, K.; Rosen, S.T.; Shanmugam, M. Glucose transporters in cancer metabolism. Curr. Opin. Oncol. 2012, 24, 650–654. [Google Scholar] [CrossRef]
- Payen, V.L.; Mina, E.; Van Hée, V.F.; Porporato, P.E.; Sonveaux, P. Monocarboxylate transporters in cancer. Mol. Metab. 2019. [Google Scholar] [CrossRef]
- Benjamin, D.; Robay, D.; Hindupur, S.K.; Pohlmann, J.; Colombi, M.; El-Shemerly, M.Y.; Maira, S.M.; Moroni, C.; Lane, H.A.; Hall, M.N. Dual Inhibition of the Lactate Transporters MCT1 and MCT4 Is Synthetic Lethal with Metformin due to NAD+ Depletion in Cancer Cells. Cell Rep. 2018, 25, 3047–3058. [Google Scholar] [CrossRef]
- Bacci, M.; Giannoni, E.; Fearns, A.; Ribas, R.; Gao, Q.; Taddei, M.L.; Pintus, G.; Dowsett, M.; Isacke, C.M.; Martin, L.A.; et al. miR-155 drives metabolic reprogramming of ER+ breast cancer cells following long-term estrogen deprivation and predicts clinical response to aromatase inhibitors. Cancer Res. 2016, 76, 1615–1626. [Google Scholar] [CrossRef] [PubMed]
- Morandi, A.; Indraccolo, S. Linking metabolic reprogramming to therapy resistance in cancer. Biochim. Biophys. Acta Rev. Cancer 2017, 1868, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.H.; Zhou, M.; Liu, H.; Ding, Y.; Khong, H.T.; Yu, D.; Fodstad, O.; Tan, M. Upregulation of lactate dehydrogenase A by ErbB2 through heat shock factor 1 promotes breast cancer cell glycolysis and growth. Oncogene 2009, 28, 3689–3701. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Liu, H.; Liu, Z.; Ding, Y.; Ledoux, S.P.; Wilson, G.L.; Voellmy, R.; Lin, Y.; Lin, W.; Nahta, R.; et al. Overcoming trastuzumab resistance in breast cancer by targeting dysregulated glucose metabolism. Cancer Res. 2011, 71, 4585–4597. [Google Scholar] [CrossRef]
- Castagnoli, L.; Iorio, E.; Dugo, M.; Koschorke, A.; Faraci, S.; Canese, R.; Casalini, P.; Nanni, P.; Vernieri, C.; Di Nicola, M.; et al. Intratumor lactate levels reflect HER2 addiction status in HER2-positive breast cancer. J. Cell Physiol. 2019, 234, 1768–1779. [Google Scholar] [CrossRef]
- Aguer, C.; Gambarotta, D.; Mailloux, R.J.; Moffat, C.; Dent, R.; McPherson, R.; Harper, M.E. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS ONE 2011, 6, e28536. [Google Scholar] [CrossRef]
- Kind, T.; Wohlgemuth, G.; Lee, D.Y.; Lu, Y.; Palazoglu, M.; Shahbaz, S.; Fiehn, O. FiehnLib: Mass spectral and retention index libraries for metabolomics based on quadrupole and time-of-flight gas chromatography/mass spectrometry. Anal. Chem. 2009, 81, 10038–10048. [Google Scholar] [CrossRef]
- Wolf, A.; Agnihotri, S.; Micallef, J.; Mukherjee, J.; Sabha, N.; Cairns, R.; Hawkins, C.; Guha, A. Hexokinase 2 is a key mediator of aerobic glycolysis and promotes tumor growth in human glioblastoma multiforme. J. Exp. Med. 2011, 208, 313–326. [Google Scholar] [CrossRef]
- Herrera-Abreu, M.T.; Palafox, M.; Asghar, U.; Rivas, M.A.; Cutts, R.J.; Garcia-Murillas, I.; Pearson, A.; Guzman, M.; Rodriguez, O.; Grueso, J.; et al. Early Adaptation and Acquired Resistance to CDK4/6 Inhibition in Estrogen Receptor-Positive Breast Cancer. Cancer Res. 2016, 76, 2301–2313. [Google Scholar] [CrossRef]
- Jansen, V.M.; Bhola, N.E.; Bauer, J.A.; Formisano, L.; Lee, K.M.; Hutchinson, K.E.; Witkiewicz, A.K.; Moore, P.D.; Estrada, M.V.; Sánchez, V.; et al. Kinome-Wide RNA Interference Screen Reveals a Role for PDK1 in Acquired Resistance to CDK4/6 Inhibition in ER-Positive Breast Cancer. Cancer Res. 2017, 77, 2488–2499. [Google Scholar] [CrossRef]
- McCartney, A.; Migliaccio, I.; Bonechi, M.; Biagioni, C.; Romagnoli, D.; De Luca, F.; Galardi, F.; Risi, E.; De Santo, I.; Benelli, M.; et al. Mechanisms of Resistance to CDK4/6 Inhibitors: Potential Implications and Biomarkers for Clinical Practice. Front. Oncol. 2019, 9, 666. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Cretella, D.; Ravelli, A.; Fumarola, C.; La Monica, S.; Digiacomo, G.; Cavazzoni, A.; Alfieri, R.; Biondi, A.; Generali, D.; Bonelli, M.; et al. The anti-tumor efficacy of CDK4/6 inhibition is enhanced by the combination with PI3K/AKT/mTOR inhibitors through impairment of glucose metabolism in TNBC cells. J. Exp. Clin. Cancer Res. 2018, 37, 72. [Google Scholar] [CrossRef]
- Warth, B.; Raffeiner, P.; Granados, A.; Huan, T.; Fang, M.; Forsberg, E.M.; Benton, H.P.; Goetz, L.; Johnson, C.H.; Siuzdak, G. Metabolomics Reveals that Dietary Xenoestrogens Alter Cellular Metabolism Induced by Palbociclib/Letrozole Combination Cancer Therapy. Cell Chem. Biol. 2018, 25, 291–300. [Google Scholar] [CrossRef]
- Corona, S.P.; Ravelli, A.; Cretella, D.; Cappelletti, M.R.; Zanotti, L.; Dester, M.; Gobbi, A.; Petronini, P.G.; Generali, D. CDK4/6 inhibitors in HER2-positive breast cancer. Crit. Rev. Oncol. Hematol. 2017, 112, 208–214. [Google Scholar] [CrossRef]
- Goel, S.; Wang, Q.; Watt, A.C.; Tolaney, S.M.; Dillon, D.A.; Li, W.; Ramm, S.; Palmer, A.C.; Yuzugullu, H.; Varadan, V.; et al. Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 2016, 29, 255–269. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameters | p Value | Hazard Ratio (HR) | |
|---|---|---|---|
| A | MKI67 | 0.909 | 0.93 (0.28–3.12) |
| ESR1 | 0.118 | 0.11 (0.01–1.75) | |
| HER2 (ERBB2) | 0.340 | 0.55 (0.16–1.88) | |
| HK2 | 0.0082 | 16.67 (2.07–134.21) | |
| B | MKI67 | 0.739 | 0.87 (0.37–2.03) |
| ESR1 | 0.0192 | 0.15 (0.03–0.73) | |
| HER2 (ERBB2) | 0.3505 | 0.66 (0.27–1.59) | |
| HK2 | 0.0188 | 2.99 (1.2–7.47) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lorito, N.; Bacci, M.; Smiriglia, A.; Mannelli, M.; Parri, M.; Comito, G.; Ippolito, L.; Giannoni, E.; Bonechi, M.; Benelli, M.; et al. Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib+. Cells 2020, 9, 668. https://doi.org/10.3390/cells9030668
Lorito N, Bacci M, Smiriglia A, Mannelli M, Parri M, Comito G, Ippolito L, Giannoni E, Bonechi M, Benelli M, et al. Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib+. Cells. 2020; 9(3):668. https://doi.org/10.3390/cells9030668
Chicago/Turabian StyleLorito, Nicla, Marina Bacci, Alfredo Smiriglia, Michele Mannelli, Matteo Parri, Giuseppina Comito, Luigi Ippolito, Elisa Giannoni, Martina Bonechi, Matteo Benelli, and et al. 2020. "Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib+" Cells 9, no. 3: 668. https://doi.org/10.3390/cells9030668
APA StyleLorito, N., Bacci, M., Smiriglia, A., Mannelli, M., Parri, M., Comito, G., Ippolito, L., Giannoni, E., Bonechi, M., Benelli, M., Migliaccio, I., Malorni, L., Chiarugi, P., & Morandi, A. (2020). Glucose Metabolic Reprogramming of ER Breast Cancer in Acquired Resistance to the CDK4/6 Inhibitor Palbociclib+. Cells, 9(3), 668. https://doi.org/10.3390/cells9030668