Overexpression of the Selective Autophagy Cargo Receptor NBR1 Modifies Plant Response to Sulfur Deficit

 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Plant Growth Conditions
2.3. Transcriptome Analysis
2.4. Reverse Transcription-Real Time Quantitative Polymerase Chain Reaction (RT-qPCR)
2.5. Western Blot Analysis
2.6. Confocal Microscopy
2.7. Yeast-Two-Hybrid (Y2H) Analysis
2.8. TAP-MS Experiment
2.9. Bioinformatic Tools and Statistical Analysis
3. Results
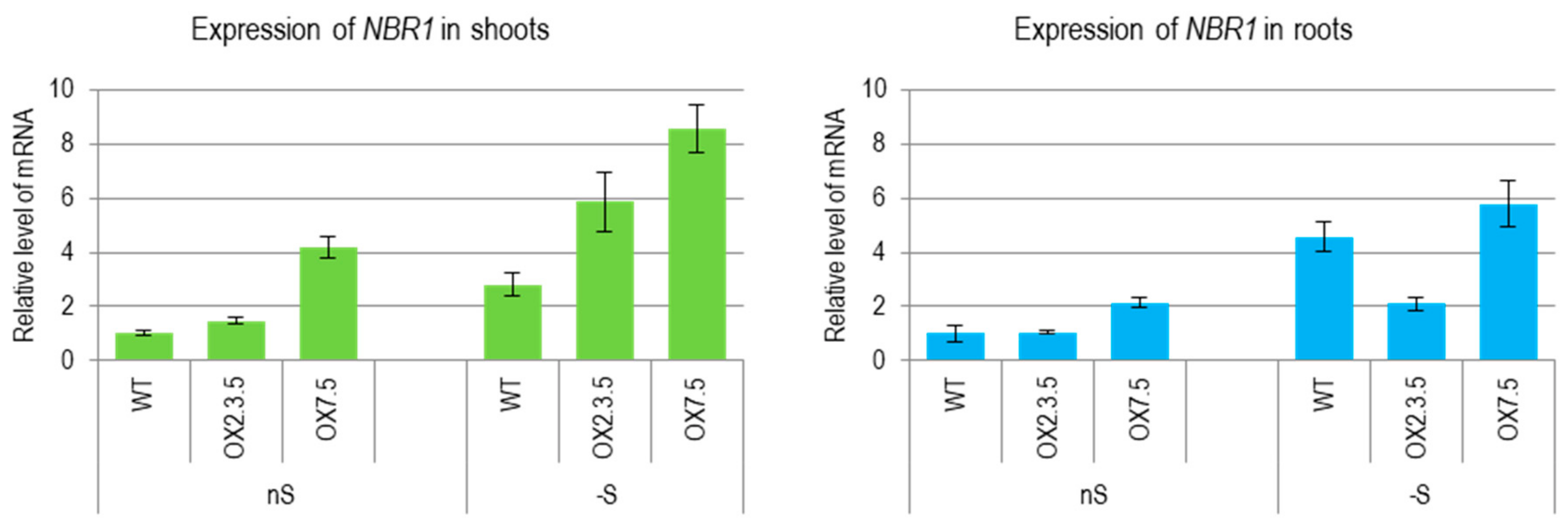
3.1. Sulfur Deficit Increases the Amount of NBR1 mRNA
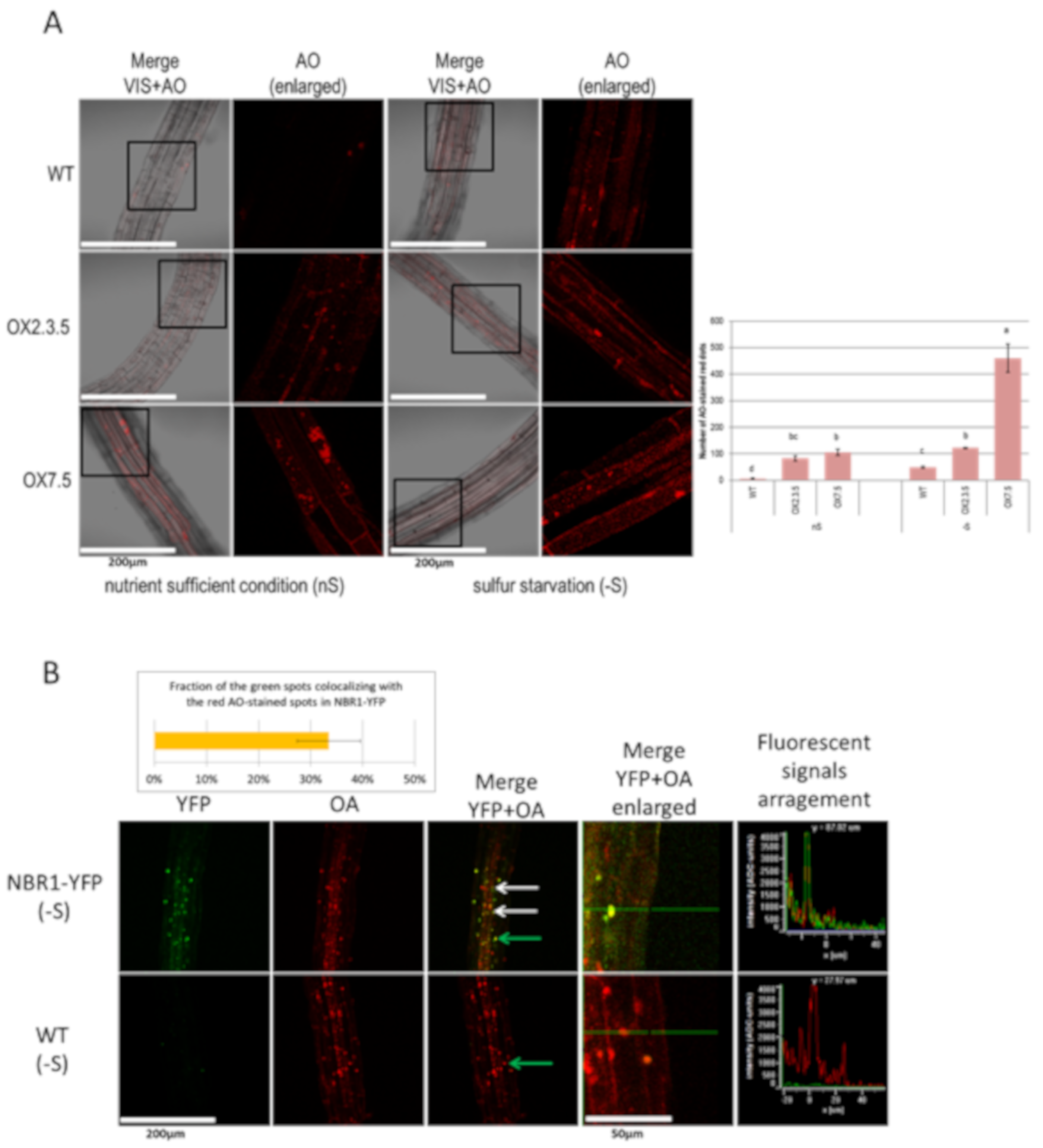
3.2. Seedlings From –S Have More Acridine Orange-Stained Red Spots
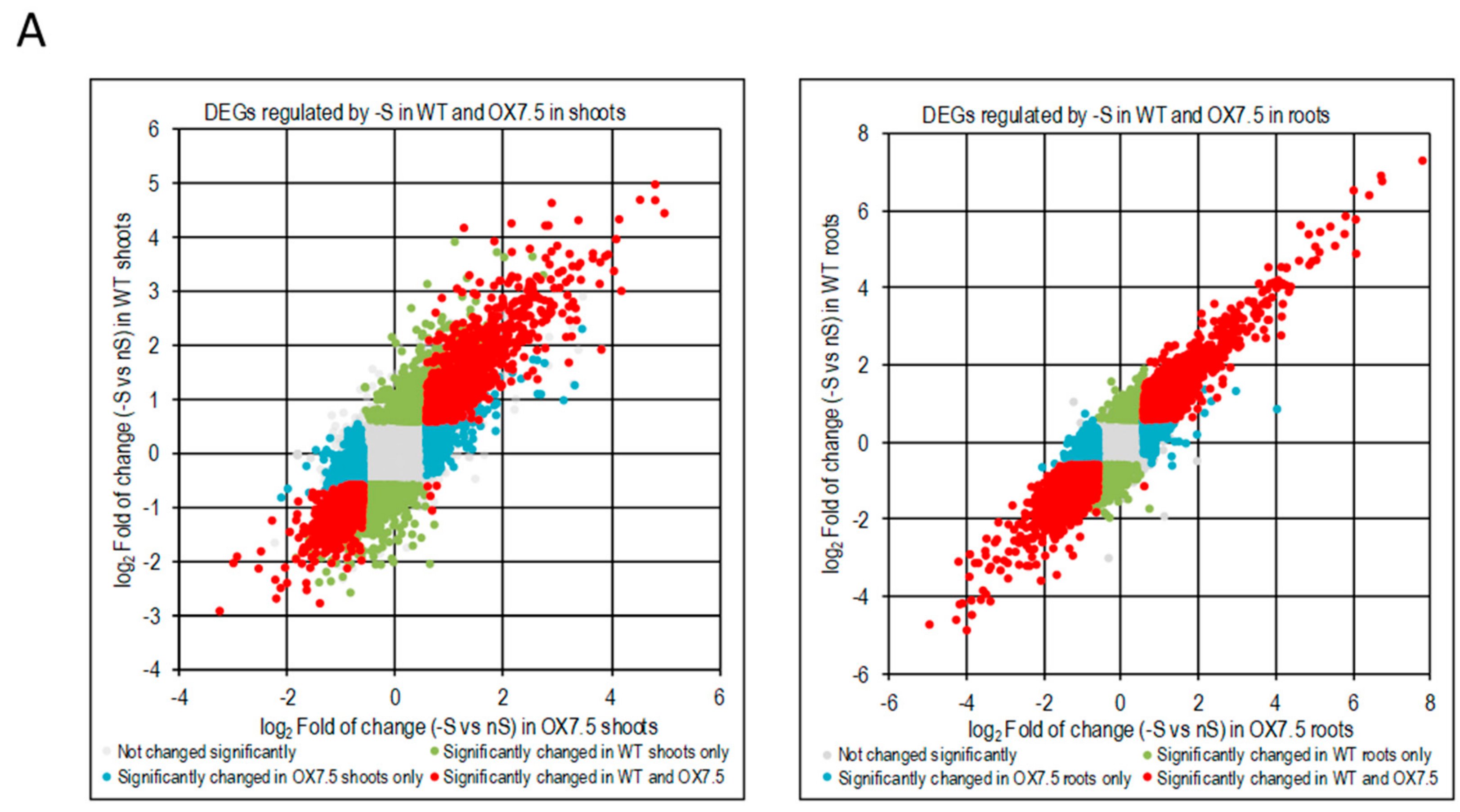
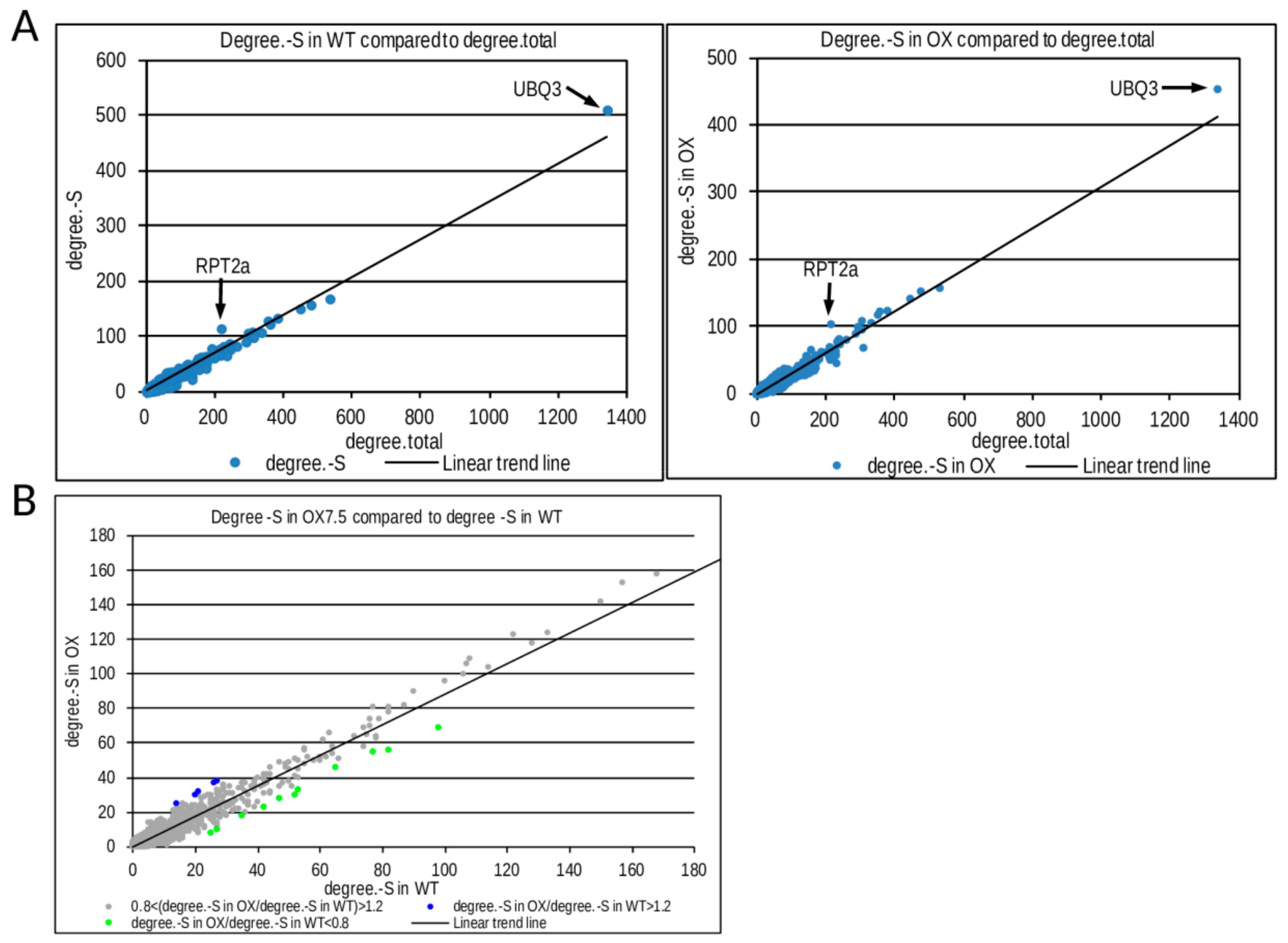
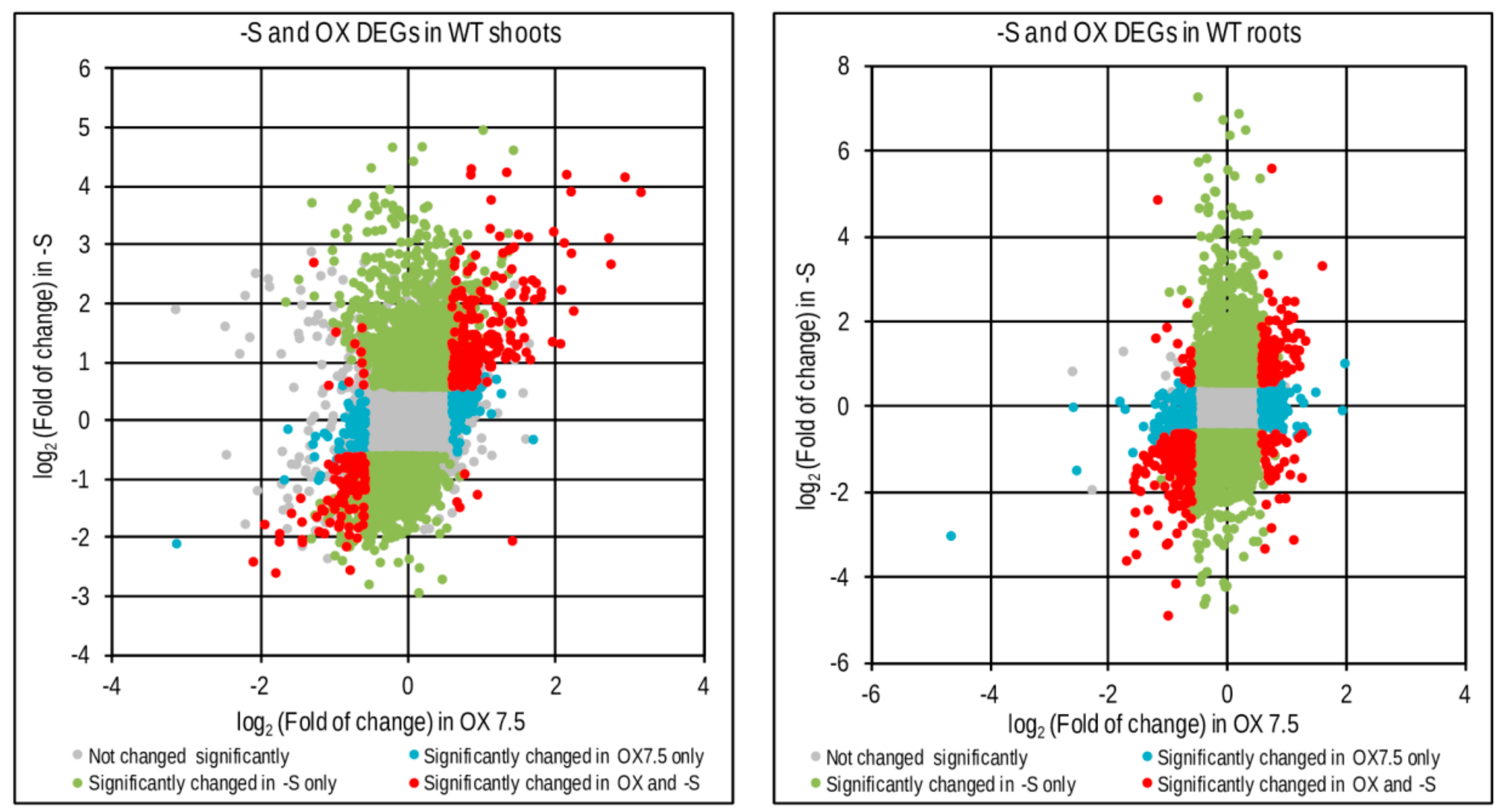
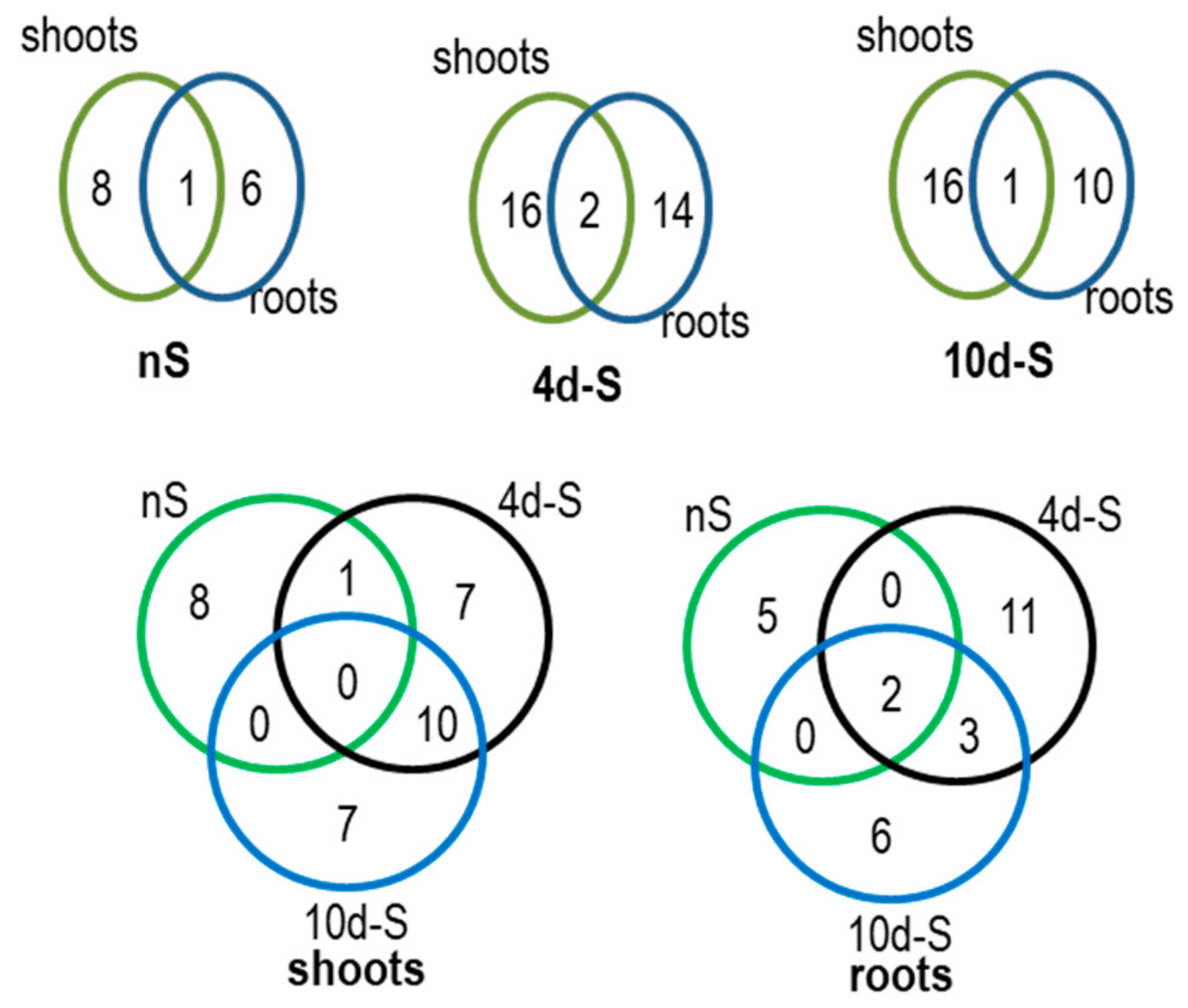
3.3. Response to –S in WT and OX7.5 at the Level of Transcription
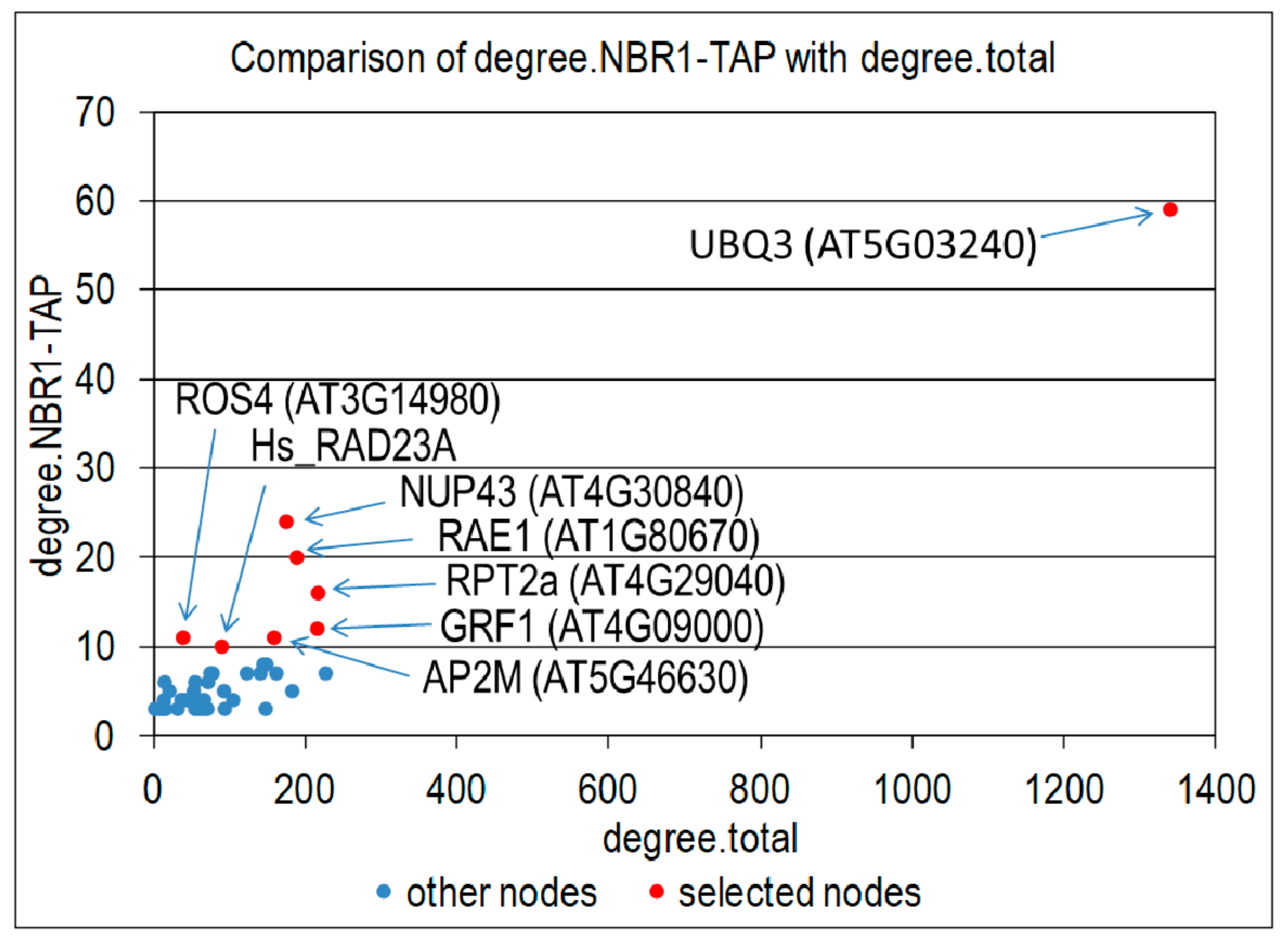
3.4. Proteins co-Purifying with NBR1-TAP
3.5. Interactions Network of Proteins Co-Purifying with NBR1-TAP
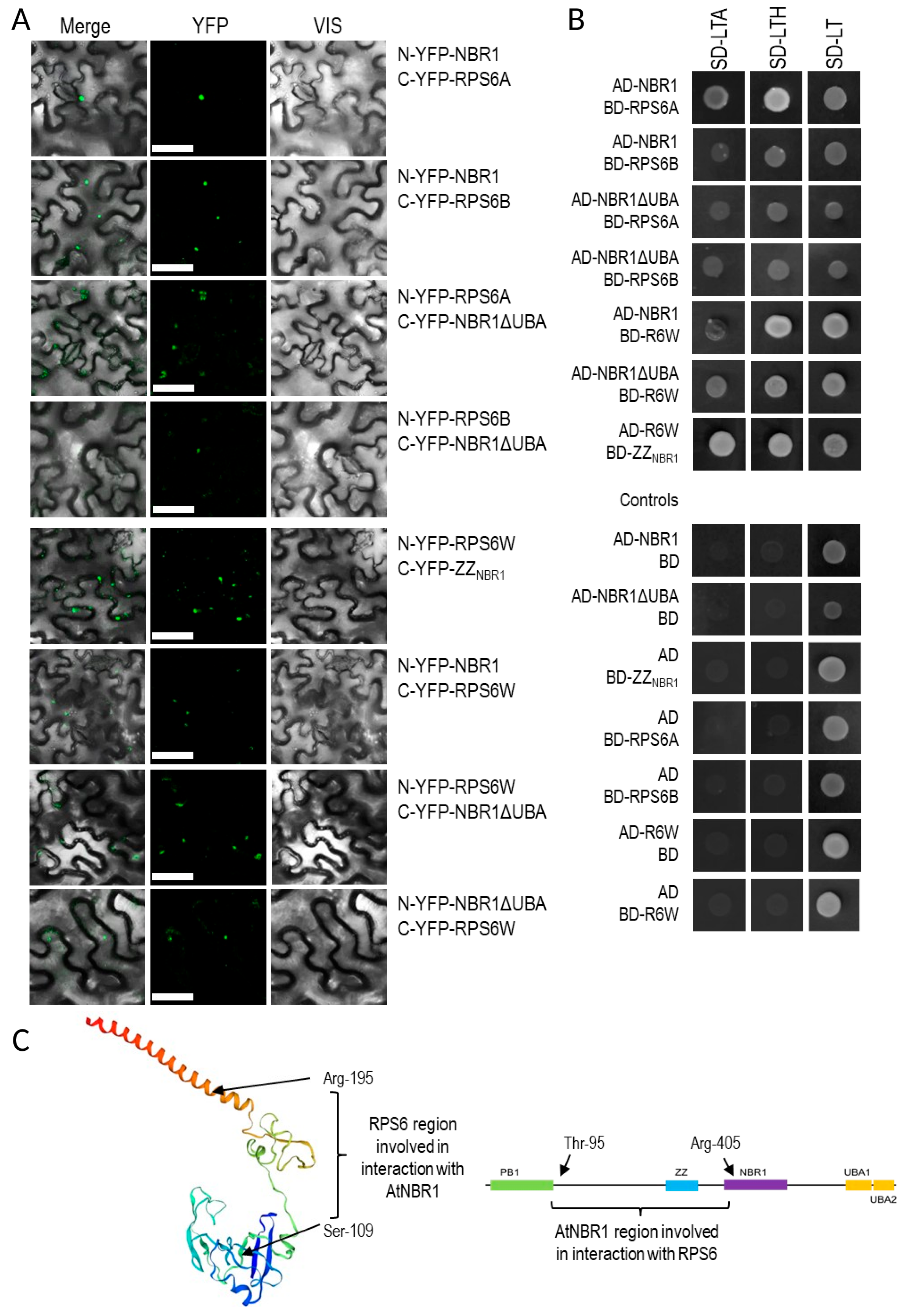
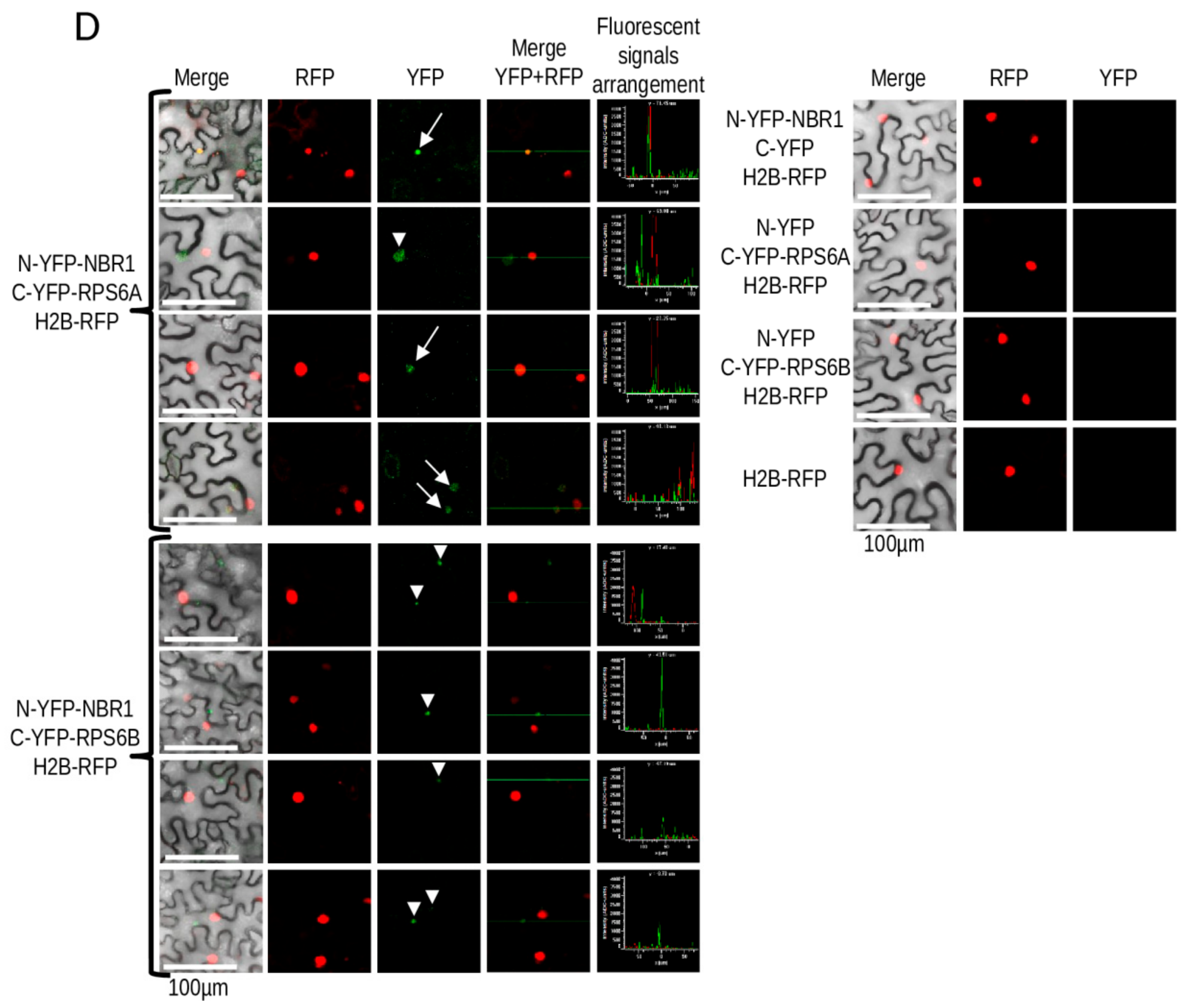
3.6. Verification of NBR1 Interaction with RPS6
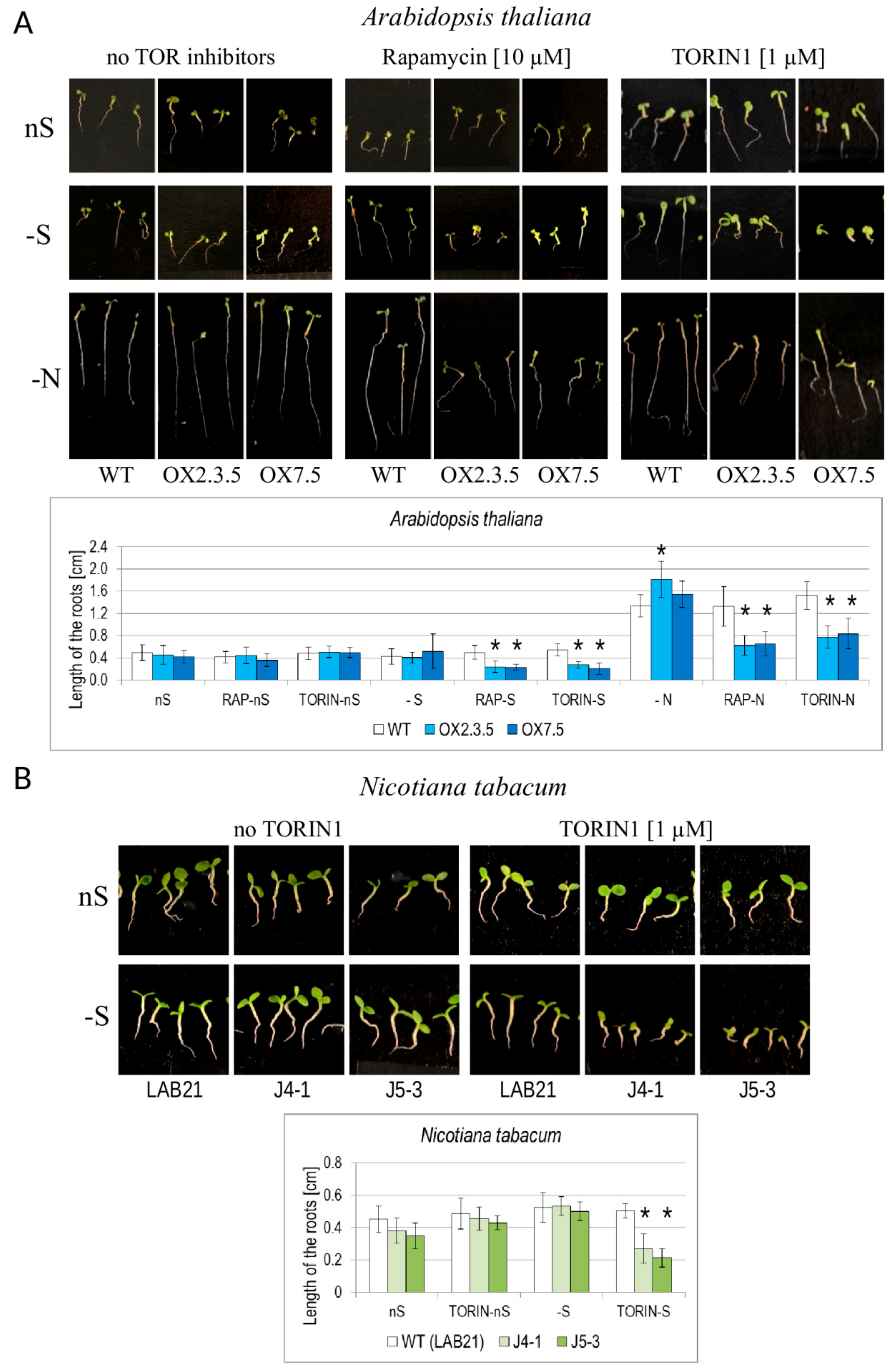
3.7. NBR1-OX Lines are Oversensitive to TOR Inhibitors During Nutrient Starvation
4. Discussion
4.1. NBR1 Expression and Autophagy Flux in NBR1-OX Lines and –S Conditions
4.2. Transcriptional Response to –S in WT and OX7.5
4.3. Proteins Co-Purifying with NBR1-TAP
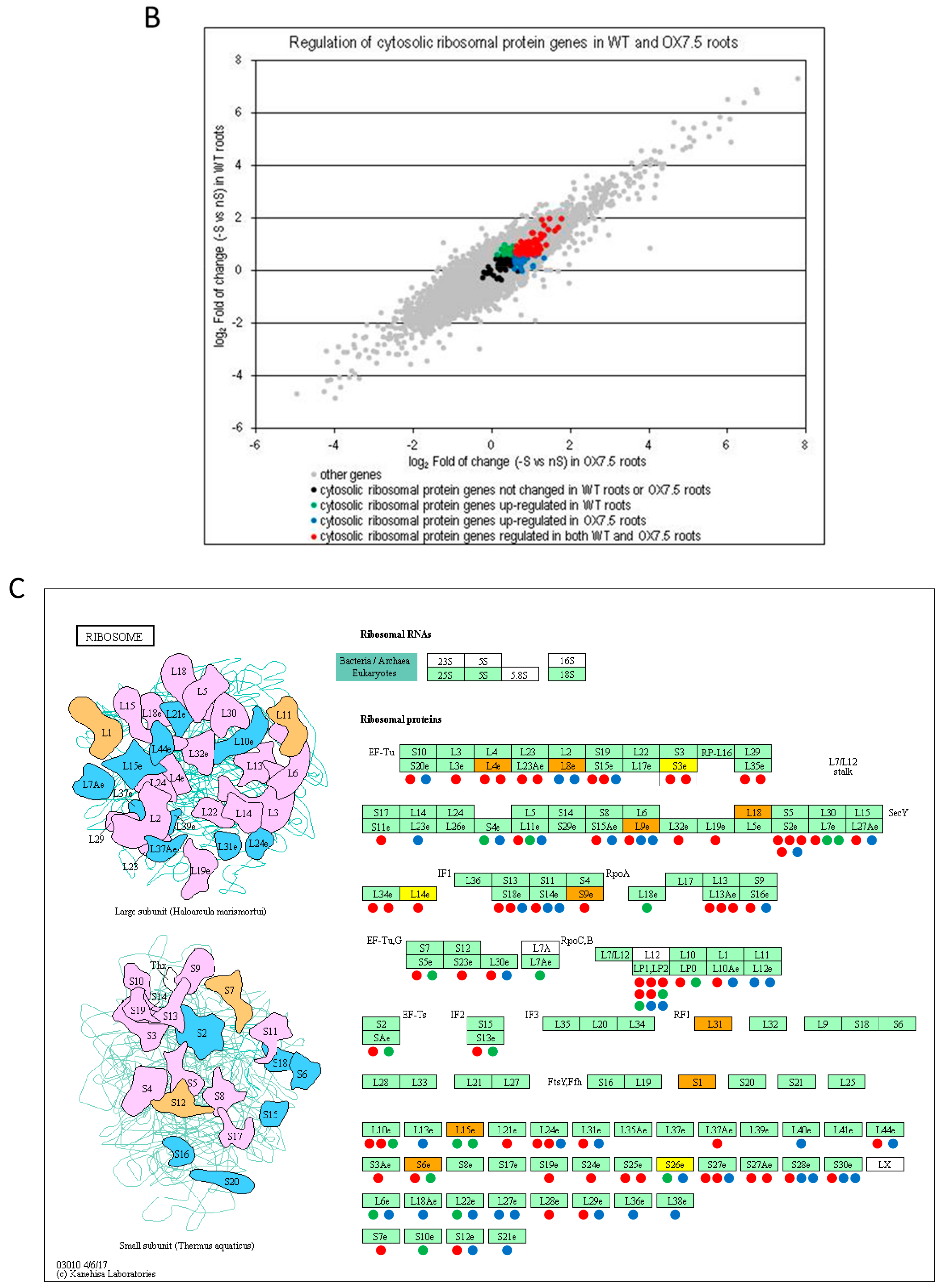
4.4. NBR1 Might be Involved in the Regulation of Ribosome Composition in -S
4.5. NBR1-OX Plants Were Oversensitive to –S and to –N in the Presence of TOR Inhibitors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Batoko, H.; Dagdas, Y.; Baluska, F.; Sirko, A. Understanding and exploiting autophagy signaling in plants. Essays Biochem. 2017, 61, 675–685. [Google Scholar] [CrossRef] [PubMed]
- Schepetilnikov, M.; Ryabova, L.A. Recent Discoveries on the Role of TOR (Target of Rapamycin) Signaling in Translation in Plants. Plant. Physiol. 2018, 176, 1095–1105. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Wu, Y.; Sheen, J. TOR signaling in plants: conservation and innovation. Development 2018, 145, dev160887. [Google Scholar] [CrossRef]
- Liu, Y.; Bassham, D.C. TOR is a negative regulator of autophagy in Arabidopsis thaliana. PLoS ONE 2010, 5, e11883. [Google Scholar] [CrossRef] [PubMed]
- Wang, P.; Mugume, Y.; Bassham, D.C. New advances in autophagy in plants: Regulation, selectivity and function. Semin. Cell Dev. Biol 2018, 80, 113–122. [Google Scholar] [CrossRef] [PubMed]
- Svenning, S.; Lamark, T.; Krause, K.; Johansen, T. Plant NBR1 is a selective autophagy substrate and a functional hybrid of the mammalian autophagic adapters NBR1 and p62/SQSTM1. Autophagy 2011, 7, 993–1010. [Google Scholar] [CrossRef] [PubMed]
- Zientara-Rytter, K.; Lukomska, J.; Moniuszko, G.; Gwozdecki, R.; Surowiecki, P.; Lewandowska, M.; Liszewska, F.; Wawrzynska, A.; Sirko, A. Identification and functional analysis of Joka2, a tobacco member of the family of selective autophagy cargo receptors. Autophagy 2011, 7, 1145–1158. [Google Scholar] [CrossRef]
- Hafren, A.; Hofius, D. NBR1-mediated antiviral xenophagy in plant immunity. Autophagy 2017, 13, 2000–2001. [Google Scholar] [CrossRef]
- Hafren, A.; Macia, J.L.; Love, A.J.; Milner, J.J.; Drucker, M.; Hofius, D. Selective autophagy limits cauliflower mosaic virus infection by NBR1-mediated targeting of viral capsid protein and particles. Proc. Natl Acad Sci USA 2017, 114, E2026–E2035. [Google Scholar] [CrossRef]
- Hafren, A.; Ustun, S.; Hochmuth, A.; Svenning, S.; Johansen, T.; Hofius, D. Turnip Mosaic Virus Counteracts Selective Autophagy of the Viral Silencing Suppressor HCpro. Plant. Physiol. 2018, 176, 649–662. [Google Scholar] [CrossRef]
- Henriquez-Valencia, C.; Arenas, M.A.; Medina, J.; Canales, J. Integrative Transcriptomic Analysis Uncovers Novel Gene Modules That Underlie the Sulfate Response in Arabidopsis thaliana. Front. Plant. Sci. 2018, 9, 470. [Google Scholar] [CrossRef] [PubMed]
- Iyer-Pascuzzi, A.S.; Jackson, T.; Cui, H.; Petricka, J.J.; Busch, W.; Tsukagoshi, H.; Benfey, P.N. Cell identity regulators link development and stress responses in the Arabidopsis root. Dev. Cell 2011, 21, 770–782. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Silbermann, M.; Speiser, A.; Forieri, I.; Linster, E.; Poschet, G.; Allboje Samami, A.; Wanatabe, M.; Sticht, C.; Teleman, A.A.; et al. Sulfur availability regulates plant growth via glucose-TOR signaling. Nat. Commun. 2017, 8, 1174. [Google Scholar] [CrossRef]
- Batool, S.; Uslu, V.V.; Rajab, H.; Ahmad, N.; Waadt, R.; Geiger, D.; Malagoli, M.; Xiang, C.B.; Hedrich, R.; Rennenberg, H.; et al. Sulfate is Incorporated into Cysteine to Trigger ABA Production and Stomatal Closure. Plant. Cell 2018, 30, 2973–2987. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.J.; Wang, Z.; Zhao, Q.; Mao, J.L.; Speiser, A.; Wirtz, M.; Hell, R.; Zhu, J.K.; Xiang, C.B. Sulfate availability affects ABA levels and germination response to ABA and salt stress in Arabidopsis thaliana. Plant. J. 2014, 77, 604–615. [Google Scholar] [CrossRef]
- Rajab, H.; Khan, M.S.; Malagoli, M.; Hell, R.; Wirtz, M. Sulfate-Induced Stomata Closure Requires the Canonical ABA Signal Transduction Machinery. Plants 2019, 8, 21. [Google Scholar] [CrossRef]
- Lewandowska, M.; Wawrzynska, A.; Moniuszko, G.; Lukomska, J.; Zientara, K.; Piecho, M.; Hodurek, P.; Zhukov, I.; Liszewska, F.; Nikiforova, V.; et al. A contribution to identification of novel regulators of plant response to sulfur deficiency: characteristics of a tobacco gene UP9C, its protein product and the effects of UP9C silencing. Mol. Plant. 2010, 3, 347–360. [Google Scholar] [CrossRef]
- Moniuszko, G.; Skoneczny, M.; Zientara-Rytter, K.; Wawrzynska, A.; Glow, D.; Cristescu, S.M.; Harren, F.J.; Sirko, A. Tobacco LSU-like protein couples sulphur-deficiency response with ethylene signalling pathway. J. Exp. Bot. 2013, 64, 5173–5182. [Google Scholar] [CrossRef]
- Sirko, A.; Wawrzynska, A.; Rodriguez, M.C.; Sektas, P. The family of LSU-like proteins. Front. Plant. Sci 2014, 5, 774. [Google Scholar] [CrossRef]
- Vandereyken, K.; Van Leene, J.; De Coninck, B.; Cammue, B.P.A. Hub Protein Controversy: Taking a Closer Look at Plant Stress Response Hubs. Front. Plant. Sci. 2018, 9, 694. [Google Scholar] [CrossRef]
- Rubio, V.; Shen, Y.; Saijo, Y.; Liu, Y.; Gusmaroli, G.; Dinesh-Kumar, S.P.; Deng, X.W. An alternative tandem affinity purification strategy applied to Arabidopsis protein complex isolation. Plant. J. 2005, 41, 767–778. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; De Meyer, B.; Hilson, P. Modular cloning in plant cells. Trends Plant. Sci. 2005, 10, 103–105. [Google Scholar] [CrossRef] [PubMed]
- Fauser, F.; Schiml, S.; Puchta, H. Both CRISPR/Cas-based nucleases and nickases can be used efficiently for genome engineering in Arabidopsis thaliana. Plant. J. 2014, 79, 348–359. [Google Scholar] [CrossRef] [PubMed]
- Tarnowski, L.; Rodiguez, M.C.; Brzywczy, J.; Piecho-Kabacik, M.; Wawrzynska, A.; Sirko, A. A selective autophagy cargo receptor NBR1 balances abscisic acid signalling in Arabidopsis thaliana. (submitted).
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Orlowska, K.P.; Klosowska, K.; Szczesny, R.J.; Cysewski, D.; Krawczyk, P.S.; Dziembowski, A. A new strategy for gene targeting and functional proteomics using the DT40 cell line. Nucleic Acids Res. 2013, 41, e167. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. agriGO v2.0: a GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. BioGRID: a general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef]
- Katari, M.S.; Nowicki, S.D.; Aceituno, F.F.; Nero, D.; Kelfer, J.; Thompson, L.P.; Cabello, J.M.; Davidson, R.S.; Goldberg, A.P.; Shasha, D.E.; et al. VirtualPlant: a software platform to support systems biology research. Plant. Physiol. 2010, 152, 500–515. [Google Scholar] [CrossRef]
- Mitou, G.; Budak, H.; Gozuacik, D. Techniques to study autophagy in plants. Int J. Plant. Genom. 2009, 2009, 451357. [Google Scholar] [CrossRef]
- Thome, M.P.; Filippi-Chiela, E.C.; Villodre, E.S.; Migliavaca, C.B.; Onzi, G.R.; Felipe, K.B.; Lenz, G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016, 129, 4622–4632. [Google Scholar] [CrossRef] [PubMed]
- Sormani, R.; Masclaux-Daubresse, C.; Daniel-Vedele, F.; Chardon, F. Transcriptional regulation of ribosome components are determined by stress according to cellular compartments in Arabidopsis thaliana. PLoS ONE 2011, 6, e28070. [Google Scholar] [CrossRef] [PubMed]
- Saracco, S.A.; Hansson, M.; Scalf, M.; Walker, J.M.; Smith, L.M.; Vierstra, R.D. Tandem affinity purification and mass spectrometric analysis of ubiquitylated proteins in Arabidopsis. Plant. J. 2009, 59, 344–358. [Google Scholar] [CrossRef]
- Jung, H.; Lee, H.N.; Marshall, R.S.; Lomax, A.W.; Yoon, M.J.; Kim, J.; Kim, J.H.; Vierstra, R.D.; Chung, T. NBR1 Mediates Selective Autophagy of Defective Proteins in Arabidopsis. J. Exp. Bot. 2019. [Google Scholar] [CrossRef]
- Sako, K.; Yanagawa, Y.; Kanai, T.; Sato, T.; Seki, M.; Fujiwara, M.; Fukao, Y.; Yamaguchi, J. Proteomic analysis of the 26S proteasome reveals its direct interaction with transit peptides of plastid protein precursors for their degradation. J. Proteome Res. 2014, 13, 3223–3230. [Google Scholar] [CrossRef] [PubMed]
- Maruyama-Nakashita, A.; Nakamura, Y.; Tohge, T.; Saito, K.; Takahashi, H. Arabidopsis SLIM1 is a central transcriptional regulator of plant sulfur response and metabolism. Plant. Cell 2006, 18, 3235–3251. [Google Scholar] [CrossRef]
- Hohmann, U.; Lau, K.; Hothorn, M. The Structural Basis of Ligand Perception and Signal Activation by Receptor Kinases. Annu. Rev. Plant. Biol. 2017, 68, 109–137. [Google Scholar] [CrossRef]
- Xi, L.; Wu, X.N.; Gilbert, M.; Schulze, W.X. Classification and Interactions of LRR Receptors and Co-receptors Within the Arabidopsis Plasma Membrane - An Overview. Front. Plant. Sci. 2019, 10, 472. [Google Scholar] [CrossRef]
- Nicolas, M.; Cubas, P. TCP factors: new kids on the signaling block. Curr Opin Plant. Biol. 2016, 33, 33–41. [Google Scholar] [CrossRef]
- Perez, M.; Guerringue, Y.; Ranty, B.; Pouzet, C.; Jauneau, A.; Robe, E.; Mazars, C.; Galaud, J.P.; Aldon, D. Specific TCP transcription factors interact with and stabilize PRR2 within different nuclear sub-domains. Plant. Sci. 2019, 287, 110197. [Google Scholar] [CrossRef]
- Spears, B.J.; Howton, T.C.; Gao, F.; Garner, C.M.; Mukhtar, M.S.; Gassmann, W. Direct Regulation of the EFR-Dependent Immune Response by Arabidopsis TCP Transcription Factors. Mol. Plant. Microbe Interact. 2019, 32, 540–549. [Google Scholar] [CrossRef] [PubMed]
- Wessling, R.; Epple, P.; Altmann, S.; He, Y.; Yang, L.; Henz, S.R.; McDonald, N.; Wiley, K.; Bader, K.C.; Glasser, C.; et al. Convergent targeting of a common host protein-network by pathogen effectors from three kingdoms of life. Cell Host Microbe 2014, 16, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Nejat, N.; Mantri, N. Plant Immune System: Crosstalk Between Responses to Biotic and Abiotic Stresses the Missing Link in Understanding Plant Defence. Curr. Issues Mol. Biol 2017, 23, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lan, P.; Gao, H.; Zheng, L.; Li, W.; Schmidt, W. Expression changes of ribosomal proteins in phosphate- and iron-deficient Arabidopsis roots predict stress-specific alterations in ribosome composition. BMC Genom. 2013, 14, 783. [Google Scholar] [CrossRef]
- Tamura, K.; Fukao, Y.; Iwamoto, M.; Haraguchi, T.; Hara-Nishimura, I. Identification and characterization of nuclear pore complex components in Arabidopsis thaliana. Plant. Cell 2010, 22, 4084–4097. [Google Scholar] [CrossRef]
- Li, D.; Zhang, L.; Li, X.; Kong, X.; Wang, X.; Li, Y.; Liu, Z.; Wang, J.; Li, X.; Yang, Y. AtRAE1 is involved in degradation of ABA receptor RCAR1 and negatively regulates ABA signalling in Arabidopsis. Plant. Cell Env. 2018, 41, 231–244. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, S.; Liu, B. 14-3-3 proteins: Macro-regulators with great potential for improving abiotic stress tolerance in plants. Biochem. Biophys. Res. Commun. 2016, 477, 9–13. [Google Scholar] [CrossRef]
- Ormancey, M.; Thuleau, P.; Mazars, C.; Cotelle, V. CDPKs and 14-3-3 Proteins: Emerging Duo in Signaling. Trends Plant. Sci. 2017, 22, 263–272. [Google Scholar] [CrossRef]
- Wilson, R.S.; Swatek, K.N.; Thelen, J.J. Regulation of the Regulators: Post-Translational Modifications, Subcellular, and Spatiotemporal Distribution of Plant 14-3-3 Proteins. Front. Plant. Sci. 2016, 7, 611. [Google Scholar] [CrossRef]
- Di Rubbo, S.; Irani, N.G.; Kim, S.Y.; Xu, Z.Y.; Gadeyne, A.; Dejonghe, W.; Vanhoutte, I.; Persiau, G.; Eeckhout, D.; Simon, S.; et al. The clathrin adaptor complex AP-2 mediates endocytosis of brassinosteroid insensitive1 in Arabidopsis. Plant. Cell 2013, 25, 2986–2997. [Google Scholar] [CrossRef]
- Ruvinsky, I.; Meyuhas, O. Ribosomal protein S6 phosphorylation: from protein synthesis to cell size. Trends Biochem. Sci. 2006, 31, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Guiboileau, A.; Avila-Ospina, L.; Yoshimoto, K.; Soulay, F.; Azzopardi, M.; Marmagne, A.; Lothier, J.; Masclaux-Daubresse, C. Physiological and metabolic consequences of autophagy deficiency for the management of nitrogen and protein resources in Arabidopsis leaves depending on nitrate availability. New Phytol. 2013, 199, 683–694. [Google Scholar] [CrossRef] [PubMed]
- Couso, I.; Perez-Perez, M.E.; Martinez-Force, E.; Kim, H.S.; He, Y.; Umen, J.G.; Crespo, J.L. Autophagic flux is required for the synthesis of triacylglycerols and ribosomal protein turnover in Chlamydomonas. J. Exp. Bot. 2018, 69, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Meyuhas, O. Ribosomal Protein S6 Phosphorylation: Four Decades of Research. Int Rev. Cell Mol. Biol. 2015, 320, 41–73. [Google Scholar] [CrossRef]
- Ferretti, M.B.; Karbstein, K. Does functional specialization of ribosomes really exist? RNA 2019, 25, 521–538. [Google Scholar] [CrossRef]
- Noda, T. Regulation of Autophagy through TORC1 and mTORC1. Biomolecules 2017, 7, 52. [Google Scholar] [CrossRef]
- Gonzalez, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef]
- Robaglia, C.; Thomas, M.; Meyer, C. Sensing nutrient and energy status by SnRK1 and TOR kinases. Curr. Opin. Plant. Biol. 2012, 15, 301–307. [Google Scholar] [CrossRef]
- Dong, P.; Xiong, F.; Que, Y.; Wang, K.; Yu, L.; Li, Z.; Ren, M. Expression profiling and functional analysis reveals that TOR is a key player in regulating photosynthesis and phytohormone signaling pathways in Arabidopsis. Front. Plant. Sci. 2015, 6, 677. [Google Scholar] [CrossRef]
- Caldana, C.; Li, Y.; Leisse, A.; Zhang, Y.; Bartholomaeus, L.; Fernie, A.R.; Willmitzer, L.; Giavalisco, P. Systemic analysis of inducible target of rapamycin mutants reveal a general metabolic switch controlling growth in Arabidopsis thaliana. Plant. J. 2013, 73, 897–909. [Google Scholar] [CrossRef]
- Dobrenel, T.; Mancera-Martinez, E.; Forzani, C.; Azzopardi, M.; Davanture, M.; Moreau, M.; Schepetilnikov, M.; Chicher, J.; Langella, O.; Zivy, M.; et al. The Arabidopsis TOR Kinase Specifically Regulates the Expression of Nuclear Genes Coding for Plastidic Ribosomal Proteins and the Phosphorylation of the Cytosolic Ribosomal Protein S6. Front. Plant. Sci. 2016, 7, 1611. [Google Scholar] [CrossRef] [PubMed]
- Sesma, A.; Castresana, C.; Castellano, M.M. Regulation of Translation by TOR, eIF4E and eIF2alpha in Plants: Current Knowledge, Challenges and Future Perspectives. Front. Plant. Sci 2017, 8, 644. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| WT-Specific –S DEGs | OX7.5-Specific –S DEGs | –S DEGs from Both Lines | |
|---|---|---|---|
| Shoots UP | (1011 genes) No significant term | (459 genes) No significant term | (1081 genes)
|
| Shoots DN | (1351 genes) No significant term | (378 genes) No significant term | (876 genes)
|
| |||
| Roots UP | (959 genes)
| (667 genes)
| (1657 genes)
|
| |||
| Roots DN | (774 genes) No significant term | (1007 genes) No significant term | (1267 genes)
|
|
| Hub No | AGI Code | Hub Degree | Gene Name/Description (from TAIR, Modified) |
|---|---|---|---|
| Hub1 | AT5G03240 | 59 | UBQ3/encodes ubiquitin that is attached to proteins destined for degradation. UBQ3 transcript level is modulated by UV-B and light/dark treatments. |
| Hub2 | AT4G30840 | 24 | NUP43/Transducin/WD40 repeat-like superfamily protein |
| Hub3 | AT1G80670 | 20 | RAE1/Encodes a protein with a DWD motif. It can bind to DDB1a in Y2H assays and may be involved in the formation of a CUL4-based E3 ubiquitin ligase |
| Hub4 | AT4G29040 | 16 | RPT2a/Encodes the 26S proteasome subunit. It is required for root meristem maintenance, and it regulates gametogenesis. It is also shown to regulate gene silencing via DNA methylation. |
| Hub5 | AT4G09000 | 12 | GRF1/Encodes a member of 14-3-3 family. The major native forms of 14-3-3s are homo- and hetero-dimers. Serving as adapters, activators, and repressors, they are involved in a vast array of processes such as the response to stress, cell-cycle control, and apoptosis. |
| Hub6 | AT5G46630 | 11 | AP2M/Clathrin adaptor complexes medium subunit family protein, contains Pfam profile: PF00928 adaptor complexes medium subunit family; it is similar to micro-adaptins of clathrin coated vesicle adaptor complexes |
| Hub7 | AT3G14980 | 11 | IDM1/ROS4/Histone H3 acetyltransferase recognizing unmethylated H3K4 through its PHD domain and methylated DNA through its MBD domain; it negatively regulates DNA demethylation |
| Hub8 | RAD23A | 11* | Homo sapiens RAD23A having interactions with many Arabidopsis proteins [34] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tarnowski, L.; Collados Rodriguez, M.; Brzywczy, J.; Cysewski, D.; Wawrzynska, A.; Sirko, A. Overexpression of the Selective Autophagy Cargo Receptor NBR1 Modifies Plant Response to Sulfur Deficit. Cells 2020, 9, 669. https://doi.org/10.3390/cells9030669
Tarnowski L, Collados Rodriguez M, Brzywczy J, Cysewski D, Wawrzynska A, Sirko A. Overexpression of the Selective Autophagy Cargo Receptor NBR1 Modifies Plant Response to Sulfur Deficit. Cells. 2020; 9(3):669. https://doi.org/10.3390/cells9030669
Chicago/Turabian StyleTarnowski, Leszek, Milagros Collados Rodriguez, Jerzy Brzywczy, Dominik Cysewski, Anna Wawrzynska, and Agnieszka Sirko. 2020. "Overexpression of the Selective Autophagy Cargo Receptor NBR1 Modifies Plant Response to Sulfur Deficit" Cells 9, no. 3: 669. https://doi.org/10.3390/cells9030669
APA StyleTarnowski, L., Collados Rodriguez, M., Brzywczy, J., Cysewski, D., Wawrzynska, A., & Sirko, A. (2020). Overexpression of the Selective Autophagy Cargo Receptor NBR1 Modifies Plant Response to Sulfur Deficit. Cells, 9(3), 669. https://doi.org/10.3390/cells9030669