Abstract
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation and subsequent proliferation of synovial tissues, which eventually leads to cartilage and bone destruction without effective treatments. Anti-citrullinated cyclic peptide/protein antibody (ACPA) and rheumatoid factor (RF) are two main characteristic autoantibodies found in RA patients and are associated with unfavorable disease outcomes. Although etiologies and causes of the disease have not been fully clarified yet, it is likely that interactive contributions of genetic and environmental factors play a main role in RA pathology. Previous works have demonstrated several genetic and environmental factors as risks of RA development and/or autoantibody productions. Among these, cigarette smoking and HLA-DRB1 are the well-established environmental and genetic risks, respectively. In this narrative review, we provide a recent update on genetic contributions to RA and the environmental risks of RA with a special focus on cigarette smoking and its impacts on RA pathology. We also describe gene–environmental interaction in RA pathogenesis with an emphasis on cigarette smoking and HLA-DRB1.
1. Introduction
Rheumatoid arthritis (RA) is an autoimmune disease characterized by chronic inflammation and subsequent proliferation of synovial tissues, which eventually leads to cartilage and bone destruction without effective treatments [1]. The prevalence of RA is reported to be 0.5–1.0% according to most epidemiologic studies [2].
Anti-citrullinated cyclic peptide/protein antibody (ACPA) and rheumatoid factor (RF) are two main characteristic autoantibodies found in 70–80% of RA patients. It is now well known that not only positivity for, but also high levels of, both of these autoantibodies have associations with joint destruction [3,4,5,6,7] and systemic bone loss even in early phases of the disease course [8,9]. Moreover, about 50–60% of patients have both ACPA and RF, and both of these autoantibodies show an additive effect on the amount and extent of bone erosion and thus disease severity [6,10]. This is why these autoantibody profiles of patients are considered to be one of the disease’s prognostic markers and are included in the 2010 ACR/European League Against Rheumatism (EULAR) classification criteria for RA [11]. In contrast, subjects who meet the classification criteria but are negative for ACPA and RF are considered to be seronegative RA patients. Several different etiopathological aspects have been observed in these patients, and thus, it is reasonable to regard seropositive and seronegative RA as distinctive subtypes. Although seronegative RA has been considered to be a milder form of the seropositive disease [5,6,12], a recent study indicated that treatment response was somewhat slower in seronegative patients, and radiographic progression was similar in seronegative and seropositive patients, suggesting that seronegative RA is not a mild form of the disease and requires intensive therapy similar to seropositive RA [13].
A growing body of evidence has accumulated, and there has been remarkable progress in understanding RA pathogenesis in the last few decades, but a lot of aspects of its precise mechanisms are still unexplained. It is now widely accepted that both environmental and genetic factors contribute to the pathogenesis of RA, and numerous previous works have found that interactive contributions of genetic and environmental factors play a main role in the development of RA [14,15]. However, again, their exact roles in the course of RA development have not been fully clarified yet.
In the following sections, we provide an overview of the genetic risks of RA, followed by the impact of cigarette smoking (CS) on RA pathology and other environmental factors whose effects can be influenced by CS. Subsequently, we describe the effects of these risks on RA pathogenesis more precisely with a special focus on gene-environmental interaction. We used MEDLINE for our literature search of the following terms: rheumatoid arthritis/RA, cigarette smoking, anti-citrullinated cyclic peptide/protein antibody/ACPA, rheumatoid factor/RF, HLA-DRB1, shared epitope allele, environmental risks, genetic risks, and single nucleotide polymorphisms/SNPs. We selected studies where the number of subjects involved was more than 100 in both the case and control for case–control studies and there were more than 100 incidences or RA subjects for cohort studies. For environmental risks other than CS, only those influenced by CS were selected. For genetic risks, only those associated with environmental risks were selected. We also selected meta-analyses, all of which were conducted using enough studies with sufficient numbers of subjects, as described above.
2. Genetic Risk Factor of RA
According to the twin study conducted by MacGregor et al., the heritability of RA was estimated to be ~60%, and there was no difference in the overall genetic contribution to RA among variables of sex, age, age at onset, and disease severity [16]. In contrast, genetic variations, mostly represented by single nucleotide polymorphisms (SNPs), also contribute to RA pathogenesis. Genome-wide association studies (GWAS) and GWAS meta-analyses with the use of in silico imputation of SNPs have reported 106 RA risk loci to date [17,18,19,20,21,22,23,24], among which, only ~20% are coding variants while the rest of the ~80% variants in non-coding regions probably regulate gene expression [20].
Of note, recent advances in a genetic analysis of juvenile idiopathic arthritis (JIA) revealed that several genetic risks in adult RA patients, such as HLA-DRB1*04 or PTPN22, also confer a risk or a protection on JIA, especially RF-positive polyarthritis, suggesting shared genetic backgrounds between adult seropositive RA and RF-positive JIA [25,26].
2.1. HLA
The major histocompatibility (MHC) region is located at chromosome 6 and contains the human leukocyte antigen (HLA) locus. The development of an HLA imputation method led to fine mapping of genetic risks of RA within the MHC region. The HLA indicates the strongest genetic risk of RA, explaining 30–50% of total genetic risk [27]. Among HLA genes, HLA-DRB1, one of the class II HLA genes, was the first identified RA risk locus [28] and confers the majority of genetic risk of RA [2]. The specific amino acid (AA) sequence at positions 70–74 of HLA-DRβ1 chains is called shared epitope (SE), and SE was reported to explain the association of HLA-DRB1 with RA susceptibility (SE hypothesis) [28]. As a result of recent advances of imputation for HLA sequences, large-scale association studies have revealed that AA positions 11 or 13, 71, and 74 of HLA-DRβ1 are strongly associated with RA in European populations [29,30]. A recent study also revealed a very similar genetic architecture in Asian populations to that in European populations, whereas AA position 57 is unique to Asian populations. The current consensus is that most risk HLA variants are shared among different populations at AA levels.
HLA-DRB1 is also associated with positivity for RF and ACPA [31] and levels of ACPA [32,33] but not for RF [34]. Non-SE alleles, such as HLA-DRB1*09:01 in Asian populations [32,33], have also been reported to be associated with RA [35] or ACPA levels, and the associations of HLA-DRB1 with ACPA levels are mainly explained by the 74th AA, alanine [32,33]. Intriguingly, differences in genetic backgrounds between ACPA-positive and ACPA-negative RA were highlighted by a clear difference in signals at the HLA region [36], and such differences can also be explained by the same HLA-DRβ1 AA positions but different risk-associated residues [30]. These findings can explain the heterogeneity in clinical manifestations between these RA subtypes and may also imply that other autoimmune-related factors contribute to ACPA-negative RA development [2].
Recent studies have also revealed that AA polymorphisms in other classical HLA genes, HLA-DPB1, HLA-B, and HLA-A [29,30,37], and a coding variant in a non-classical HLA gene, HLA-DOA, which alters the expression levels of several genes, also indicate the risk of ACPA-positive RA [38].
2.2. PTPN22
PTPN22 encodes a protein tyrosine phosphatase that is exclusively expressed in immune cells [39]. The SNP R620W, and the resultant risk allele, lymphoid tyrosine phosphatase (LYP) allele, is the most well-characterized risk variant of RA, as well as multiple autoimmune diseases, including type I diabetes, systemic lupus erythematosus, Hashimoto thyroiditis, Graves’ disease, Addison’s disease, myasthenia gravis, vitiligo, systemic sclerosis, juvenile idiopathic arthritis, and psoriatic arthritis [40]. Intriguingly, the association of the risk variant with RA susceptibility is only found in Caucasians, but not in Asian populations according to the meta-analysis conducted by Nabi et al. [41]. The LYP allele is a gain-of-function variant leading to decreased TCR and BCR signaling, followed by a breakdown of both central and peripheral tolerance [42,43]. Impaired regulatory T cell function [44] and frequency [45] reduced Toll-like receptor 7-induced type I interferon signal [46], and hypercitrullination of peripheral blood mononuclear cells via physical interaction with peptidylarginine deaminase type 4 (PADI4) [47] was also reported in relation to this variant. Each of these effects probably work in a cell-specific manner with respect to the pathogenesis of RA.
2.3. PADI4
PADI4 was identified as the first non-MHC RA risk locus in the Japanese population [48] and was later confirmed in European populations [19]. PADI4 was expressed in myeloid lineage cells and rheumatoid arthritis synovial tissues [48], as well as in Porphyromonas gingivalis and Aggregatibacter actinomycetemcomitans in gingival tissue [49], and it post-translationally converts peptide-bound arginine residues into citrulline, leading to citrullinated epitope generation, which is recognized by ACPAs [50].
2.4. Important Considerations for a Genetic Study
Because allelic variants presenting in more than 1–5% of a given population are identified in GWAS, a number of unusual or rare variants are missed (missing heritability). Missing heritability is hard to analyze with the same statistical methods used in GWAS, and thus, specific statistics for analysis are needed. Moreover, GWAS usually investigate SNPs that are in strong linkage disequilibrium (LD) with other SNPs and serve as proxies for them, and thus, the identified SNPs by GWAS are regarded merely as tags for the yet-to-be-identified causal allele. Next-generation sequencing (NGS) is one of the promising tools for future fine-mapping studies. With the use of NGS, several HLA-related genes, including non-classical HLA genes, HLA-like genes, and pseudo-HLA genes, as well as key immune-related genes, can be incorporated into current reference panels, which will enable us to identify disease-related variants with higher resolution.
3. Cigarette Smoking as the Most Robust Environmental Risk of RA
Previous studies have shown the contribution of various environmental factors to RA pathogenesis. Inhaled pollutants, especially CS, have been the most extensively studied and a topic of this article (Table 1).

Table 1.
The risks of cigarette smoking for rheumatoid arthritis (RA) development, RA-related pathologies, and comorbidities.
Regardless of the autoantibody status, CS increases the risk of RA development by 26% in those who smoked 1–10 pack-years (a lifelong CS exposure defined by the following formula; pack-years = [number of cigarettes smoked per day/20] × [number of years smoked]) and by 94% in those with more than 20 pack-years according to the meta-analysis conducted by Di Giuseppe et al. [51]. It has been reported that males are more susceptible to CS than females with respect to RA development [52]. CS can even affect treatment response to disease-modifying anti-rheumatic drugs (DMARDs) [53] and thus can be a risk of future joint destruction [54,55,56].
3.1. Effects of Intensity and Duration of Cigarette Smoking and Smoking Cessation
Importantly, both CS intensity and duration are directly related to the risk of RA development with prolonged increased risk even after CS cessation [51]. Di Giuseppe et al. conducted a meta-analysis of the association between pack-years and the risk of RA development. Three prospective and seven case–control studies were included in the analyses, and they found that smokers had a higher risk of RA development than never-smokers in a dose-dependent manner up to 20 pack-years, after which the risk did not increase further. Among smokers, RF-positive cases had higher a risk than RF-negative cases [51]. In contrast, Hedström et al. conducted a case–control study with 3655 cases and 5883 controls from a Swedish population [52], in which they found a dose-dependent increase of RA risk in both ACPA-positive and -negative cases, with greater effects in ACPA-positive cases. Interestingly, CS duration had a higher influence on the association of CS and RA than did CS intensity. They also found different effects of CS cessation on ACPA-positive and -negative cases; the association of CS with RA no longer persisted after 20 years of cessation in ACPA-negative cases, while the association persisted with cumulative dose dependency in ACPA-positive cases.
3.2. Effects of Passive Smoking
While direct smoking has become an established risk of RA and its disease course, Seror et al. reported that passive smoking during childhood affected susceptibility to RA in the French E3N cohort (98,995 women born between 1925 and 1950) [57,58]. Disease activity was also affected by passive smoking in Korean (191 cases) [59] and Egyptian (100 cases) [60] female RA patients, implicating the importance of avoiding any CS-exposing environment. In contrast, Hedström et al. found that there was no association between passive smoking and RA risk in the EIRA cohort (589 cases and 1764 controls aged 18–70 years) from Sweden, which might be explained by a threshold of smoking intensity below which an association between smoking exposure and RA does not occur [61]. A recent study conducted by Kronzer et al. (1198 cases and 3061 controls) also did not show a clear association between passive smoking and RA risk [62]. However, because those two studies did not measure the effect of childhood exposure to passive smoking and there seemed to be a linear trend between pack-years and ORs in the latter study [62], it may be of interest to know not only whether there is a true association between childhood exposure to passive smoking and RA risk, but also if the association is just a consequence of intensity of smoking exposure or a specific effect of childhood exposure.
3.3. Effects of Cigarette Smoking on RA-Related Autoantibody Production
As has been implicated in the studies above and others, the association between CS and RA risk is stronger in seropositive cases than in seronegative cases. It has also been suggested that CS may affect RA-associated autoantibody formation. A study conducted by van Wasemael et al. showed that CS was associated with multiple autoantibody positivity (RF, ACPA, anti-carbamylated protein (CarP) antibody) not only in RA patients of European descent but also in Japanese non-RA subjects [63]. Furthermore, the study also indicated that CS might have a stronger association with RF than with ACPA or anti-CarP antibody. CS may break tolerance to autoantigens in RA, which might be one of the triggers of RA onset in subsets of patients. We also recently reported that CS affected both positivity and levels of ACPA and RF with greater effects on RF using 6239 Asian RA cases, the largest Asian study ever [64]. The study also implicated the dose-dependent effects of CS and the effects of CS cessation on autoantibody levels, the latter of which lasted for up to 20 years both in ACPA and RF cases.
3.4. Other Environmetal Risks Augmented by Cigarette Smoking
Several environmental risks have been reported to be influenced by CS. Among them, occupational silica exposure seems to be convincing, while the rest need to be investigated further with well-powered studies for confirmation of the associations.
3.4.1. Occupational Silica Exposure
Occupational exposure to crystalline silica (SiO2), especially in male workers, is a well-known example of environmental risk. Two Swedish studies, a population-based case–control study comprising 577 incident RA cases and 659 randomly selected controls from EIRA [65] and an independent Swedish construction health examination cohort study comprising a total of 240,983 participants [66], reported that risk of RA by silica exposure exceeded that expected from the separate effects of silica and CS among smokers. It was suggested that silica-induced inflammation and fibrosis may be mechanistically separate, because the steps in the development of silicosis, including acute and chronic inflammation and fibrosis, have different molecular and cellular requirements [67]. Autoimmunity would probably start with activation of the innate immune system, leading to proinflammatory cytokine production, pulmonary inflammation, subsequent activation of adaptive immunity, breaking of tolerance, and autoantibody production leading to tissue damage. It also suggests substantial genetic involvement and gene/environment interaction in silica-induced autoimmunity [67].
3.4.2. Alcohol Consumption
Consumption of moderate amount of alcohol has been reported for the beneficial effect on RA development [68,69,70]. It is well accepted that CS and alcohol consumption are common in RA patients, thus both need to be adjusted when the association of each risk with RA is to be investigated, because CS is more prevalent among alcohol drinkers [71]. The study conducted by Källberg et al. comprising the Swedish EIRA cohort (1204 cases and 871 controls) and the Danish CACORA (444 cases and 533 controls) indicated a greater alcohol-related risk reduction for ACPA-positive RA observed in ever-smokers carrying SE alleles compared with never-smokers [72].
3.4.3. Sugar-Sweetened Soda Consumption
Regular consumption of sugar-sweetened soda has been reported to be associated with increased risk of seropositive RA in women, independent of other dietary and lifestyle factors [73]. The study population comprised two cohorts, the Nurses’ Health Study (NHS), comprising 121,700 female nurses, and NHS II, consisting of 116,671 female nurses, and an effect modification of CS was found among smokers with > 10 pack-years in the NHS cohort, but not in the NHS II cohort. Thus, further studies in independent populations are necessary for validation.
3.4.4. High Salt Intake
According to the case-control study (386 cases and 1886 matched controls) conducted by Sundström et al., sodium intake more than doubled the risk of RA among smokers, which was not observed among non-smokers. Moreover, the risk was further increased in the development of ACPA-positive and/or SE-positive RA cases, indicating a possible interactive effect between CS and high sodium intake on ACPA-positive RA.
4. Impacts of Cigarette Smoking on RA Pathogenesis
4.1. Effects of Cigarette Smoking on Immune Systems
CS affects both innate and adaptive immune responses, leading to altered cellular and humoral immunity to cause a systemic inflammation [74].
Skewed helper T (Th) cell subsets (Th1, Th2, and Th17) were observed depending on the relation to specific diseases [75,76,77], and Th1-skewness was reported among RA patients with CS [78]. In contrast, asthma, a Th2-skewed disease, has been reported to be associated with risk of RA [62,79,80,81,82,83,84,85,86], and CS is one of the well-established risks of asthma [87]. Activation of Th17 cells via aryl hydrocarbon receptor (AHR) [88] was also reported. Because Th-skewness is flexible and dynamic in the same individual with RA depending on various factors, such as medications, which, in turn, may influence disease activity or clinical course [89], it will be intriguing to further investigate how Th-skewness contributes to the pathogenesis of RA in relation to CS.
Increased levels of several pro-inflammatory cytokines (tumor necrosis factor (TNF)-α; interleukin (IL)-1α, IL-1β, IL-5, IL-6, IL-8, IL-13, IL-15, and IL-21; and interferon (IFN)-γ) in smokers with systemic autoimmune diseases including RA have also been well-documented [74,90,91,92,93,94,95]. Indeed, drugs targeting several of these cytokines, TNF-α, IL-6, and IL-1β, are currently used as biologic DMARDs for the treatment of RA [96] as well as JIA [97].
According to the study conducted by Glossop et al., CS increases TNF-α production from T cells, and both intensity and duration of CS are correlated with higher TNF-α/soluble TNF receptor (sTNFR) ratios in RA patients. Furthermore, smokers had higher ratios of TNF-α/sTNFR than non-smokers, suggesting that higher levels of TNF-α or ratios of TNF-α/sTNFR in smokers might be associated with TNF-α antagonist treatment resistance [94].
It was reported that serum levels of soluble IL-2 receptor (sIL-2R) were higher in smokers [98,99]. Furthermore, sIL-2R can affect the response to infliximab in RA patients, and a low serum sIL-2R level predicts rapid response to infliximab [100], suggesting that IL-2-sIL-2R activation may affect the response to anti-TNF-α treatment in RA patients with CS.
Elevated serum IFN-γ levels [95] and IFN-γ secretion both from effector CD4 and CD8 T cells have also been documented in RA patients [101]. As for the effects of CS, Bidkar et al. showed that CS exposure induced IFN-γ secretion from splenocytes of humanized transgenic (Tg)-mice carrying RA-susceptible HLA-DRB1*0401, while CS exposure augmented Th2 response in Tg-mice carrying RA-resistant HLA-DRB1*04:02. Despite the limitations of a mouse study, this implied a possible interaction of CS with the host HLA genes, leading to modulation of host immunity [102]. As mentioned above, CS can promote both Th1 and Th2 polarization. CD8 T cells, which are another major source of IFN-γ, have been reported to increase in number and be more prone to secreting cytokines due to CS in patients with chronic obstructive pulmonary disease (COPD) [103,104,105,106,107], and low soluble programmed death protein 1 ligand (sPD-L1) levels [108] and increased activated cytotoxic CD8 T cells [109] were also reported in RA patients. In contrast, the effects of CS on natural killer (NK) cells, which have similar cytotoxic functions as CD8 T cells, were variable in terms of the numbers and functions depending on an individual’s health status [110,111,112,113,114,115,116]. Thus, the effect of CS on IFN-γ production might be cell-type specific under the influence of the genetic background of an individual [117], which can eventually contribute to form a specific disease phenotype including RA.
Other mechanisms of CS suggested to affect immune systems with regard to RA development, such as autoimmunity to vimentin including induction of carbamylated vimentin [118], are also intriguing and are thus expected to be studied further.
4.2. Interactive Effects of Cigarette Smoking and Genetic Components
The most rheumatic diseases show complex traits with interactions between multiple genetic and environmental factors. Likewise, gene–environment interactions also play a critical role for RA pathogenesis (Figure 1).
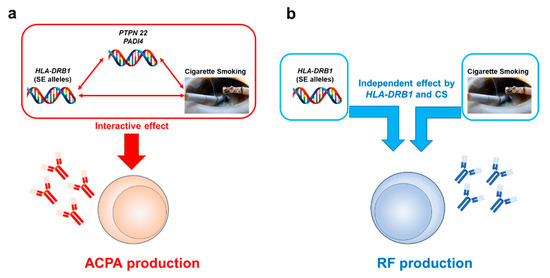
Figure 1.
The differential effects of gene and environmental factors (cigarette smoking) on ACPA and RF production. The interactive effects of cigarette smoking (CS) and genetic components, especially HLA-DRB1, contribute to ACPA production and subsequently predispose subjects to ACPA-positive RA development. PTPN22 and PADI4 are also implicated as additional genetic components (a). For RF production, an interactive effect between genetic and CS is much less clear; rather, they are independent risk factors (b).
4.2.1. Interactive Effects between CS and the HLA-DRB1 Gene on RA Development
CS has been implicated for its interactive effect, especially in relation to HLA-DRB1 in seropositive RA cases. Several studies, not limited to Caucasian populations and including two Asian cohort studies, have investigated the interactive association between CS and ACPA formation in the context of HLA-DRB1 alleles [119,120,121,122,123,124,125]. These studies, with the exception of a single North American cohort (SONORA), reported an interactive effect between CS and SE on ACPA-positive RA development. While most of the recent studies have focused on ACPA positivity, there have also been several studies that have investigated the interaction between CS and SE on RF-positive RA cases. Padyukov et al. observed that both CS and SE alleles conferred increased risk of RF-positive RA development, and there was a strong interaction between these two risks in the Swedish cohort (858 cases and 1048 controls) [126]. In contrast, the intra-case analysis conducted by Mattey et al. in 371 northern European white RA cases revealed that CS and HLA-DRB1*04:01 were independently associated with RF production [127]. Thus far, the effect of the interaction between CS and SE on ACPA production is clear, while that on RF production is still debated—partly due to lack of recent evidence—and thus requires more research.
4.2.2. Interactive Effects between CS and the HLA-DRB1 Gene on RA-Related Autoantibody Production
While the interactive association of SE and CS with ACPA positivity is well documented, the effects on levels of ACPA as well as RF have not been studied well. In our recent study of Japanese RA cases, CS affected not only positivity but also levels of both ACPA and RF. The effect of CS was dependent on SE presence for ACPA but independent of SE status for RF [64]. Hedström et al. also reported similar findings in a case-control study with 3645 cases and 5883 controls. In the subset of patients positive for both RF and ACPA and the subjects only with positive ACPA, both CS and SE conferred independent risks, and there was a strong interaction between CS and SE. In the subset of patients with only positive RF, there was an increased risk of disease among smokers, which was only marginally affected by SE, and no interaction between CS and SE was observed. In the subset of patients negative for both RF and ACPA, neither CS nor SE conferred an increased RA risk [128]. These studies strongly indicate the different effect of CS on the development of ACPA and RF with regard to the interaction with SE alleles, highlighting the distinctive pathogeneses in different subsets of RA patients.
Furthermore, the study conducted by van der Helm-van Mil et al. suggested that interactive effects with CS were different among SE subsets; the interaction was strongest for the HLA-DRB1*01:01 or *01:02 and HLA-DRB1*10:01 SE alleles [124]. In contrast, Lundström et al. also reported that all SE alleles tested (*01, *04:01, *04:04, *04:05, *04:08, and *10:01) strongly interacted with CS in conferring an increased risk of ACPA-positive RA, regardless of the fine specificity of SE in the EIRA cohort (1319 cases and 943 controls) [129]. We also did not observe a difference between HLA-DRB1*04:05 and non-*04:05 SE alleles in the interactive effect with CS on ACPA levels, and *09:01 did not show an interactive effect with CS on ACPA levels in Japanese RA cases [64]. In contrast, Bang et al. observed the different effect of interaction of HLA-DRB1 alleles with CS on ACPA-positive RA development in a Korean case–control study (1924 cases and 1119 controls) [130]. Among the HLA-DRB1 alleles they tested (five SE alleles, *01:01, *04:01, *04:04, *04:05, and *10:01, and a non-SE allele, *09:01, frequently observed in Asian populations), *10:01 showed the strongest interaction with CS, and the genotype heterozygous for *04:05 and *09:01 conferred the highest risk of both ACPA-positive and -negative RA development in the interaction with CS. These discrepant results may be partly due to the different frequencies of each SE allele among different ethnicities, and thus, meta-analyses or well-powered multi-ethnic studies are necessary to draw a solid conclusion.
4.2.3. Interactive Effects between CS and the PTPN22 Gene on RA Pathology
Mahdi et al. reported specific interactive effects of CS and SE, or PTPN22 (620W allele), one of the major GWAS genes and a potential causal variant in RA [131], on citrullinated α-enolase in a case-control study (1000 cases and 872 controls) [132]. The same group further extended the study by stratifying ACPA-positive RA patients into 17 subsets based on their profiles of different ACPA specificities (α-enolase, vimentin, fibrinogen, and collagen type II), which revealed the strongest association of SE, PTPN22, and CS in the subset of patients with antibodies to citrullinated α-enolase and vimentin [133]. In contrast, Willemze et al. showed that SE and CS promoted nonspecific citrullination rather than citrullination of specific antigens (α-enolase, vimentin, fibrinogen, and myelin basic protein) in Dutch RA patients with ACPA (661 cases) [134]. Fisher et al. also reported that the interaction between SE and CS was not exclusive to any of the specific citrullinated peptides (α-enolase, vimentin, and fibrinogen) in Korean RA patients (513 cases and 1101 controls) [135]. These studies strongly indicated the possible interactive effect of CS and SE on protein citrullination, with the specificity still remaining unknown, which in turn leads to ACPA formation.
4.2.4. Interactive Effects between CS and the PADI4 Gene on RA Pathology
Kochi et al. found that PADI4 polymorphism (rs1748033) predisposed male smokers to RA in a total of 2018 cases and 2035 controls from Japanese samples and also observed similar trends in a total of 635 cases and 391 controls from Dutch samples [136]. The study also showed that PADI4 polymorphism, rs11203367, was significantly associated with ACPA status in ever-smokers in a recessive model, suggesting that PADI4 polymorphism may be involved in the appearance of ACPA in smokers.
In summary, the gene-environment interaction, especially SE and CS, is strongly indicated in ACPA formation, while the effect on RF formation, if any, may be weaker than that on ACPA, although recent studies focusing on RF positivity are relatively lacking. As we mentioned in a previous section, seropositive polyarthritis JIA shares genetic components that confer a risk or protection with adult seropositive RA [25,26], and thus, it will be of interest to examine if the same effects seen in adult RA patients can also be found in the subset of JIA, especially with regard to passive smoking.
4.3. Effects of CS on Epigenetic Changes
CS causes wide-spread genome-scale changes in DNA methylation. In the epigenome-wide association study (EWAS) of the Swedish EIRA cohort (354 cases and 337 controls), Liu et al. identified two clusters of differentially methylated regions within the MHC region. By correcting cellular heterogeneity to adjust for cell-type proportions and with the use of analysis to filter out associations likely to be a consequence of disease, four CpGs also showed an association between genotype and variance of methylation, one of which was significantly associated with both clusters and the rest of which also showed suggestive association [76].
Zeilinger et al. conducted an independent EWAS with the use of large German populations, a total of 1814 for discovery and a total of 479 for replication, and found wide-spread differences in the degree of site-specific methylation as a function of CS in each of the 22 autosomes, confirming the broad effect of CS on epigenetic changes. Among the observed changes, methylation-specific protein binding patterns observed for cg05575921 within aryl hydrocarbon receptor (AHR) repressor (AHRR) had the highest level of changes in DNA methylation associated with CS, suggesting a regulatory role for gene expression. Importantly, methylation levels in past-smokers were close to the ones seen in never-smokers depending on cessation time and pack-years [137].
As for the interactive effects of CS with gene polymorphisms on RA epigenome, Meng et al. identified a significant interaction between rs6933349 of mucin 22 (MUC22) and CS in DNA methylation of cg21325723 in terms of the risk of developing ACPA-positive RA in both Caucasian and Asian populations [138].
Although data for epigenetic phenomena in RA are currently limited in terms of study scale and power [139], it is highly likely that epigenetic changes play crucial roles in the development of RA via gene regulation. Further study will be needed with considerations of sample throughput methods and genome coverage and resolution, such as the use of whole-genome bisulfite sequencing [140]. Longitudinal cohorts will also be essential for establishing the temporal origin of deleterious events and distinguishing causal from consequential effects [141].
4.4. Cigarette Smoking Modulates Periodontal Disease Leading to a Higher Risk of RA Development
It is well established that CS is one of the risks of periodontal disease (PD) [142], and there have been many studies that have linked CS-affected PD development with a higher risk of RA development. As we mentioned in the previous section, P. gingivalis, a major periodontal pathogen [143,144], can induce ACPA by bacterial PAD, and worsen the severity of RA [145,146,147]. Another major pathogen, A. actinomycetemcomitans is also known to be associated with hypercitrullination in the gingival tissues [148]. Furthermore, periodontitis is known to correlate with ACPA levels in healthy individuals [149] and often precedes RA development [150,151]. Notably, RA and periodontitis also shared genetic risks, such as SE alleles [152]. Not only ACPA but also RF positivity is associated with periodontitis [142,153], although the underlying mechanisms might be different between these autoantibodies [64,128]. Collectively, CS can also be a strong risk modifier of RA development via its effect on PD pathology, especially in individuals with SE alleles.
4.5. Airway Inflammation
CS significantly increases the number of alveolar macrophages and other monocytes, which, in turn, increases levels of lysosomal enzymes and secrete elastase responsible for parenchymal and connective tissue damage [74]. Demoruelle et al. demonstrated that airway inflammation is common in healthy ACPA-positive subjects before clinically apparent RA development [154]. Klareskog et al. also demonstrated that CS induces protein citrullination in the lungs [119]. Thus, one interesting possible hypothesis is that initial inflammation and immune abnormality of RA may generate from the lungs [154].
Matrix metalloproteinases (MMP)-12 has been implicated in the pathogenesis of RA [155], and animal experiments have suggested MMP-12 as one potential mediator of airway inflammation. For example, MMP-12 expression was increased in macrophages and dendritic cells in the lungs of CS-exposed mice [156]. Other MMPs, proMMP-2 and proMMP-9, have also been reported to be increased in the sera of smokers [157]. Although RA synovial fibroblasts-derived MMP-9 may directly contribute to joint destruction in RA [158], further investigation as to whether the effects of these MMPs on RA pathogenesis are dependent on airway inflammation or not is required.
4.6. The Role of Passive Smoking in RA Pathogenesis and the Role of Nicotine
Although nicotine is a main toxic substance in both direct smoking and passive smoking, passive smoking in childhood may have distinctive mechanistic impacts on RA pathogenesis from direct CS considering that the smoke from the burning end of a cigarette has more toxins than the smoke inhaled by smokers, and passive smoking in childhood increases the risk of asthma [159], which may be associated with RA risk [62].
Interestingly, Jian et al. reported that the risk of CS on both ACPA-positive and -negative RA development increased only with the use of inhaling cigarettes but not nicotine-contained chewed cigarettes (OR, 1.0; 95% CI, 0.8–1.2), suggesting that nicotine is not directly involved in RA pathogenesis [160]. Indeed, nicotine inhibits TNF-α-induced IL-6 and IL-8 secretion in fibroblast-like synoviocytes (FLS) from RA patients [161]. Moreover, several animal experiments showed an immunosuppressive effect of nicotine, such as the loss of antibody response and T-cell proliferation [162,163,164]. In contrast, the paradoxical effect of nicotine on RA has also been reported; nicotine pretreatment aggravated adjuvant-induced arthritis (AIA), whereas post-treatment with nicotine suppressed the disease in rats [165]. Furthermore, blood cotinine, a nicotine metabolite, was positively correlated with the prevalence of naive CD3+ T cells among non-smokers exposed to passive smoking [166]. Thus far, the role of nicotine in the pathogenesis of RA is still questionable and needs further investigation with regard to the effect of passive smoking on RA.
5. Concluding Remarks and Future Directions of Studies
Numerous efforts have been made to clarify the pathogenesis of RA in relation to CS. Despite such efforts, the underlying mechanisms still have not been clarified due to the complex nature of the effects of CS on RA pathology. However, a growing body of data has suggested that environment–environment or gene-environment interactions are key mechanisms to trigger the onset and modify the course of the disease, and thus, future studies should take into consideration these interactions in association studies of CS and RA. In addition, most studies have found that the effect of CS on RA pathogenesis is limited or much stronger in seropositive RA, and effects of CS seem to be different between ACPA- and RF-positive subsets, which will also be an important subject of future studies. The different effect of CS among different ethnicities will also be of interest.
In summary, the effect of CS on RA pathology has multiple aspects; interaction with genetic components as well as other environmental factors, effects on immune systems including both innate and acquired immunity, and epigenetic changes by several key chemical compounds or reactive oxygen species (ROS), which altogether might contribute to pathogenesis. Well-powered studies with consideration of several key points mentioned above will further clarify the precise pathogenic role of CS, which will lead to our better understanding of RA pathogenesis and the development of better treatment options.
Author Contributions
Conceptualization, Y.I. and C.T.; writing—original draft preparation, Y.I. and C.T.; writing—review and editing, Y.I. and C.T.; visualization, Y.I. and C.T.; supervision, C.T.; project administration, C.T. All authors have read and agreed to the published version of the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Calabresi, E.; Petrelli, F.; Bonifacio, A.F.; Puxeddu, I.; Alunno, A. One year in review 2018: Pathogenesis of rheumatoid arthritis. Clin. Exp. Rheumatol. 2018, 36, 175–184. [Google Scholar] [PubMed]
- Okada, Y.; Eyre, S.; Suzuki, A.; Kochi, Y.; Yamamoto, K. Genetics of rheumatoid arthritis: 2018 status. Ann. Rheum. Dis. 2019, 78, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Bukhari, M.; Lunt, M.; Harrison, B.J.; Scott, D.G.; Symmons, D.P.; Silman, A.J. Rheumatoid factor is the major predictor of increasing severity of radiographic erosions in rheumatoid arthritis: Results from the Norfolk Arthritis Register Study, a large inception cohort. Arthritis Rheum. 2002, 46, 906–912. [Google Scholar] [CrossRef]
- Terao, C.; Yamakawa, N.; Yano, K.; Markusse, I.M.; Ikari, K.; Yoshida, S.; Furu, M.; Hashimoto, M.; Ito, H.; Fujii, T.; et al. Rheumatoid Factor Is Associated with the Distribution of Hand Joint Destruction in Rheumatoid Arthritis. Arthritis Rheumatol. 2015, 67, 3113–3123. [Google Scholar] [CrossRef] [PubMed]
- Syversen, S.W.; Goll, G.L.; van der Heijde, D.; Landewé, R.; Lie, B.A.; Odegård, S.; Uhlig, T.; Gaarder, P.I.; Kvien, T.K. Prediction of radiographic progression in rheumatoid arthritis and the role of antibodies against mutated citrullinated vimentin: Results from a 10-year prospective study. Ann. Rheum. Dis. 2010, 69, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Hecht, C.; Englbrecht, M.; Rech, J.; Schmidt, S.; Araujo, E.; Engelke, K.; Finzel, S.; Schett, G. Additive effect of anti-citrullinated protein antibodies and rheumatoid factor on bone erosions in patients with RA. Ann. Rheum. Dis. 2015, 74, 2151–2156. [Google Scholar] [CrossRef]
- Akdemir, G.; Verheul, M.K.; Heimans, L.; Wevers-de Boer, K.V.; Goekoop-Ruiterman, Y.P.; van Oosterhout, M.; Harbers, J.B.; Bijkerk, C.; Steup-Beekman, G.M.; Lard, L.R.; et al. Predictive factors of radiological progression after 2 years of remission-steered treatment in early arthritis patients: A post hoc analysis of the IMPROVED study. RMD Open 2016, 2, e000172. [Google Scholar] [CrossRef]
- Orsolini, G.; Caimmi, C.; Viapiana, O.; Idolazzi, L.; Fracassi, E.; Gatti, D.; Adami, G.; Rossini, M. Titer-Dependent Effect of Anti-Citrullinated Protein Antibodies on Systemic Bone Mass in Rheumatoid Arthritis Patients. Calcif. Tissue Int. 2017, 101, 17–23. [Google Scholar] [CrossRef]
- Bugatti, S.; Bogliolo, L.; Vitolo, B.; Manzo, A.; Montecucco, C.; Caporali, R. Anti-citrullinated protein antibodies and high levels of rheumatoid factor are associated with systemic bone loss in patients with early untreated rheumatoid arthritis. Arthritis Res. Ther. 2016, 18, 226. [Google Scholar] [CrossRef]
- Katchamart, W.; Koolvisoot, A.; Aromdee, E.; Chiowchanwesawakit, P.; Muengchan, C. Associations of rheumatoid factor and anti-citrullinated peptide antibody with disease progression and treatment outcomes in patients with rheumatoid arthritis. Rheumatol. Int. 2015, 35, 1693–1699. [Google Scholar] [CrossRef]
- Aletaha, D.; Neogi, T.; Silman, A.J.; Funovits, J.; Felson, D.T.; Bingham, C.O.; Birnbaum, N.S.; Burmester, G.R.; Bykerk, V.P.; Cohen, M.D.; et al. 2010 Rheumatoid arthritis classification criteria: An American College of Rheumatology/European League Against Rheumatism collaborative initiative. Arthritis Rheum. 2010, 62, 2569–2581. [Google Scholar] [CrossRef] [PubMed]
- Rönnelid, J.; Wick, M.C.; Lampa, J.; Lindblad, S.; Nordmark, B.; Klareskog, L.; van Vollenhoven, R.F. Longitudinal analysis of citrullinated protein/peptide antibodies (anti-CP) during 5 year follow up in early rheumatoid arthritis: Anti-CP status predicts worse disease activity and greater radiological progression. Ann. Rheum. Dis. 2005, 64, 1744–1749. [Google Scholar] [CrossRef] [PubMed]
- Nordberg, L.B.; Lillegraven, S.; Aga, A.B.; Sexton, J.; Olsen, I.C.; Lie, E.; Berner Hammer, H.; Uhlig, T.; van der Heijde, D.; Kvien, T.K.; et al. Comparing the disease course of patients with seronegative and seropositive rheumatoid arthritis fulfilling the 2010 ACR/EULAR classification criteria in a treat-to-target setting: 2-year data from the ARCTIC trial. RMD Open 2018, 4, e000752. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Raychaudhuri, S.; Gregersen, P.K. Recent Advances in Defining the Genetic Basis of Rheumatoid Arthritis. Annu. Rev. Genom. Hum. Genet. 2016, 17, 273–301. [Google Scholar] [CrossRef]
- Terao, C.; Ikari, K.; Nakayamada, S.; Takahashi, Y.; Yamada, R.; Ohmura, K.; Hashimoto, M.; Furu, M.; Ito, H.; Fujii, T.; et al. A twin study of rheumatoid arthritis in the Japanese population. Mod. Rheumatol. 2016, 26, 685–689. [Google Scholar] [CrossRef] [PubMed]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.; Zhernakova, A.; Hinks, A.; et al. Genome-wide association study meta-analysis identifies seven new rheumatoid arthritis risk loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Okada, Y.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Kawaguchi, T.; Stahl, E.A.; Kurreeman, F.A.; Nishida, N.; et al. Meta-analysis identifies nine new loci associated with rheumatoid arthritis in the Japanese population. Nat. Genet. 2012, 44, 511–516. [Google Scholar] [CrossRef]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-density genetic mapping identifies new susceptibility loci for rheumatoid arthritis. Nat. Genet. 2012, 44, 1336–1340. [Google Scholar] [CrossRef]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2014, 506, 376–381. [Google Scholar] [CrossRef]
- Kurreeman, F.A.; Stahl, E.A.; Okada, Y.; Liao, K.; Diogo, D.; Raychaudhuri, S.; Freudenberg, J.; Kochi, Y.; Patsopoulos, N.A.; Gupta, N.; et al. Use of a multiethnic approach to identify rheumatoid- arthritis-susceptibility loci, 1p36 and 17q12. Am. J. Hum. Genet. 2012, 90, 524–532. [Google Scholar] [CrossRef]
- Kim, K.; Bang, S.Y.; Ikari, K.; Yoo, D.H.; Cho, S.K.; Choi, C.B.; Sung, Y.K.; Kim, T.H.; Jun, J.B.; Kang, Y.M.; et al. Association-heterogeneity mapping identifies an Asian-specific association of the GTF2I locus with rheumatoid arthritis. Sci. Rep. 2016, 6, 27563. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Muramatsu, T.; Suita, N.; Kanai, M.; Kawakami, E.; Iotchkova, V.; Soranzo, N.; Inazawa, J.; Tanaka, T. Significant impact of miRNA-target gene networks on genetics of human complex traits. Sci. Rep. 2016, 6, 22223. [Google Scholar] [CrossRef] [PubMed]
- Sakaue, S.; Hirata, J.; Maeda, Y.; Kawakami, E.; Nii, T.; Kishikawa, T.; Ishigaki, K.; Terao, C.; Suzuki, K.; Akiyama, M.; et al. Integration of genetics and miRNA-target gene network identified disease biology implicated in tissue specificity. Nucleic Acids Res. 2018, 46, 11898–11909. [Google Scholar] [CrossRef]
- De Silvestri, A.; Capittini, C.; Poddighe, D.; Marseglia, G.L.; Mascaretti, L.; Bevilacqua, E.; Scotti, V.; Rebuffi, C.; Pasi, A.; Martinetti, M.; et al. HLA-DRB1 alleles and juvenile idiopathic arthritis: Diagnostic clues emerging from a meta-analysis. Autoimmun. Rev. 2017, 16, 1230–1236. [Google Scholar] [CrossRef] [PubMed]
- Nigrovic, P.A.; Raychaudhuri, S.; Thompson, S.D. Review: Genetics and the Classification of Arthritis in Adults and Children. Arthritis Rheumatol. 2018, 70, 7–17. [Google Scholar] [CrossRef]
- van der Woude, D.; Houwing-Duistermaat, J.J.; Toes, R.E.; Huizinga, T.W.; Thomson, W.; Worthington, J.; van der Helm-van Mil, A.H.; de Vries, R.R. Quantitative heritability of anti-citrullinated protein antibody-positive and anti-citrullinated protein antibody-negative rheumatoid arthritis. Arthritis Rheum. 2009, 60, 916–923. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. An approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef]
- Han, B.; Diogo, D.; Eyre, S.; Kallberg, H.; Zhernakova, A.; Bowes, J.; Padyukov, L.; Okada, Y.; González-Gay, M.A.; Rantapää-Dahlqvist, S.; et al. Fine mapping seronegative and seropositive rheumatoid arthritis to shared and distinct HLA alleles by adjusting for the effects of heterogeneity. Am. J. Hum. Genet. 2014, 94, 522–532. [Google Scholar] [CrossRef]
- Ohmura, K.; Terao, C.; Maruya, E.; Katayama, M.; Matoba, K.; Shimada, K.; Murasawa, A.; Honjo, S.; Takasugi, K.; Tohma, S.; et al. Anti-citrullinated peptide antibody-negative RA is a genetically distinct subset: A definitive study using only bone-erosive ACPA-negative rheumatoid arthritis. Rheumatology 2010, 49, 2298–2304. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Ikari, K.; Ohmura, K.; Suzuki, T.; Iwamoto, T.; Takasugi, K.; Saji, H.; Taniguchi, A.; Momohara, S.; Yamanaka, H.; et al. Quantitative effect of HLA-DRB1 alleles to ACPA levels in Japanese rheumatoid arthritis: No strong genetic impact of shared epitope to ACPA levels after stratification of HLA-DRB1*09:01. Ann. Rheum. Dis. 2012, 71, 1095–1097. [Google Scholar] [CrossRef] [PubMed]
- Terao, C.; Suzuki, A.; Ikari, K.; Kochi, Y.; Ohmura, K.; Katayama, M.; Nakabo, S.; Yamamoto, N.; Suzuki, T.; Iwamoto, T.; et al. An association between amino acid position 74 of HLA-DRB1 and anti-citrullinated protein antibody levels in Japanese patients with anti-citrullinated protein antibody-positive rheumatoid arthritis. Arthritis Rheumatol. 2015, 67, 2038–2045. [Google Scholar] [CrossRef] [PubMed]
- Hiwa, R.; Ikari, K.; Ohmura, K.; Nakabo, S.; Matsuo, K.; Saji, H.; Yurugi, K.; Miura, Y.; Maekawa, T.; Taniguchi, A.; et al. Analysis Identified a Genetically Unique Subset within Rheumatoid Arthritis and Distinct Genetic Background of Rheumatoid Factor Levels from Anticyclic Citrullinated Peptide Antibodies. J. Rheumatol. 2018, 45, 470–480. [Google Scholar] [CrossRef]
- Kochi, Y.; Yamada, R.; Kobayashi, K.; Takahashi, A.; Suzuki, A.; Sekine, A.; Mabuchi, A.; Akiyama, F.; Tsunoda, T.; Nakamura, Y.; et al. Analysis of single-nucleotide polymorphisms in Japanese rheumatoid arthritis patients shows additional susceptibility markers besides the classic shared epitope susceptibility sequences. Arthritis Rheum. 2004, 50, 63–71. [Google Scholar] [CrossRef]
- Padyukov, L.; Seielstad, M.; Ong, R.T.; Ding, B.; Rönnelid, J.; Seddighzadeh, M.; Alfredsson, L.; Klareskog, L.; Epidemiological Investigation of Rheumatoid Arthritis (EIRA) Study Group. A genome-wide association study suggests contrasting associations in ACPA-positive versus ACPA-negative rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 259–265. [Google Scholar] [CrossRef]
- Okada, Y.; Kim, K.; Han, B.; Pillai, N.E.; Ong, R.T.; Saw, W.Y.; Luo, M.; Jiang, L.; Yin, J.; Bang, S.Y.; et al. Risk for ACPA-positive rheumatoid arthritis is driven by shared HLA amino acid polymorphisms in Asian and European populations. Hum. Mol. Genet. 2014, 23, 6916–6926. [Google Scholar] [CrossRef]
- Okada, Y.; Suzuki, A.; Ikari, K.; Terao, C.; Kochi, Y.; Ohmura, K.; Higasa, K.; Akiyama, M.; Ashikawa, K.; Kanai, M.; et al. Contribution of a Non-classical HLA Gene, HLA-DOA, to the Risk of Rheumatoid Arthritis. Am. J. Hum. Genet. 2016, 99, 366–374. [Google Scholar] [CrossRef]
- Vang, T.; Miletic, A.V.; Bottini, N.; Mustelin, T. Protein tyrosine phosphatase PTPN22 in human autoimmunity. Autoimmunity 2007, 40, 453–461. [Google Scholar] [CrossRef]
- Burn, G.L.; Svensson, L.; Sanchez-Blanco, C.; Saini, M.; Cope, A.P. Why is PTPN22 a good candidate susceptibility gene for autoimmune disease? FEBS Lett. 2011, 585, 3689–3698. [Google Scholar] [CrossRef]
- Nabi, G.; Akhter, N.; Wahid, M.; Bhatia, K.; Mandal, R.K.; Dar, S.A.; Jawed, A.; Haque, S. Meta-analysis reveals PTPN22 1858C/T polymorphism confers susceptibility to rheumatoid arthritis in Caucasian but not in Asian population. Autoimmunity 2016, 49, 197–210. [Google Scholar] [CrossRef] [PubMed]
- Gregersen, P.K.; Lee, H.S.; Batliwalla, F.; Begovich, A.B. PTPN22: Setting thresholds for autoimmunity. Semin. Immunol. 2006, 18, 214–223. [Google Scholar] [CrossRef] [PubMed]
- Rieck, M.; Arechiga, A.; Onengut-Gumuscu, S.; Greenbaum, C.; Concannon, P.; Buckner, J.H. Genetic variation in PTPN22 corresponds to altered function of T and B lymphocytes. J. Immunol. 2007, 179, 4704–4710. [Google Scholar] [CrossRef] [PubMed]
- Vang, T.; Landskron, J.; Viken, M.K.; Oberprieler, N.; Torgersen, K.M.; Mustelin, T.; Tasken, K.; Tautz, L.; Rickert, R.C.; Lie, B.A. The autoimmune-predisposing variant of lymphoid tyrosine phosphatase favors T helper 1 responses. Hum. Immunol. 2013, 74, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Milla, V.; Mahfuz, G.A.; Tyyne, V.; Emmi-Leena, I.; Ilse, E.; Jorma, T.; Mikael, K.; Riitta, V.; Jorma, I.; Johanna, L.; et al. Type 1 diabetes linked PTPN22 gene polymorphism is associated with the frequency of circulating regulatory T cells. Eur. J. Immunol. 2019. [Google Scholar] [CrossRef]
- Wang, Y.; Ewart, D.; Crabtree, J.N.; Yamamoto, A.; Baechler, E.C.; Fazeli, P.; Peterson, E.J. PTPN22 Variant R620W Is Associated with Reduced Toll-like Receptor 7-Induced Type I Interferon in Systemic Lupus Erythematosus. Arthritis Rheumatol. 2015, 67, 2403–2414. [Google Scholar] [CrossRef]
- Chang, H.H.; Dwivedi, N.; Nicholas, A.P.; Ho, I.C. The W620 Polymorphism in PTPN22 Disrupts Its Interaction with Peptidylarginine Deiminase Type 4 and Enhances Citrullination and NETosis. Arthritis Rheumatol. 2015, 67, 2323–2334. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 34, 395–402. [Google Scholar] [CrossRef]
- Engström, M.; Eriksson, K.; Lee, L.; Hermansson, M.; Johansson, A.; Nicholas, A.P.; Gerasimcik, N.; Lundberg, K.; Klareskog, L.; Catrina, A.I.; et al. Increased citrullination and expression of peptidylarginine deiminases independently of P. gingivalis and A. actinomycetemcomitans in gingival tissue of patients with periodontitis. J. Transl. Med. 2018, 16, 214. [Google Scholar] [CrossRef]
- Cantaert, T.; De Rycke, L.; Bongartz, T.; Matteson, E.L.; Tak, P.P.; Nicholas, A.P.; Baeten, D. Citrullinated proteins in rheumatoid arthritis: Crucial … but not sufficient! Arthritis Rheum. 2006, 54, 3381–3389. [Google Scholar] [CrossRef]
- Di Giuseppe, D.; Discacciati, A.; Orsini, N.; Wolk, A. Cigarette smoking and risk of rheumatoid arthritis: A dose-response meta-analysis. Arthritis Res. Ther. 2014, 16, R61. [Google Scholar] [CrossRef] [PubMed]
- Hedström, A.K.; Stawiarz, L.; Klareskog, L.; Alfredsson, L. Smoking and susceptibility to rheumatoid arthritis in a Swedish population-based case-control study. Eur. J. Epidemiol. 2018, 33, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Torrente-Segarra, V.; Bergstra, S.A.; Solomon-Escoto, K.; Da Silva, J.; Veale, D.J.; Al-Emadi, S.; Huizinga, T. Is current smoking status and its relationship to anti-cyclic citrullinated peptide antibodies a predictor of worse response to biological therapies in rheumatoid arthritis patients? Scand. J. Rheumatol. 2018, 47, 360–363. [Google Scholar] [CrossRef] [PubMed]
- Rydell, E.; Forslind, K.; Nilsson, J.; Jacobsson, L.T.H.; Turesson, C. Smoking, body mass index, disease activity, and the risk of rapid radiographic progression in patients with early rheumatoid arthritis. Arthritis Res. Ther. 2018, 20, 82. [Google Scholar] [CrossRef] [PubMed]
- Sivas, F.; Yurdakul, F.G.; Kiliçarslan, A.; Duran, S.; Başkan, B.; Bodur, H. Relationship Between Smoking and Structural Damage, Autoimmune Antibodies, and Disability in Rheumatoid Arthritis Patients. Arch. Rheumatol. 2018, 33, 45–51. [Google Scholar] [CrossRef]
- Vittecoq, O.; Richard, L.; Banse, C.; Lequerré, T. The impact of smoking on rheumatoid arthritis outcomes. Joint Bone Spine 2018, 85, 135–138. [Google Scholar] [CrossRef]
- Hedström, A.K.; Klareskog, L.; Alfredsson, L. Exposure to passive smoking and rheumatoid arthritis risk: Results from the Swedish EIRA study. Ann. Rheum. Dis. 2018, 77, 970–972. [Google Scholar] [CrossRef]
- Seror, R.; Henry, J.; Gusto, G.; Aubin, H.J.; Boutron-Ruault, M.C.; Mariette, X. Passive smoking in childhood increases the risk of developing rheumatoid arthritis. Rheumatology 2018. [Google Scholar] [CrossRef]
- Kim, S.K.; Choe, J.Y. Passive Smoking is Responsible for Disease Activity in Female Patients with Rheumatoid Arthritis. Arch. Rheumatol. 2018, 33, 143–149. [Google Scholar] [CrossRef]
- Hammam, N.; Gheita, T.A. Impact of secondhand smoking on disease activity in women with rheumatoid arthritis. Clin. Rheumatol. 2017, 36, 2415–2420. [Google Scholar] [CrossRef]
- Stolt, P.; Bengtsson, C.; Nordmark, B.; Lindblad, S.; Lundberg, I.; Klareskog, L.; Alfredsson, L. Quantification of the influence of cigarette smoking on rheumatoid arthritis: Results from a population based case-control study, using incident cases. Ann. Rheum. Dis. 2003, 62, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Kronzer, V.L.; Crowson, C.S.; Sparks, J.A.; Vassallo, R.; Davis, J.M. Investigating Asthma, Allergic Disease, Passive Smoke Exposure, and Risk of Rheumatoid Arthritis. Arthritis Rheumatol. 2019, 71, 1217–1224. [Google Scholar] [CrossRef] [PubMed]
- van Wesemael, T.J.; Ajeganova, S.; Humphreys, J.; Terao, C.; Muhammad, A.; Symmons, D.P.; MacGregor, A.J.; Hafström, I.; Trouw, L.A.; van der Helm-van Mil, A.H.; et al. Smoking is associated with the concurrent presence of multiple autoantibodies in rheumatoid arthritis rather than with anti-citrullinated protein antibodies per se: A multicenter cohort study. Arthritis Res. Ther. 2016, 18, 285. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Ikari, K.; Hashimoto, M.; Ohmura, K.; Tanaka, M.; Ito, H.; Taniguchi, A.; Yamanaka, H.; Mimori, T.; Terao, C. Shared epitope defines distinct associations of cigarette smoking with levels of anticitrullinated protein antibody and rheumatoid factor. Ann. Rheum. Dis. 2019, 78, 1480–1487. [Google Scholar] [CrossRef] [PubMed]
- Stolt, P.; Yahya, A.; Bengtsson, C.; Källberg, H.; Rönnelid, J.; Lundberg, I.; Klareskog, L.; Alfredsson, L.; Group, E.S. Silica exposure among male current smokers is associated with a high risk of developing ACPA-positive rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1072–1076. [Google Scholar] [CrossRef]
- Blanc, P.D.; Järvholm, B.; Torén, K. Prospective risk of rheumatologic disease associated with occupational exposure in a cohort of male construction workers. Am. J. Med. 2015, 128, 1094–1101. [Google Scholar] [CrossRef]
- Pollard, K.M. Silica, Silicosis, and Autoimmunity. Front. Immunol. 2016, 7, 97. [Google Scholar] [CrossRef]
- Jonsson, I.M.; Verdrengh, M.; Brisslert, M.; Lindblad, S.; Bokarewa, M.; Islander, U.; Carlsten, H.; Ohlsson, C.; Nandakumar, K.S.; Holmdahl, R.; et al. Ethanol prevents development of destructive arthritis. Proc. Natl. Acad. Sci. USA 2007, 104, 258–263. [Google Scholar] [CrossRef]
- Scott, I.C.; Tan, R.; Stahl, D.; Steer, S.; Lewis, C.M.; Cope, A.P. The protective effect of alcohol on developing rheumatoid arthritis: A systematic review and meta-analysis. Rheumatology 2013, 52, 856–867. [Google Scholar] [CrossRef]
- Nissen, M.J.; Gabay, C.; Scherer, A.; Finckh, A.; Swiss Clinical Quality Management Project in Rheumatoid Arthritis. The effect of alcohol on radiographic progression in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 1265–1272. [Google Scholar] [CrossRef]
- Di Giuseppe, D.; Wallin, A.; Bottai, M.; Askling, J.; Wolk, A. Long-term intake of dietary long-chain n-3 polyunsaturated fatty acids and risk of rheumatoid arthritis: A prospective cohort study of women. Ann. Rheum. Dis. 2014, 73, 1949–1953. [Google Scholar] [CrossRef] [PubMed]
- Källberg, H.; Jacobsen, S.; Bengtsson, C.; Pedersen, M.; Padyukov, L.; Garred, P.; Frisch, M.; Karlson, E.W.; Klareskog, L.; Alfredsson, L. Alcohol consumption is associated with decreased risk of rheumatoid arthritis: Results from two Scandinavian case-control studies. Ann. Rheum. Dis. 2009, 68, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Costenbader, K.H.; Gao, X.; Al-Daabil, M.; Sparks, J.A.; Solomon, D.H.; Hu, F.B.; Karlson, E.W.; Lu, B. Sugar-sweetened soda consumption and risk of developing rheumatoid arthritis in women. Am. J. Clin. Nutr. 2014, 100, 959–967. [Google Scholar] [CrossRef] [PubMed]
- Sopori, M. Effects of cigarette smoke on the immune system. Nat. Rev. Immunol. 2002, 2, 372–377. [Google Scholar] [CrossRef] [PubMed]
- Harrison, O.J.; Foley, J.; Bolognese, B.J.; Long, E.; Podolin, P.L.; Walsh, P.T. Airway infiltration of CD4+ CCR6+ Th17 type cells associated with chronic cigarette smoke induced airspace enlargement. Immunol. Lett. 2008, 121, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Cozen, W.; Diaz-Sanchez, D.; James Gauderman, W.; Zadnick, J.; Cockburn, M.G.; Gill, P.S.; Masood, R.; Hamilton, A.S.; Jyrala, M.; Mack, T.M. Th1 and Th2 cytokines and IgE levels in identical twins with varying levels of cigarette consumption. J. Clin. Immunol. 2004, 24, 617–622. [Google Scholar] [CrossRef]
- Whetzel, C.A.; Corwin, E.J.; Klein, L.C. Disruption in Th1/Th2 immune response in young adult smokers. Addict. Behav. 2007, 32, 1–8. [Google Scholar] [CrossRef]
- Reckner Olsson, A.; Skogh, T.; Wingren, G. Comorbidity and lifestyle, reproductive factors, and environmental exposures associated with rheumatoid arthritis. Ann. Rheum. Dis. 2001, 60, 934–939. [Google Scholar] [CrossRef]
- Kero, J.; Gissler, M.; Hemminki, E.; Isolauri, E. Could TH1 and TH2 diseases coexist? Evaluation of asthma incidence in children with coeliac disease, type 1 diabetes, or rheumatoid arthritis: A register study. J. Allergy Clin. Immunol. 2001, 108, 781–783. [Google Scholar] [CrossRef]
- Hemminki, K.; Li, X.; Sundquist, J.; Sundquist, K. Subsequent autoimmune or related disease in asthma patients: Clustering of diseases or medical care? Ann. Epidemiol. 2010, 20, 217–222. [Google Scholar] [CrossRef]
- Karatay, S.; Yildirim, K.; Ugur, M.; Senel, K.; Erdal, A.; Durmus, B.; Baysal, O.; Altay, Z.; Sarac, A.J.; Gur, A.; et al. Prevalence of atopic disorders in rheumatic diseases. Mod. Rheumatol. 2013, 23, 351–356. [Google Scholar] [CrossRef] [PubMed]
- de Roos, A.J.; Cooper, G.S.; Alavanja, M.C.; Sandler, D.P. Personal and family medical history correlates of rheumatoid arthritis. Ann. Epidemiol. 2008, 18, 433–439. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hassan, W.U.; Keaney, N.P.; Holland, C.D.; Kelly, C.A. Bronchial reactivity and airflow obstruction in rheumatoid arthritis. Ann. Rheum. Dis. 1994, 53, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Provenzano, G.; Donato, G.; Brai, G.; Rinaldi, F. Prevalence of allergic respiratory diseases in patients with RA. Ann. Rheum. Dis. 2002, 61, 281. [Google Scholar] [CrossRef]
- Choi, I.A.; Park, S.H.; Cha, H.S.; Park, W.; Kim, H.A.; Yoo, D.H.; Baek, H.J.; Lee, S.G.; Lee, Y.J.; Park, Y.B.; et al. Prevalence of co-morbidities and evaluation of their monitoring in Korean patients with rheumatoid arthritis: Comparison with the results of an international, cross-sectional study (COMORA). Int. J. Rheum. Dis. 2018, 21, 1414–1422. [Google Scholar] [CrossRef]
- Dougados, M.; Soubrier, M.; Antunez, A.; Balint, P.; Balsa, A.; Buch, M.H.; Casado, G.; Detert, J.; El-Zorkany, B.; Emery, P.; et al. Prevalence of comorbidities in rheumatoid arthritis and evaluation of their monitoring: Results of an international, cross-sectional study (COMORA). Ann. Rheum. Dis. 2014, 73, 62–68. [Google Scholar] [CrossRef]
- Hekking, P.P.; Bel, E.H. Developing and emerging clinical asthma phenotypes. J. Allergy Clin. Immunol. Pract. 2014, 2, 671–680. [Google Scholar] [CrossRef]
- Talbot, J.; Peres, R.S.; Pinto, L.G.; Oliveira, R.D.R.; Lima, K.A.; Donate, P.B.; Silva, J.R.; Ryffel, B.; Cunha, T.M.; Alves-Filho, J.C.; et al. Smoking-induced aggravation of experimental arthritis is dependent of aryl hydrocarbon receptor activation in Th17 cells. Arthritis Res. Ther. 2018, 20, 119. [Google Scholar] [CrossRef]
- Ruschpler, P.; Stiehl, P. Shift in Th1 (IL-2 and IFN-gamma) and Th2 (IL-10 and IL-4) cytokine mRNA balance within two new histological main-types of rheumatoid-arthritis (RA). Cell. Mol. Biol. 2002, 48, 285–293. [Google Scholar]
- Harel-Meir, M.; Sherer, Y.; Shoenfeld, Y. Tobacco smoking and autoimmune rheumatic diseases. Nat. Clin. Pract. Rheumatol. 2007, 3, 707–715. [Google Scholar] [CrossRef]
- Klareskog, L.; Padyukov, L.; Alfredsson, L. Smoking as a trigger for inflammatory rheumatic diseases. Curr. Opin. Rheumatol. 2007, 19, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, Y.; Tanaka, T. Interleukin 6 and rheumatoid arthritis. Biomed. Res. Int. 2014, 2014, 698313. [Google Scholar] [CrossRef] [PubMed]
- Arnson, Y.; Shoenfeld, Y.; Amital, H. Effects of tobacco smoke on immunity, inflammation and autoimmunity. J. Autoimmun. 2010, 34, J258–J265. [Google Scholar] [CrossRef] [PubMed]
- Glossop, J.R.; Dawes, P.T.; Mattey, D.L. Association between cigarette smoking and release of tumour necrosis factor alpha and its soluble receptors by peripheral blood mononuclear cells in patients with rheumatoid arthritis. Rheumatology 2006, 45, 1223–1229. [Google Scholar] [CrossRef]
- Reyes-Pérez, I.V.; Sánchez-Hernández, P.E.; Muñoz-Valle, J.F.; Martínez-Bonilla, G.E.; García-Iglesias, T.; González-Díaz, V.; García-Arellano, S.; Cerpa-Cruz, S.; Polanco-Cruz, J.; Ramírez-Dueñas, M.G. Cytokines (IL-15, IL-21, and IFN-γ) in rheumatoid arthritis: Association with positivity to autoantibodies (RF, anti-CCP, anti-MCV, and anti-PADI4) and clinical activity. Clin. Rheumatol. 2019, 38, 3061–3071. [Google Scholar] [CrossRef]
- Smolen, J.S.; Landewé, R.B.M.; Bijlsma, J.W.J.; Burmester, G.R.; Dougados, M.; Kerschbaumer, A.; McInnes, I.B.; Sepriano, A.; van Vollenhoven, R.F.; de Wit, M.; et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2019 Update. Ann. Rheum. Dis. 2020. [Google Scholar] [CrossRef]
- Vanoni, F.; Minoia, F.; Malattia, C. Biologics in juvenile idiopathic arthritis: A narrative review. Eur. J. Pediatr. 2017, 176, 1147–1153. [Google Scholar] [CrossRef]
- Tollerud, D.J.; Weiss, S.T.; Leung, D.Y. Elevated soluble interleukin-2 receptors in young healthy cigarette smokers: Lack of association with atopy or airways hyperresponsiveness. Int. Arch. Allergy Immunol. 1992, 97, 25–30. [Google Scholar] [CrossRef]
- Tollerud, D.J.; Kurman, C.C.; Nelson, D.L.; Brown, L.M.; Maloney, E.M.; Blattner, W.A. Racial variation in serum-soluble interleukin-2 receptor levels: A population-based study of healthy smokers and nonsmokers. Clin. Immunol. Immunopathol. 1994, 70, 274–279. [Google Scholar] [CrossRef]
- Kuuliala, A.; Nissinen, R.; Kautiainen, H.; Repo, H.; Leirisalo-Repo, M. Low circulating soluble interleukin 2 receptor level predicts rapid response in patients with refractory rheumatoid arthritis treated with infliximab. Ann. Rheum. Dis. 2006, 65, 26–29. [Google Scholar] [CrossRef]
- Davis, J.M.; Crowson, C.S.; Knutson, K.L.; Achenbach, S.J.; Strausbauch, M.A.; Therneau, T.M.; Matteson, E.L.; Gabriel, S.E.; Wettstein, P.J. Longitudinal relationships between rheumatoid factor and cytokine expression by immunostimulated peripheral blood lymphocytes from patients with rheumatoid arthritis: New insights into B-cell activation. Clin. Immunol. 2020, 108342. [Google Scholar] [CrossRef] [PubMed]
- Bidkar, M.; Vassallo, R.; Luckey, D.; Smart, M.; Mouapi, K.; Taneja, V. Cigarette Smoke Induces Immune Responses to Vimentin in both, Arthritis-Susceptible and -Resistant Humanized Mice. PLoS ONE 2016, 11, e0162341. [Google Scholar] [CrossRef] [PubMed]
- Nadigel, J.; Préfontaine, D.; Baglole, C.J.; Maltais, F.; Bourbeau, J.; Eidelman, D.H.; Hamid, Q. Cigarette smoke increases TLR4 and TLR9 expression and induces cytokine production from CD8(+) T cells in chronic obstructive pulmonary disease. Respir. Res. 2011, 12, 149. [Google Scholar] [CrossRef] [PubMed]
- Saetta, M.; Di Stefano, A.; Turato, G.; Facchini, F.M.; Corbino, L.; Mapp, C.E.; Maestrelli, P.; Ciaccia, A.; Fabbri, L.M. CD8+ T-lymphocytes in peripheral airways of smokers with chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 1998, 157, 822–826. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.Q.; Liu, X.S.; Wang, J.M.; Xu, Y.J. CD8(+) Tc-lymphocytes immunodeviation in peripheral blood and airway from patients of chronic obstructive pulmonary disease and changes after short-term smoking cessation. Chin. Med. J. 2013, 126, 3608–3615. [Google Scholar]
- Chen, G.; Zhou, M.; Chen, L.; Meng, Z.J.; Xiong, X.Z.; Liu, H.J.; Xin, J.B.; Zhang, J.C. Cigarette Smoke Disturbs the Survival of CD8+ Tc/Tregs Partially through Muscarinic Receptors-Dependent Mechanisms in Chronic Obstructive Pulmonary Disease. PLoS ONE 2016, 11, e0147232. [Google Scholar] [CrossRef]
- Koch, A.; Gaczkowski, M.; Sturton, G.; Staib, P.; Schinköthe, T.; Klein, E.; Rubbert, A.; Bacon, K.; Wassermann, K.; Erdmann, E. Modification of surface antigens in blood CD8+ T-lymphocytes in COPD: Effects of smoking. Eur. Respir. J. 2007, 29, 42–50. [Google Scholar] [CrossRef]
- Wasén, C.; Erlandsson, M.C.; Bossios, A.; Ekerljung, L.; Malmhäll, C.; Töyrä Silfverswärd, S.; Pullerits, R.; Lundbäck, B.; Bokarewa, M.I. Smoking Is Associated with Low Levels of Soluble PD-L1 in Rheumatoid Arthritis. Front. Immunol. 2018, 9, 1677. [Google Scholar] [CrossRef]
- Wasén, C.; Turkkila, M.; Bossios, A.; Erlandsson, M.; Andersson, K.M.; Ekerljung, L.; Malmhäll, C.; Brisslert, M.; Töyrä Silfverswärd, S.; Lundbäck, B.; et al. Smoking activates cytotoxic CD8. J. Autoimmun. 2017, 78, 101–110. [Google Scholar] [CrossRef]
- Wang, J.; Urbanowicz, R.A.; Tighe, P.J.; Todd, I.; Corne, J.M.; Fairclough, L.C. Differential activation of killer cells in the circulation and the lung: A study of current smoking status and chronic obstructive pulmonary disease (COPD). PLoS ONE 2013, 8, e58556. [Google Scholar] [CrossRef]
- Stolberg, V.R.; Martin, B.; Mancuso, P.; Olszewski, M.A.; Freeman, C.M.; Curtis, J.L.; Chensue, S.W. Role of CC chemokine receptor 4 in natural killer cell activation during acute cigarette smoke exposure. Am. J. Pathol. 2014, 184, 454–463. [Google Scholar] [CrossRef] [PubMed]
- Tollerud, D.J.; Clark, J.W.; Brown, L.M.; Neuland, C.Y.; Mann, D.L.; Pankiw-Trost, L.K.; Blattner, W.A.; Hoover, R.N. Association of cigarette smoking with decreased numbers of circulating natural killer cells. Am. Rev. Respir. Dis. 1989, 139, 194–198. [Google Scholar] [CrossRef] [PubMed]
- Moszczyński, P.; Rutowski, J.; Słowiński, S. The effect of cigarettes smoking on the blood counts of T and NK cells in subjects with occupational exposure to organic solvents. Cent. Eur. J. Public Health 1996, 4, 164–168. [Google Scholar] [PubMed]
- Mian, M.F.; Lauzon, N.M.; Stämpfli, M.R.; Mossman, K.L.; Ashkar, A.A. Impairment of human NK cell cytotoxic activity and cytokine release by cigarette smoke. J. Leukoc. Biol. 2008, 83, 774–784. [Google Scholar] [CrossRef]
- Arimilli, S.; Damratoski, B.E.; Prasad, G.L. Combustible and non-combustible tobacco product preparations differentially regulate human peripheral blood mononuclear cell functions. Toxicol. In Vitro 2013, 27, 1992–2004. [Google Scholar] [CrossRef]
- Mian, M.F.; Pek, E.A.; Mossman, K.L.; Stämpfli, M.R.; Ashkar, A.A. Exposure to cigarette smoke suppresses IL-15 generation and its regulatory NK cell functions in poly I:C-augmented human PBMCs. Mol. Immunol. 2009, 46, 3108–3116. [Google Scholar] [CrossRef]
- de Lima, C.A.D.; Rushansky, E.; Adelino, J.E.; de Oliveira Souza, A.P.; d’Emery Alves Santos, P.; de Araújo Mariano, M.H.Q.; Crovella, S.; de Azevêdo Silva, J.; Sandrin-Garcia, P. Are key cytokines genetic and serum levels variations related to rheumatoid arthritis clinical severity? Gene 2020, 722, 144098. [Google Scholar] [CrossRef]
- Ospelt, C.; Bang, H.; Feist, E.; Camici, G.; Keller, S.; Detert, J.; Krämer, A.; Gay, S.; Ghannam, K.; Burmester, G.R. Carbamylation of vimentin is inducible by smoking and represents an independent autoantigen in rheumatoid arthritis. Ann. Rheum. Dis. 2017, 76, 1176–1183. [Google Scholar] [CrossRef]
- Klareskog, L.; Stolt, P.; Lundberg, K.; Källberg, H.; Bengtsson, C.; Grunewald, J.; Rönnelid, J.; Harris, H.E.; Ulfgren, A.K.; Rantapää-Dahlqvist, S.; et al. A new model for an etiology of rheumatoid arthritis: Smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006, 54, 38–46. [Google Scholar] [CrossRef]
- Too, C.L.; Yahya, A.; Murad, S.; Dhaliwal, J.S.; Larsson, P.T.; Muhamad, N.A.; Abdullah, N.A.; Mustafa, A.N.; Klareskog, L.; Alfredsson, L.; et al. Smoking interacts with HLA-DRB1 shared epitope in the development of anti-citrullinated protein antibody-positive rheumatoid arthritis: Results from the Malaysian Epidemiological Investigation of Rheumatoid Arthritis (MyEIRA). Arthritis Res. Ther. 2012, 14, R89. [Google Scholar] [CrossRef]
- Lee, H.S.; Irigoyen, P.; Kern, M.; Lee, A.; Batliwalla, F.; Khalili, H.; Wolfe, F.; Lum, R.F.; Massarotti, E.; Weisman, M.; et al. Interaction between smoking, the shared epitope, and anti-cyclic citrullinated peptide: A mixed picture in three large North American rheumatoid arthritis cohorts. Arthritis Rheum. 2007, 56, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Bang, S.Y.; Lee, K.H.; Cho, S.K.; Lee, H.S.; Lee, K.W.; Bae, S.C. Smoking increases rheumatoid arthritis susceptibility in individuals carrying the HLA-DRB1 shared epitope, regardless of rheumatoid factor or anti-cyclic citrullinated peptide antibody status. Arthritis Rheum. 2010, 62, 369–377. [Google Scholar] [CrossRef] [PubMed]
- Murphy, D.; Mattey, D.; Hutchinson, D. Anti-citrullinated protein antibody positive rheumatoid arthritis is primarily determined by rheumatoid factor titre and the shared epitope rather than smoking per se. PLoS ONE 2017, 12, e0180655. [Google Scholar] [CrossRef] [PubMed]
- van der Helm-van Mil, A.H.; Verpoort, K.N.; le Cessie, S.; Huizinga, T.W.; de Vries, R.R.; Toes, R.E. The HLA-DRB1 shared epitope alleles differ in the interaction with smoking and predisposition to antibodies to cyclic citrullinated peptide. Arthritis Rheum. 2007, 56, 425–432. [Google Scholar] [CrossRef]
- Pedersen, M.; Jacobsen, S.; Garred, P.; Madsen, H.O.; Klarlund, M.; Svejgaard, A.; Pedersen, B.V.; Wohlfahrt, J.; Frisch, M. Strong combined gene-environment effects in anti-cyclic citrullinated peptide-positive rheumatoid arthritis: A nationwide case-control study in Denmark. Arthritis Rheum. 2007, 56, 1446–1453. [Google Scholar] [CrossRef]
- Padyukov, L.; Silva, C.; Stolt, P.; Alfredsson, L.; Klareskog, L. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004, 50, 3085–3092. [Google Scholar] [CrossRef]
- Mattey, D.L.; Dawes, P.T.; Clarke, S.; Fisher, J.; Brownfield, A.; Thomson, W.; Hajeer, A.H.; Ollier, W.E. Relationship among the HLA-DRB1 shared epitope, smoking, and rheumatoid factor production in rheumatoid arthritis. Arthritis Rheum. 2002, 47, 403–407. [Google Scholar] [CrossRef]
- Hedström, A.K.; Rönnelid, J.; Klareskog, L.; Alfredsson, L. Complex Relationships of Smoking, HLA-DRB1 Genes, and Serologic Profiles in Patients with Early Rheumatoid Arthritis: Update from a Swedish Population-Based Case-Control Study. Arthritis Rheumatol. 2019. [Google Scholar] [CrossRef]
- Lundström, E.; Källberg, H.; Alfredsson, L.; Klareskog, L.; Padyukov, L. Gene-environment interaction between the DRB1 shared epitope and smoking in the risk of anti-citrullinated protein antibody-positive rheumatoid arthritis: All alleles are important. Arthritis Rheum. 2009, 60, 1597–1603. [Google Scholar] [CrossRef]
- Bang, S.Y.; Lee, H.S.; Lee, K.W.; Bae, S.C. Interaction of HLA-DRB1*09:01 and *04:05 with smoking suggests distinctive mechanisms of rheumatoid arthritis susceptibility beyond the shared epitope. J. Rheumatol. 2013, 40, 1054–1062. [Google Scholar] [CrossRef]
- Westra, H.J.; Martínez-Bonet, M.; Onengut-Gumuscu, S.; Lee, A.; Luo, Y.; Teslovich, N.; Worthington, J.; Martin, J.; Huizinga, T.; Klareskog, L.; et al. Fine-mapping and functional studies highlight potential causal variants for rheumatoid arthritis and type 1 diabetes. Nat. Genet. 2018, 50, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Mahdi, H.; Fisher, B.A.; Källberg, H.; Plant, D.; Malmström, V.; Rönnelid, J.; Charles, P.; Ding, B.; Alfredsson, L.; Padyukov, L.; et al. Specific interaction between genotype, smoking and autoimmunity to citrullinated alpha-enolase in the etiology of rheumatoid arthritis. Nat. Genet. 2009, 41, 1319–1324. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, K.; Bengtsson, C.; Kharlamova, N.; Reed, E.; Jiang, X.; Kallberg, H.; Pollak-Dorocic, I.; Israelsson, L.; Kessel, C.; Padyukov, L.; et al. Genetic and environmental determinants for disease risk in subsets of rheumatoid arthritis defined by the anticitrullinated protein/peptide antibody fine specificity profile. Ann. Rheum. Dis. 2013, 72, 652–658. [Google Scholar] [CrossRef]
- Willemze, A.; van der Woude, D.; Ghidey, W.; Levarht, E.W.; Stoeken-Rijsbergen, G.; Verduyn, W.; de Vries, R.R.; Houwing-Duistermaat, J.J.; Huizinga, T.W.; Trouw, L.A.; et al. The interaction between HLA shared epitope alleles and smoking and its contribution to autoimmunity against several citrullinated antigens. Arthritis Rheum. 2011, 63, 1823–1832. [Google Scholar] [CrossRef]
- Fisher, B.A.; Bang, S.Y.; Chowdhury, M.; Lee, H.S.; Kim, J.H.; Charles, P.; Venables, P.; Bae, S.C. Smoking, the HLA-DRB1 shared epitope and ACPA fine-specificity in Koreans with rheumatoid arthritis: Evidence for more than one pathogenic pathway linking smoking to disease. Ann. Rheum. Dis. 2014, 73, 741–747. [Google Scholar] [CrossRef]
- Kochi, Y.; Thabet, M.M.; Suzuki, A.; Okada, Y.; Daha, N.A.; Toes, R.E.; Huizinga, T.W.; Myouzen, K.; Kubo, M.; Yamada, R.; et al. PADI4 polymorphism predisposes male smokers to rheumatoid arthritis. Ann. Rheum. Dis. 2011, 70, 512–515. [Google Scholar] [CrossRef]
- Zeilinger, S.; Kühnel, B.; Klopp, N.; Baurecht, H.; Kleinschmidt, A.; Gieger, C.; Weidinger, S.; Lattka, E.; Adamski, J.; Peters, A.; et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS ONE 2013, 8, e63812. [Google Scholar] [CrossRef]
- Meng, W.; Zhu, Z.; Jiang, X.; Too, C.L.; Uebe, S.; Jagodic, M.; Kockum, I.; Murad, S.; Ferrucci, L.; Alfredsson, L.; et al. DNA methylation mediates genotype and smoking interaction in the development of anti-citrullinated peptide antibody-positive rheumatoid arthritis. Arthritis Res. Ther. 2017, 19, 71. [Google Scholar] [CrossRef]
- Ballestar, E. Epigenetic alterations in autoimmune rheumatic diseases. Nat. Rev. Rheumatol. 2011, 7, 263–271. [Google Scholar] [CrossRef]
- Viatte, S.; Plant, D.; Raychaudhuri, S. Genetics and epigenetics of rheumatoid arthritis. Nat. Rev. Rheumatol. 2013, 9, 141–153. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef]
- Eriksson, K.; Nise, L.; Alfredsson, L.; Catrina, A.I.; Askling, J.; Lundberg, K.; Klareskog, L.; Yucel-Lindberg, T. Seropositivity combined with smoking is associated with increased prevalence of periodontitis in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2018, 77, 1236–1238. [Google Scholar] [CrossRef]
- Albandar, J.M.; Streckfus, C.F.; Adesanya, M.R.; Winn, D.M. Cigar, pipe, and cigarette smoking as risk factors for periodontal disease and tooth loss. J. Periodontol. 2000, 71, 1874–1881. [Google Scholar] [CrossRef]
- Potempa, J.; Mydel, P.; Koziel, J. The case for periodontitis in the pathogenesis of rheumatoid arthritis. Nat. Rev. Rheumatol. 2017, 13, 606–620. [Google Scholar] [CrossRef]
- Janssen, K.M.; Vissink, A.; de Smit, M.J.; Westra, J.; Brouwer, E. Lessons to be learned from periodontitis. Curr. Opin. Rheumatol. 2013, 25, 241–247. [Google Scholar] [CrossRef]
- Wegner, N.; Wait, R.; Sroka, A.; Eick, S.; Nguyen, K.A.; Lundberg, K.; Kinloch, A.; Culshaw, S.; Potempa, J.; Venables, P.J. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: Implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010, 62, 2662–2672. [Google Scholar] [CrossRef]
- Lappin, D.F.; Apatzidou, D.; Quirke, A.M.; Oliver-Bell, J.; Butcher, J.P.; Kinane, D.F.; Riggio, M.P.; Venables, P.; McInnes, I.B.; Culshaw, S. Influence of periodontal disease, Porphyromonas gingivalis and cigarette smoking on systemic anti-citrullinated peptide antibody titres. J. Clin. Periodontol. 2013, 40, 907–915. [Google Scholar] [CrossRef]
- Konig, M.F.; Abusleme, L.; Reinholdt, J.; Palmer, R.J.; Teles, R.P.; Sampson, K.; Rosen, A.; Nigrovic, P.A.; Sokolove, J.; Giles, J.T.; et al. Aggregatibacter actinomycetemcomitans-induced hypercitrullination links periodontal infection to autoimmunity in rheumatoid arthritis. Sci. Transl. Med. 2016, 8, 369ra176. [Google Scholar] [CrossRef]
- Terao, C.; Asai, K.; Hashimoto, M.; Yamazaki, T.; Ohmura, K.; Yamaguchi, A.; Takahashi, K.; Takei, N.; Ishii, T.; Kawaguchi, T.; et al. Significant association of periodontal disease with anti-citrullinated peptide antibody in a Japanese healthy population—The Nagahama study. J. Autoimmun. 2015, 59, 85–90. [Google Scholar] [CrossRef]
- Bello-Gualtero, J.M.; Lafaurie, G.I.; Hoyos, L.X.; Castillo, D.M.; De-Avila, J.; Munevar, J.C.; Unriza, S.; Londoño, J.; Valle-Oñate, R.; Romero-Sánchez, C. Periodontal Disease in Individuals with a Genetic Risk of Developing Arthritis and Early Rheumatoid Arthritis: A Cross-Sectional Study. J. Periodontol. 2016, 87, 346–356. [Google Scholar] [CrossRef]
- Golub, L.M.; Payne, J.B.; Reinhardt, R.A.; Nieman, G. Can systemic diseases co-induce (not just exacerbate) periodontitis? A hypothetical “two-hit” model. J. Dent. Res. 2006, 85, 102–105. [Google Scholar] [CrossRef] [PubMed]
- Marotte, H.; Farge, P.; Gaudin, P.; Alexandre, C.; Mougin, B.; Miossec, P. The association between periodontal disease and joint destruction in rheumatoid arthritis extends the link between the HLA-DR shared epitope and severity of bone destruction. Ann. Rheum. Dis. 2006, 65, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Gargiulo, A.V.; Robinson, J.; Toto, P.D.; Gargiulo, A.W. Identification of rheumatoid factor in periodontal disease. J. Periodontol. 1982, 53, 568–577. [Google Scholar] [CrossRef] [PubMed]
- Demoruelle, M.K.; Weisman, M.H.; Simonian, P.L.; Lynch, D.A.; Sachs, P.B.; Pedraza, I.F.; Harrington, A.R.; Kolfenbach, J.R.; Striebich, C.C.; Pham, Q.N.; et al. Brief report: Airways abnormalities and rheumatoid arthritis-related autoantibodies in subjects without arthritis: Early injury or initiating site of autoimmunity? Arthritis Rheum. 2012, 64, 1756–1761. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Sun, H.; Wang, X.; Koike, T.; Mishima, H.; Ikeda, K.; Watanabe, T.; Ochiai, N.; Fan, J. Association of increased expression of macrophage elastase (matrix metalloproteinase 12) with rheumatoid arthritis. Arthritis Rheum. 2004, 50, 3112–3117. [Google Scholar] [CrossRef] [PubMed]
- Bracke, K.; Cataldo, D.; Maes, T.; Gueders, M.; Noël, A.; Foidart, J.M.; Brusselle, G.; Pauwels, R.A. Matrix metalloproteinase-12 and cathepsin D expression in pulmonary macrophages and dendritic cells of cigarette smoke-exposed mice. Int. Arch. Allergy Immunol. 2005, 138, 169–179. [Google Scholar] [CrossRef] [PubMed]
- Raitio, A.; Tuomas, H.; Kokkonen, N.; Salo, T.; Sorsa, T.; Hanemaaijer, R.; Oikarinen, A. Levels of matrix metalloproteinase-2, -9 and -8 in the skin, serum and saliva of smokers and non-smokers. Arch. Dermatol. Res. 2005, 297, 242–248. [Google Scholar] [CrossRef]
- Xue, M.; McKelvey, K.; Shen, K.; Minhas, N.; March, L.; Park, S.Y.; Jackson, C.J. Endogenous MMP-9 and not MMP-2 promotes rheumatoid synovial fibroblast survival, inflammation and cartilage degradation. Rheumatology 2014, 53, 2270–2279. [Google Scholar] [CrossRef]
- Burke, H.; Leonardi-Bee, J.; Hashim, A.; Pine-Abata, H.; Chen, Y.; Cook, D.G.; Britton, J.R.; McKeever, T.M. Prenatal and passive smoke exposure and incidence of asthma and wheeze: Systematic review and meta-analysis. Pediatrics 2012, 129, 735–744. [Google Scholar] [CrossRef]
- Jiang, X.; Alfredsson, L.; Klareskog, L.; Bengtsson, C. Smokeless tobacco (moist snuff) use and the risk of developing rheumatoid arthritis: Results from a case-control study. Arthritis Care Res. 2014, 66, 1582–1586. [Google Scholar] [CrossRef]
- Zhou, Y.; Zuo, X.; Li, Y.; Wang, Y.; Zhao, H.; Xiao, X. Nicotine inhibits tumor necrosis factor-α induced IL-6 and IL-8 secretion in fibroblast-like synoviocytes from patients with rheumatoid arthritis. Rheumatol. Int. 2012, 32, 97–104. [Google Scholar] [CrossRef]
- Wu, S.; Luo, H.; Xiao, X.; Zhang, H.; Li, T.; Zuo, X. Attenuation of collagen induced arthritis via suppression on Th17 response by activating cholinergic anti-inflammatory pathway with nicotine. Eur. J. Pharmacol. 2014, 735, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Yang, H.; Ulloa, L.; Al-Abed, Y.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef] [PubMed]
- van Maanen, M.A.; Lebre, M.C.; van der Poll, T.; LaRosa, G.J.; Elbaum, D.; Vervoordeldonk, M.J.; Tak, P.P. Stimulation of nicotinic acetylcholine receptors attenuates collagen-induced arthritis in mice. Arthritis Rheum. 2009, 60, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yang, Y.H.; Rajaiah, R.; Moudgil, K.D. Nicotine-induced differential modulation of autoimmune arthritis in the Lewis rat involves changes in interleukin-17 and anti-cyclic citrullinated peptide antibodies. Arthritis Rheum. 2011, 63, 981–991. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Plada, M.; Tzatzarakis, M.; Marcos, A.; Warnberg, J.; Gomez-Martinez, S.; Breidenassel, C.; Gonzalez-Gross, M.; Tsatsakis, A.M.; Saris, W.H.; et al. Passive smoking alters circulating naïve/memory lymphocyte T-cell subpopulations in children. Pediatr. Allergy Immunol. 2010, 21, 1171–1178. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).