HMGB1 Translocation in Neurons after Ischemic Insult: Subcellular Localization in Mitochondria and Peroxisomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Procedures
2.2. Immunohistochemical Staining
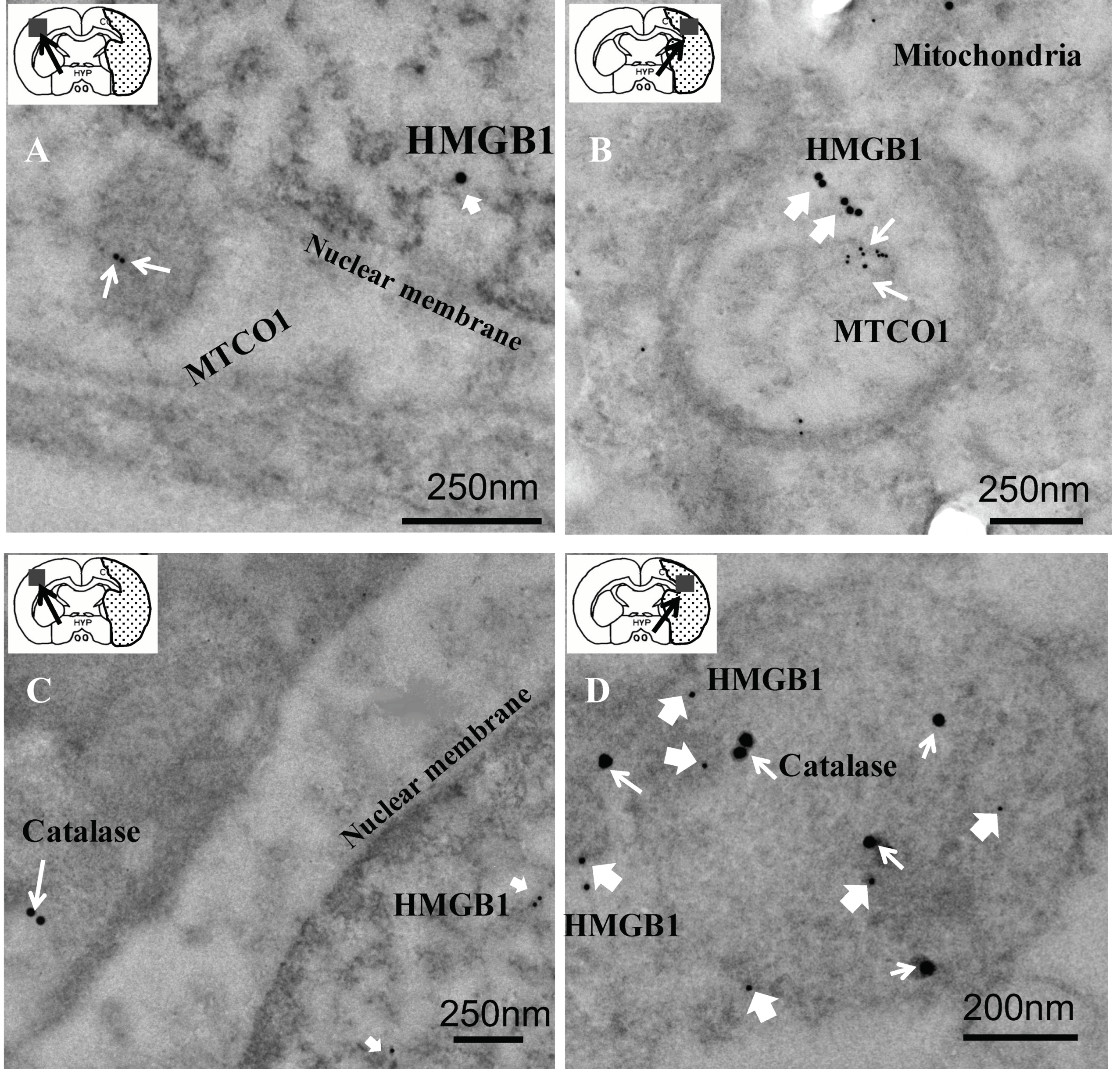
2.3. Transmission Electron Microscopic (TEM) Examination
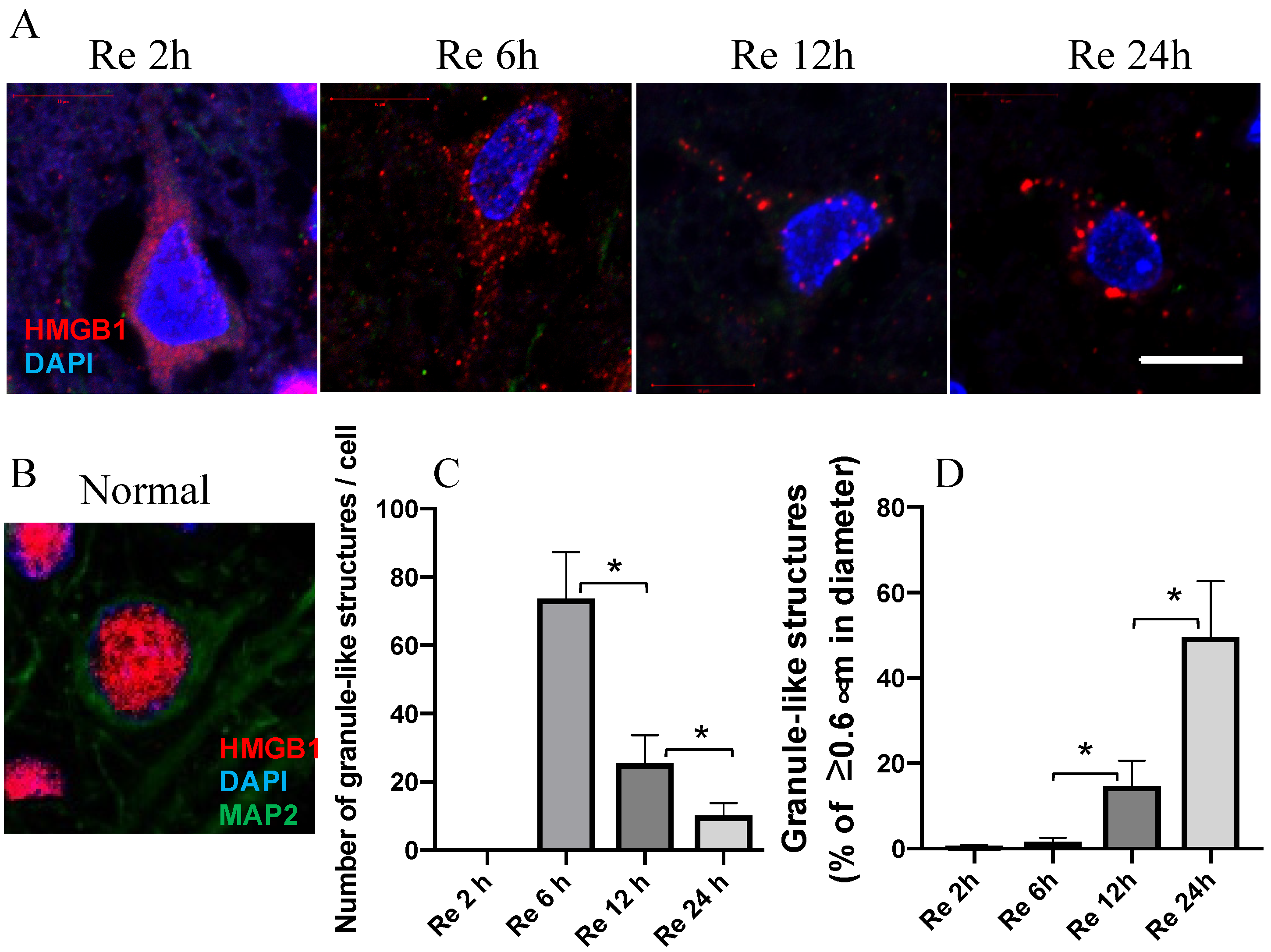
2.4. Granule-Like Structures Count
2.5. Statistical Analysis
3. Results
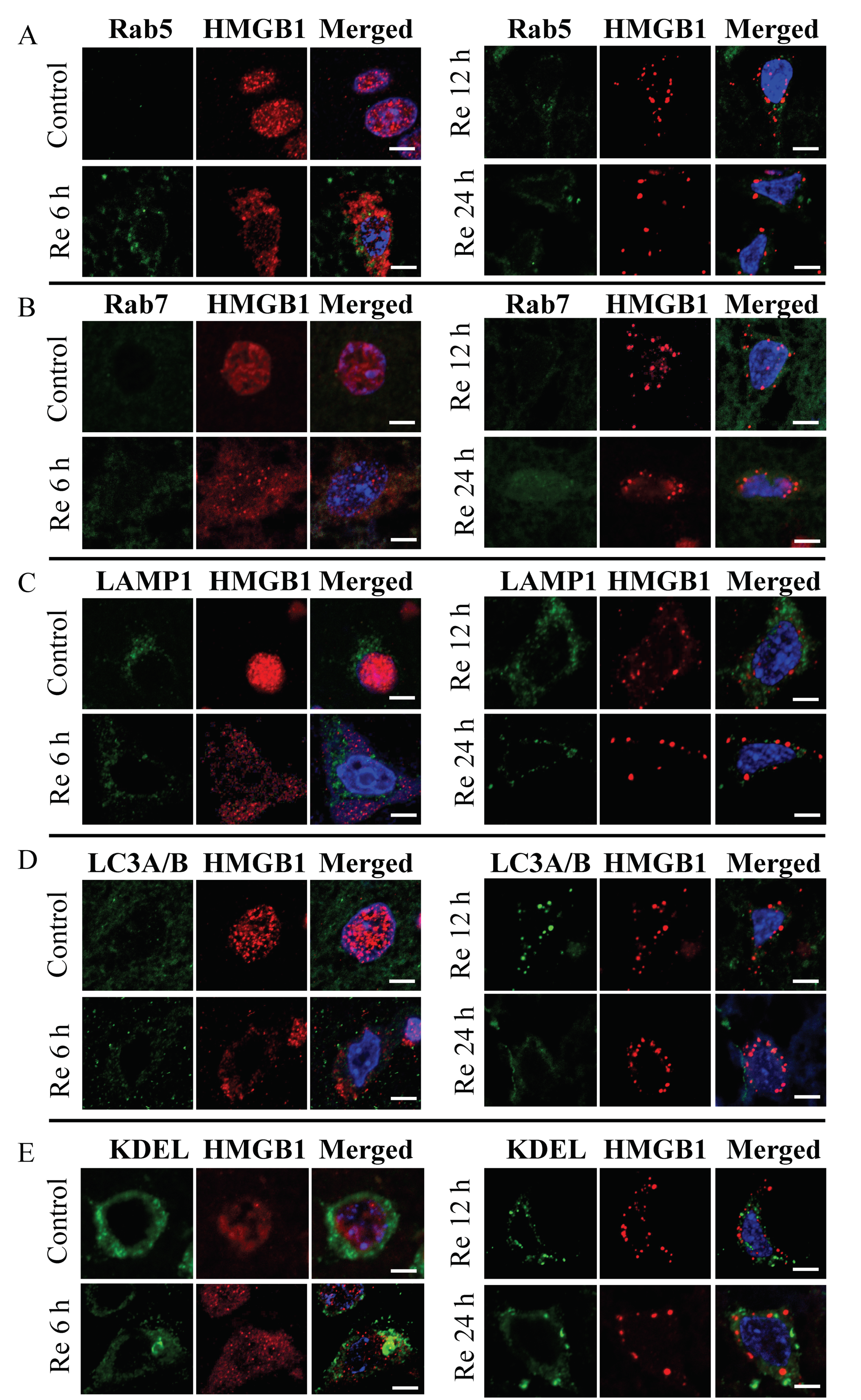
3.1. HMGB1 Translocation in a Time-Dependent Manner in Neurons
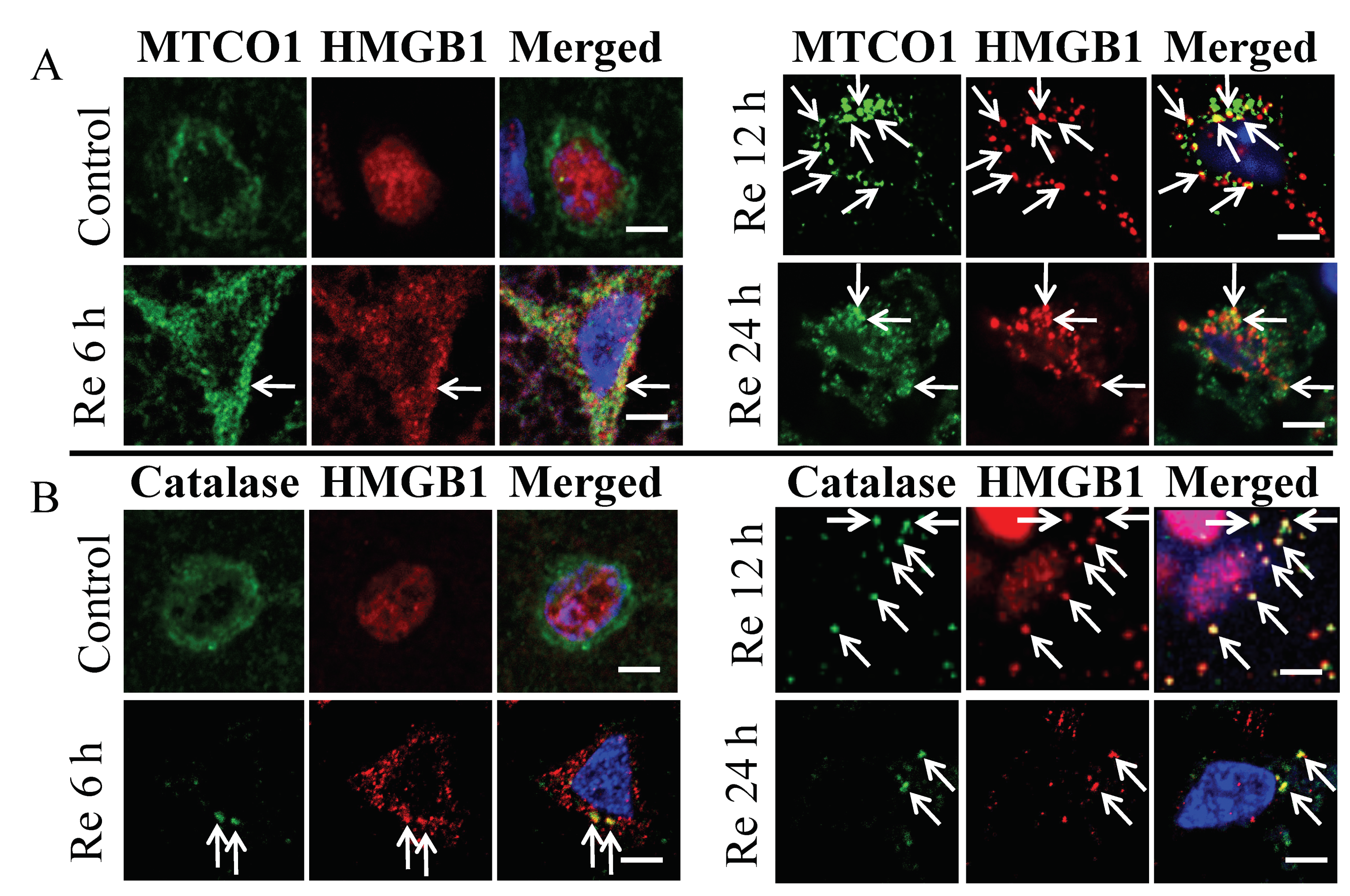
3.2. Colocalization of HMGB1 and MTCO1 and Catalase
3.3. Relationship of HMGB1 with MTCO1 and Catalase
3.4. Morphologies of Mitochondria Related to Mitochondrial Fission after MCAO
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Liu, K.; Mori, S.; Takahashi, H.K.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef]
- Zhang, J.; Takahashi, H.K.; Liu, K.; Wake, H.; Liu, R.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Mori, S.; et al. Anti-high mobility group box-1 monoclonal antibody protects the blood-brain barrier from ischemia-induced disruption in rats. Stroke 2011, 42, 1420–1428. [Google Scholar] [CrossRef]
- Okuma, Y.; Liu, K.; Wake, H.; Zhang, J.; Maruo, T.; Date, I.; Yoshino, T.; Ohtsuka, A.; Otani, N.; Tomura, S.; et al. Anti-high mobility group box-1 antibody therapy for traumatic brain injury. Ann. Neurol. 2012, 72, 373–384. [Google Scholar] [CrossRef]
- Wang, D.; Liu, K.; Wake, H.; Teshigawara, K.; Mori, S.; Nishibori, M. Anti-high mobility group box-1 (HMGB1) antibody inhibits hemorrhage-induced brain injury and improved neurological deficits in rats. Sci. Rep. 2017, 7, 46243. [Google Scholar] [CrossRef] [PubMed]
- Okuma, Y.; Liu, K.; Wake, H.; Liu, R.; Nishimura, Y.; Hui, Z.; Teshigawara, K.; Haruma, J.; Yamamoto, Y.; Yamamoto, H.; et al. Glycyrrhizin inhibits traumatic brain injury by reducing HMGB1-RAGE interaction. Neuropharmacology 2014, 85, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Uezono, N.; Zhu, Y.; Fujimoto, Y.; Yasui, T.; Matsuda, T.; Nakajo, M.; Abematsu, M.; Setoguchi, T.; Mori, S.; Takahashi, H.K.; et al. Prior Treatment with Anti-High Mobility Group Box-1 Antibody Boosts Human Neural Stem Cell Transplantation-Mediated Functional Recovery After Spinal Cord Injury. Stem Cells 2018, 36, 737–750. [Google Scholar] [CrossRef] [PubMed]
- Faraco, G.; Fossati, S.; Bianchi, M.; Patrone, M.; Pedrazzi, M.; Sparatore, B.; Moroni, F.; Chiarugi, A. High mobility group box 1 protein is released by neural cells upon different stresses and worsens ischemic neurodegeneration in vitro and in vivo. J. Neurochem. 2007, 103, 590–603. [Google Scholar] [CrossRef]
- Qiu, J.; Nishimura, M.; Wang, Y.; Sims, J.R.; Qiu, S.; Savitz, S.I.; Salomone, S.; Moskowitz, M.A. Early release of HMGB-1 from neurons after the onset of brain ischemia. J. Cereb. Blood Flow Metab. 2008, 28, 927–938. [Google Scholar] [CrossRef]
- Kim, J.B.; Sig Choi, J.; Yu, Y.M.; Nam, K.; Piao, C.S.; Kim, S.W.; Lee, M.H.; Han, P.L.; Park, J.S.; Lee, J.K. HMGB1, a novel cytokine-like mediator linking acute neuronal death and delayed neuroinflammation in the postischemic brain. J. Neurosci. 2006, 26, 6413–6421. [Google Scholar] [CrossRef]
- Gardella, S.; Andrei, C.; Ferrera, D.; Lotti, L.V.; Torrisi, M.R.; Bianchi, M.E.; Rubartelli, A. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002, 3, 995–1001. [Google Scholar] [CrossRef]
- Yamasoba, D.; Tsubota, M.; Domoto, R.; Sekiguchi, F.; Nishikawa, H.; Liu, K.; Nishibori, M.; Ishikura, H.; Yamamoto, T.; Taga, A.; et al. Peripheral HMGB1-induced hyperalgesia in mice: Redox state-dependent distinct roles of RAGE and TLR4. J. Pharm. Sci. 2016, 130, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Owens, K.; Park, J.H.; Gourley, S.; Jones, H.; Kristian, T. Mitochondrial dynamics: Cell-type and hippocampal region specific changes following global cerebral ischemia. J. Bioenerg. Biomembr. 2015, 47, 13–31. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Aoki, M.; Kawagoe, J.; Yoshida, T.; Hattori, A.; Kogure, K.; Itoyama, Y. Ischemic delayed neuronal death. A mitochondrial hypothesis. Stroke 1995, 26, 1478–1489. [Google Scholar] [CrossRef]
- Solenski, N.J.; DiPierro, C.G.; Trimmer, P.A.; Kwan, A.L.; Helm, G.A. Ultrastructural changes of neuronal mitochondria after transient and permanent cerebral ischemia. Stroke 2002, 33, 816–824. [Google Scholar] [CrossRef]
- Van der Bliek, A.M.; Shen, Q.; Kawajiri, S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harb. Perspect. Biol. 2013, 5, a011072. [Google Scholar] [CrossRef]
- Kaneko, Y.; Pappas, C.; Malapira, T.; Vale, F.L.; Tajiri, N.; Borlongan, C.V. Extracellular HMGB1 modulates glutamate metabolism associated with kainic acid-induced epilepsy-like hyperactivity in primary rat neural cells. Cell Physiol. Biochem. 2017, 41, 947–959. [Google Scholar] [CrossRef]
- Fu, L.; Liu, K.; Wake, H.; Teshigawara, K.; Yoshino, T.; Takahashi, H.; Mori, S.; Nishibori, M. Therapeutic effects of anti-HMGB1 monoclonal antibody on pilocarpine-induced status epilepticus in mice. Sci. Rep. 2017, 7, 1179. [Google Scholar] [CrossRef]
- Liu, F.; Lu, J.; Manaenko, A.; Tang, J.; Hu, Q. Mitochondria in ischemic stroke: New insight and implications. Aging Dis. 2018, 9, 924. [Google Scholar] [CrossRef]
- Hyun, H.W.; Ko, A.R.; Kang, T.C. Mitochondrial translocation of high mobility group box 1 facilitates LIM kinase 2-mediated programmed necrotic neuronal death. Front. Cell Neurosci. 2016, 10, 99. [Google Scholar] [CrossRef]
- Liu, Y.; Yan, W.; Tohme, S.; Chen, M.; Fu, Y.; Tian, D.; Lotze, M.; Tang, D.; Tsung, A. Hypoxia induced HMGB1 and mitochondrial DNA interactions mediate tumor growth in hepatocellular carcinoma through Toll-like receptor 9. J. Hepatol. 2015, 63, 114–121. [Google Scholar] [CrossRef]
- Ito, H.; Fujita, K.; Tagawa, K.; Chen, X.; Homma, H.; Sasabe, T.; Shimizu, J.; Shimizu, S.; Tamura, T.; Muramatsu, S.; et al. HMGB1 facilitates repair of mitochondrial DNA damage and extends the lifespan of mutant ataxin-1 knock-in mice. EMBO Mol. Med. 2015, 7, 78–101. [Google Scholar] [CrossRef] [PubMed]
- Di Cara, F.; Andreoletti, P.; Trompier, D.; Vejux, A.; Bülow, M.H.; Sellin, J.; Lizard, G.; Cherkaoui-Malki, M.; Savary, S. Peroxisomes in immune response and inflammation. Int. J. Mol. Sci. 2019, 20, 3877. [Google Scholar] [CrossRef] [PubMed]
- Lismont, C.; Revenco, I.; Fransen, M. Peroxisomal hydrogen peroxide metabolism and signaling in health and disease. Int. J. Mol. Sci. 2019, 20, 3673. [Google Scholar] [CrossRef] [PubMed]
- Islinger, M.; Voelkl, A.; Fahimi, H.; Schrader, M. The peroxisome: An update on mysteries 2.0. Histo. Cell Biol. 2018, 150, 443–471. [Google Scholar] [CrossRef] [PubMed]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef]
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873. [Google Scholar] [CrossRef]
- Ito, I.; Fukazawa, J.; Yoshida, M. Post-translational methylation of high mobility group box 1 (HMGB1) causes its cytoplasmic localization in neutrophils. J. Biol. Chem. 2007, 282, 16336–16344. [Google Scholar] [CrossRef]
- Lu, B.; Antoine, D.J.; Kwan, K.; Lundbäck, P.; Wähämaa, H.; Schierbeck, H.; Robinson, M.; Van Zoelen, M.A.; Yang, H.; Li, J. JAK/STAT1 signaling promotes HMGB1 hyperacetylation and nuclear translocation. Proc. Natl. Acad. Sci. USA 2014, 111, 3068–3073. [Google Scholar] [CrossRef]
- Shin, J.H.; Kim, I.D.; Kim, S.W.; Lee, H.K.; Jin, Y.; Park, J.H.; Kim, T.K.; Suh, C.K.; Kwak, J.; Lee, K.H.; et al. Ethyl pyruvate inhibits HMGB1 phosphorylation and release by chelating calcium. Mol. Med. 2015, 20, 649–657. [Google Scholar] [CrossRef]
- Shin, J.H.; Lee, H.K.; Lee, H.B.; Jin, Y.; Lee, J.K. Ethyl pyruvate inhibits HMGB1 phosphorylation and secretion in activated microglia and in the postischemic brain. Neurosci. Lett. 2014, 558, 159–163. [Google Scholar] [CrossRef]
- Janko, C.; Filipovic, M.; Munoz, L.E.; Schorn, C.; Schett, G.; Ivanovic-Burmazovic, I.; Herrmann, M. Redox modulation of HMGB1-related signaling. Antioxid. Redox Signal. 2014, 20, 1075–1085. [Google Scholar] [CrossRef]
- Richard, S.A.; Jiang, Y.; Xiang, L.H.; Zhou, S.; Wang, J.; Su, Z.; Xu, H. Post-translational modifications of high mobility group box 1 and cancer. Am. J. Transl. Res. 2017, 9, 5181–5196. [Google Scholar] [PubMed]
- Tang, Y.; Zhao, X.; Antoine, D.; Xiao, X.; Wang, H.; Andersson, U.; Billiar, T.R.; Tracey, K.J.; Lu, B. Regulation of posttranslational modifications of HMGB1 during immune responses. Antioxid. Redox Signal. 2016, 24, 620–634. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Kang, R.; Livesey, K.M.; Cheh, C.-W.; Farkas, A.; Loughran, P.; Hoppe, G.; Bianchi, M.E.; Tracey, K.J.; Zeh, H.J. Endogenous HMGB1 regulates autophagy. J. Cell Biol. 2010, 190, 881–892. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, D.; Liu, K.; Fukuyasu, Y.; Teshigawara, K.; Fu, L.; Wake, H.; Ohtsuka, A.; Nishibori, M. HMGB1 Translocation in Neurons after Ischemic Insult: Subcellular Localization in Mitochondria and Peroxisomes. Cells 2020, 9, 643. https://doi.org/10.3390/cells9030643
Wang D, Liu K, Fukuyasu Y, Teshigawara K, Fu L, Wake H, Ohtsuka A, Nishibori M. HMGB1 Translocation in Neurons after Ischemic Insult: Subcellular Localization in Mitochondria and Peroxisomes. Cells. 2020; 9(3):643. https://doi.org/10.3390/cells9030643
Chicago/Turabian StyleWang, Dengli, Keyue Liu, Yusuke Fukuyasu, Kiyoshi Teshigawara, Li Fu, Hidenori Wake, Aiji Ohtsuka, and Masahiro Nishibori. 2020. "HMGB1 Translocation in Neurons after Ischemic Insult: Subcellular Localization in Mitochondria and Peroxisomes" Cells 9, no. 3: 643. https://doi.org/10.3390/cells9030643
APA StyleWang, D., Liu, K., Fukuyasu, Y., Teshigawara, K., Fu, L., Wake, H., Ohtsuka, A., & Nishibori, M. (2020). HMGB1 Translocation in Neurons after Ischemic Insult: Subcellular Localization in Mitochondria and Peroxisomes. Cells, 9(3), 643. https://doi.org/10.3390/cells9030643