Curcumin Derivative GT863 Inhibits Amyloid-Beta Production via Inhibition of Protein N-Glycosylation

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Cell Lines and Cell Culture
2.3. Cell Treatment
2.4. Assessment of Cell Viability
2.5. ELISA for Aβ
2.6. Preparation of Cell Samples and Immunoblotting
2.7. In Vitro Secretase Assay
2.8. Statistical Analysis
3. Results
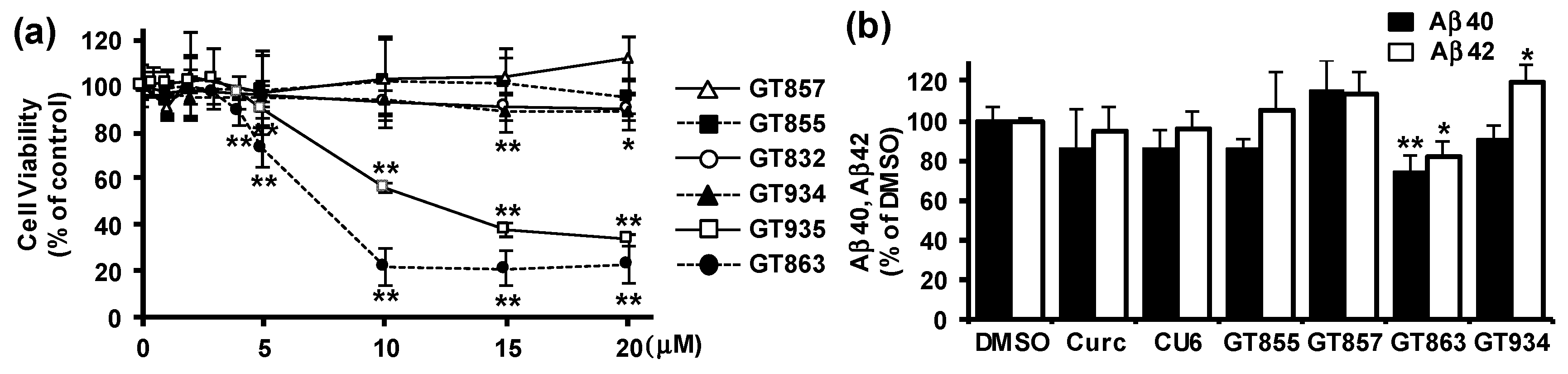
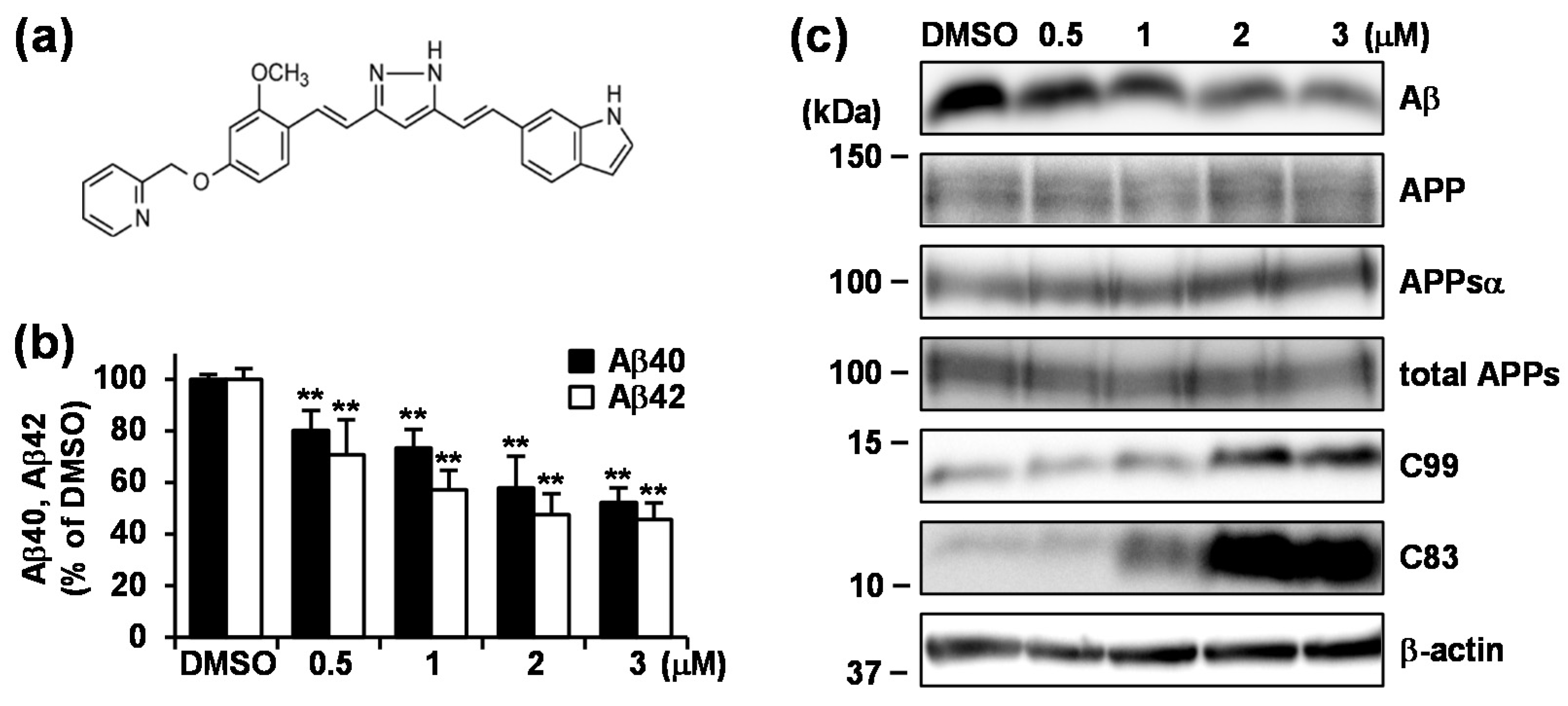
3.1. GT863 was Identified as a Potent Inhibitor of Aβ40 and Aβ42 Production in CHO-APP Cells.
3.2. Treatment with GT863 for 48 h Reduced Aβ40 and Aβ42 Production and Increased C83 and C99 Levels in CHO-APP Cells
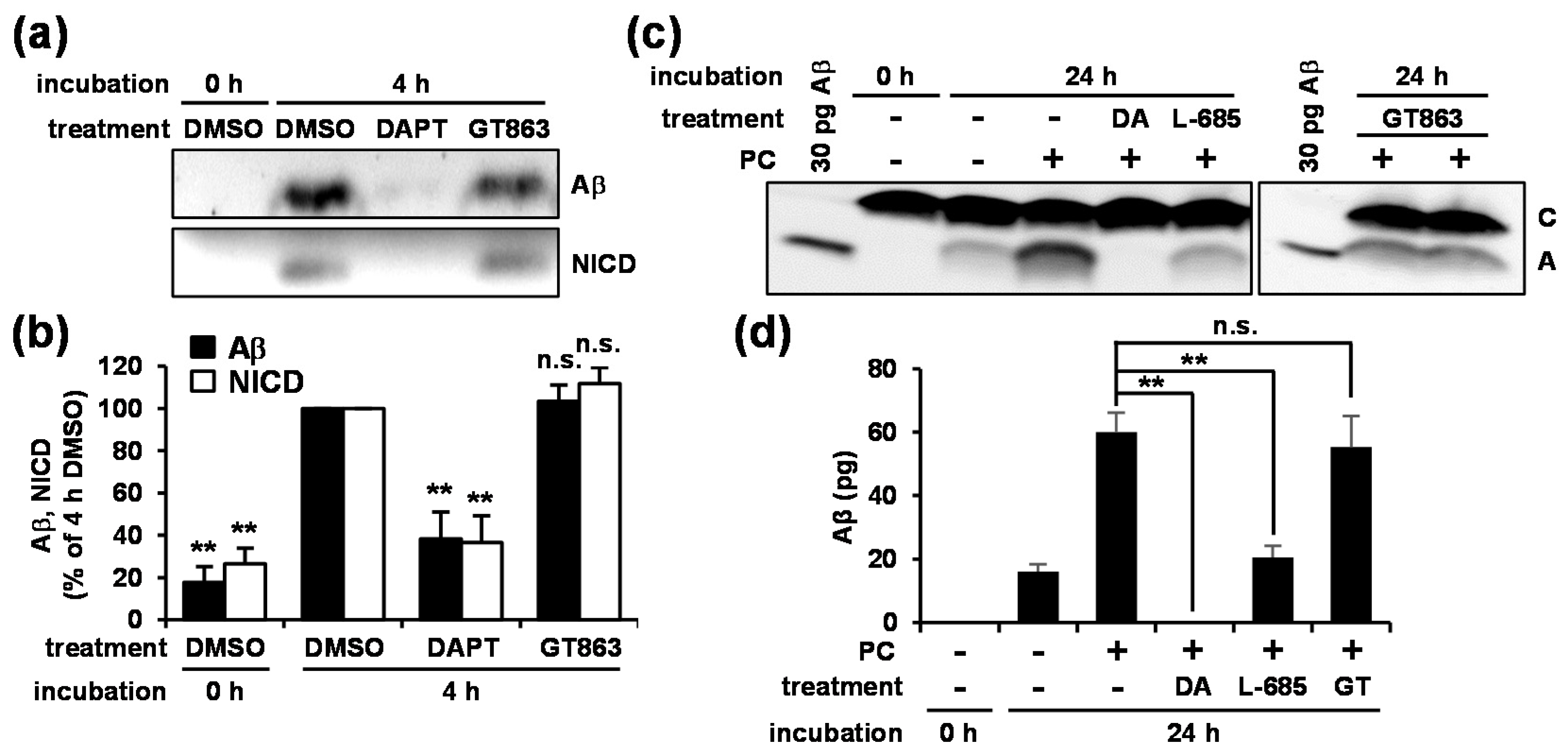
3.3. GT863 Did Not Inhibit γ-Secretase Activity in Vitro
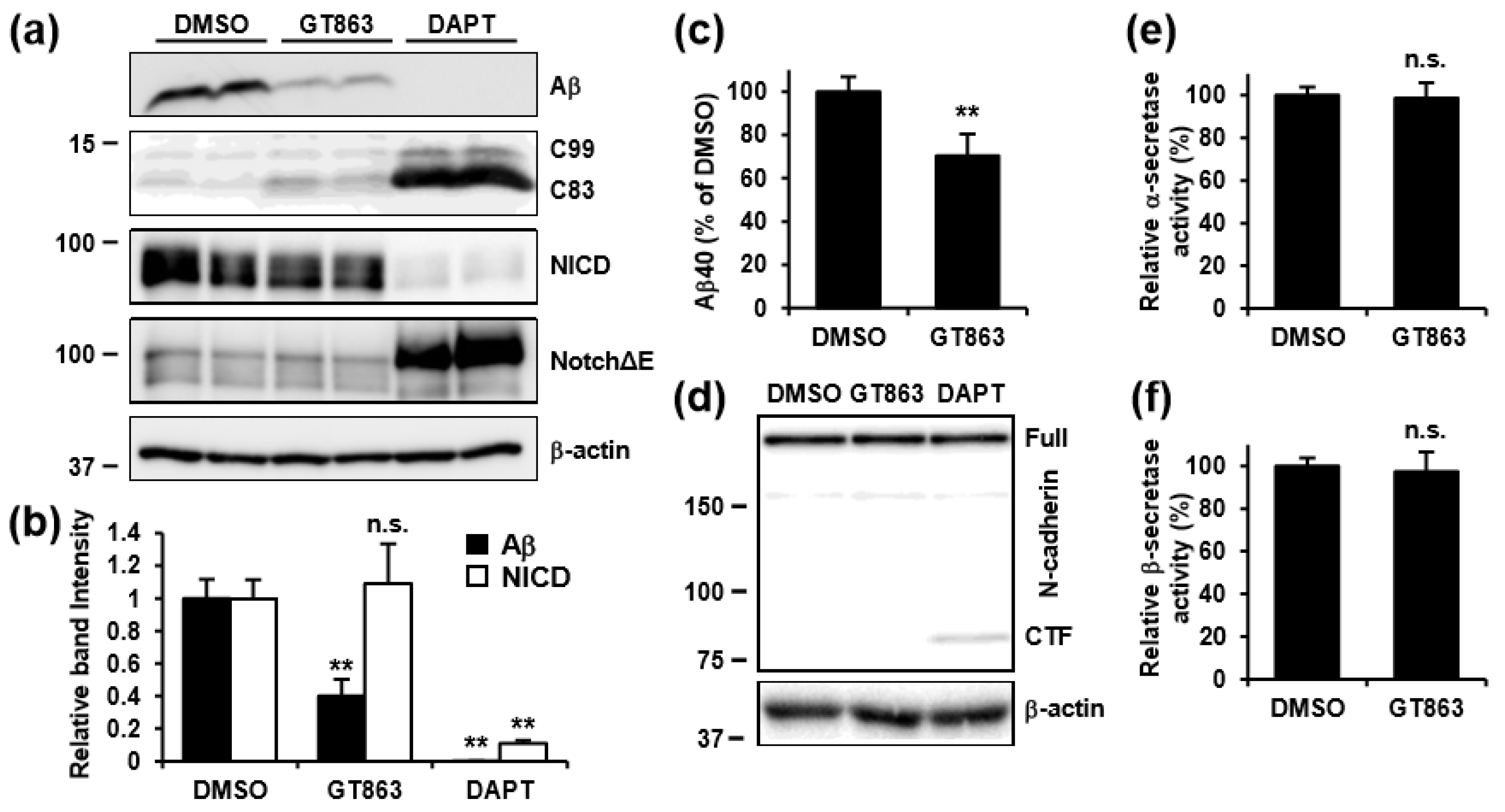
3.4. GT863 Inhibited γ-Cleavage in γ-Ssecretase Substrate-Selective Fashion in CHO-APP/NotchΔE and SH-SY5Y Cells
3.5. GT863 Suppressed N-Glycosylation of Proteins Including Nicastrin
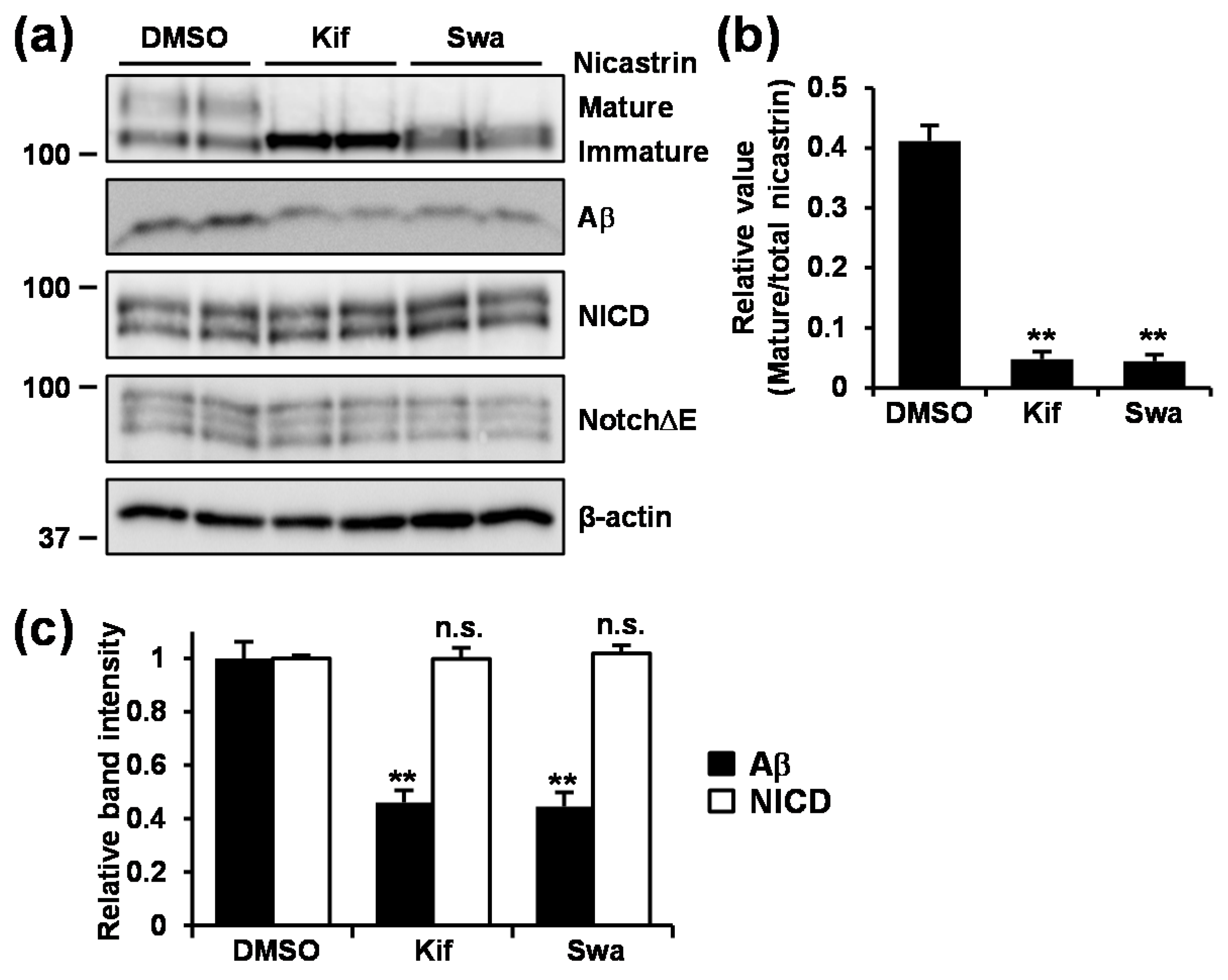
3.6. Inhibition of Mannosidase Suppressed Aβ Production Without Affecting NotchΔE Cleavage.
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef] [PubMed]
- Thinakaran, G.; Koo, E.H. Amyloid precursor protein trafficking, processing, and function. J. Biol. Chem. 2008, 283, 29615–29619. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Lessons from a failed γ-secretase Alzheimer trial. Cell 2014, 159, 721–726. [Google Scholar] [CrossRef]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Kim, J.; Lee, H.J.; Lee, K.W. Naturally occurring phytochemicals for the prevention of Alzheimer’s disease. J. Neurochem. 2010, 112, 1415–1430. [Google Scholar] [CrossRef]
- Reddy, P.H.; Manczak, M.; Yin, X.; Grady, M.C.; Mitchell, A.; Tonk, S.; Kuruva, C.S.; Bhatti, J.S.; Kandimalla, R.; Vijayan, M.; et al. Protective Effects of Indian Spice Curcumin Against Amyloid-β in Alzheimer’s Disease. J. Alzheimers Dis. 2018, 61, 843–866. [Google Scholar] [CrossRef]
- Zhang, C.; Browne, A.; Child, D.; Tanzi, R.E. Curcumin decreases amyloid-β peptide levels by attenuating the maturation of amyloid-β precursor protein. J. Biol. Chem. 2010, 285, 28472–28480. [Google Scholar] [CrossRef]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem. 2005, 280, 5892–5901. [Google Scholar] [CrossRef]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem. 2007, 102, 1095–1104. [Google Scholar] [CrossRef] [PubMed]
- Baum, L.; Lam, C.W.; Cheung, S.K.; Kwok, T.; Lui, V.; Tsoh, J.; Lam, L.; Leung, V.; Hui, E.; Ng, C.; et al. Six-month randomized, placebo-controlled, double-blind, pilot clinical trial of curcumin in patients with Alzheimer disease. J. Clin. Psychopharmacol. 2008, 28, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Hamaguchi, T.; Ono, K.; Yamada, M. Curcumin and Alzheimer’s disease. CNS Neurosci. Ther. 2010, 16, 285–297. [Google Scholar] [CrossRef]
- Belkacemi, A.; Doggui, S.; Dao, L.; Ramassamy, C. Challenges associated with curcumin therapy in Alzheimer disease. Expert Rev. Mol. Med. 2011, 13, e34. [Google Scholar] [CrossRef] [PubMed]
- Kotani, R.; Urano, Y.; Sugimoto, H.; Noguchi, N. Decrease of Amyloid-β Levels by Curcumin Derivative via Modulation of Amyloid-β Protein Precursor Trafficking. J. Alzheimers Dis. 2017, 56, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Valera, E.; Dargusch, R.; Maher, P.A.; Schubert, D. Modulation of 5-lipoxygenase in proteotoxicity and Alzheimer’s disease. J. Neurosci. 2013, 33, 10512–10525. [Google Scholar] [CrossRef]
- Okuda, M.; Hijikuro, I.; Fujita, Y.; Wu, X.; Nakayama, S.; Sakata, Y.; Noguchi, Y.; Ogo, M.; Akasofu, S.; Ito, Y.; et al. PE859, a novel tau aggregation inhibitor, reduces aggregated tau and prevents onset and progression of neural dysfunction in vivo. PLoS ONE 2015, 10, e0117511. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Hijikuro, I.; Fujita, Y.; Teruya, T.; Kawakami, H.; Takahashi, T.; Sugimoto, H. Design and synthesis of curcumin derivatives as tau and amyloid β dual aggregation inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 5024–5028. [Google Scholar] [CrossRef] [PubMed]
- Okuda, M.; Fujita, Y.; Hijikuro, I.; Wada, M.; Uemura, T.; Kobayashi, Y.; Waku, T.; Tanaka, N.; Nishimoto, T.; Izumi, Y.; et al. A Novel Curcumin Derivative, Inhibits Amyloid-β and Tau Aggregation, and Ameliorates Cognitive Dysfunction in Senescence-Accelerated Mouse Prone 8. J. Alzheimers Dis. 2017, 59, 313–328. [Google Scholar] [CrossRef]
- Yanagisawa, D.; Ibrahim, N.F.; Taguchi, H.; Morikawa, S.; Hirao, K.; Shirai, N.; Sogabe, T.; Tooyama, I. Curcumin derivative with the substitution at C-4 position, but not curcumin, is effective against amyloid pathology in APP/PS1 mice. Neurobiol. Aging 2015, 36, 201–210. [Google Scholar] [CrossRef]
- Serafini, M.M.; Catanzaro, M.; Rosini, M.; Racchi, M.; Lanni, C. Curcumin in Alzheimer’s disease: Can we think to new strategies and perspectives for this molecule? Pharmacol. Res. 2017, 124, 146–155. [Google Scholar] [CrossRef]
- Koo, E.H.; Squazzo, S.L. Evidence that production and release of amyloid beta-protein involves the endocytic pathway. J. Biol. Chem. 1994, 269, 17386–17389. [Google Scholar]
- Funamoto, S.; Sasaki, T.; Ishihara, S.; Nobuhara, M.; Nakano, M.; Watanabe-Takahashi, M.; Saito, T.; Kakuda, N.; Miyasaka, T.; Nishikawa, K.; et al. Substrate ectodomain is critical for substrate preference and inhibition of γ-secretase. Nat. Commun. 2013, 4, 2529. [Google Scholar] [CrossRef] [PubMed]
- Urano, Y.; Ho, V.D.K.; Araki, H.; Noguchi, N. 24(S)-Hydroxycholesterol induces ER dysfunction-mediated unconventional cell death. Cell Death Discov. 2019, 113. [Google Scholar] [CrossRef] [PubMed]
- Urano, Y.; Ochiai, S.; Noguchi, N. Suppression of amyloid-β production by 24S-hydroxycholesterol via inhibition of intracellular amyloid precursor protein trafficking. FASEB J. 2013, 27, 4305–4315. [Google Scholar] [CrossRef] [PubMed]
- Moniruzzaman, M.; shihara, S.; Nobuhara, M.; Higashide, H.; Funamoto, S. Glycosylation status of nicastrin influences catalytic activity and substrate preference of γ-secretase. Biochem. Biophys. Res. Commun. 2018, 502, 98–103. [Google Scholar] [CrossRef]
- Yagishita, S.; Futai, E.; Ishiura, S. In vitro reconstitution of gamma-secretase activity using yeast microsomes. Biochem. Biophys. Res. Commun. 2008, 377, 141–145. [Google Scholar] [CrossRef]
- Marambaud, P.; Wen, P.H.; Dutt, A.; Shioi, J.; Takashima, A.; Siman, R.; Robakis, N.K. A CBP binding transcriptional repressor produced by the PS1/epsilon-cleavage of N-cadherin is inhibited by PS1 FAD mutations. Cell 2003, 114, 635–645. [Google Scholar] [CrossRef]
- Shah, S.; Lee, S.F.; Tabuchi, K.; Hao, Y.H.; Yu, C.; LaPlant, Q.; Ball, H.; Dann, C.E., 3rd; Südhof, T.; Yu, G. Nicastrin functions as a gamma-secretase-substrate receptor. Cell 2005, 122, 435–447. [Google Scholar] [CrossRef]
- Zhang, X.; Hoey, R.J.; Lin, G.; Koide, A.; Leung, B.; Ahn, K.; Dolios, G.; Paduch, M.; Ikeuchi, T.; Wang, R.; et al. Iden tification of a tetratricopeptide repeat-like domain in the nicastrin subunit of γ-secretase using synthetic antibodies. Proc. Natl. Acad. Sci. USA 2012, 109, 8534–8539. [Google Scholar] [CrossRef]
- Yang, D.S.; Tandon, A.; Chen, F.; Yu, G.; Yu, H.; Arawaka, S.; Hasegawa, H.; Duthie, M.; Schmidt, S.D.; Ramabhadran, T.V.; et al. Mature glycosylation and trafficking of nicastrin modulate its binding to presenilins. J. Biol. Chem. 2002, 277, 28135–28142. [Google Scholar] [CrossRef]
- Schedin-Weiss, S.; Winblad, B.; Tjernberg, L.O. The role of protein glycosylation in Alzheimer disease. FEBS J. 2014, 281, 46–62. [Google Scholar] [CrossRef]
- Kizuka, Y.; Kitazume, S.; Taniguchi, N. N-glycan and Alzheimer’s disease. Biochim. Biophys. Acta. Gen. Subj. 2017, 1861, 2447–2454. [Google Scholar] [CrossRef]
- Bolduc, D.M.; Montagna, D.R.; Gu, Y.; Selkoe, D.J.; Wolfe, M.S. Nicastrin functions to sterically hinder γ-secretase-substrate interactions driven by substrate transmembrane domain. Proc. Natl. Acad. Sci. USA 2016, 113, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, M.S. Substrate recognition and processing by γ-secretase. Biochim. Biophys. Acta. Biomembr. 2019, 183016. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Complex N-linked glycosylated nicastrin associates with active gamma-secretase and undergoes tight cellular regulation. J. Biol. Chem. 2002, 277, 35113–35117. [Google Scholar] [CrossRef]
- Herreman, A.; Van Gassen, G.; Bentahir, M.; Nyabi, O.; Craessaerts, K.; Mueller, U.; Annaert, W.; De Strooper, B. gamma-Secretase activity requires the presenilin-dependent trafficking of nicastrin through the Golgi apparatus but not its complex glycosylation. J. Cell Sci. 2003, 116, 1127–1136. [Google Scholar] [CrossRef]
- Shirotani, K.; Edbauer, D.; Capell, A.; Schmitz, J.; Steiner, H.; Haass, C. Gamma-secretase activity is associated with a conformational change of nicastrin. J. Biol. Chem. 2003, 278, 16474–16477. [Google Scholar] [CrossRef]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by β-secretase cleavage within the secretory pathway. Nat. Med. 1995, 12, 1291–1296. [Google Scholar] [CrossRef]
- Thinakaran, G.; Teplow, D.B.; Siman, R.; Greenberg, B.; Sisodia, S.S. Metabolism of the “Swedish” amyloid precursor protein variant in neuro2a (N2a) cells. Evidence that cleavage at the “beta-secretase” site occurs in the golgi apparatus. J. Biol. Chem. 1996, 271, 9390–9397. [Google Scholar] [CrossRef]
- Yamakawa, H.; Yagishita, S.; Futai, E.; Ishiura, S. beta-Secretase inhibitor potency is decreased by aberrant beta-cleavage location of the “Swedish mutant” amyloid precursor protein. J. Biol. Chem. 2010, 285, 1634–1642. [Google Scholar] [CrossRef]
- Riddell, D.R.; Christie, G.; Hussain, I.; Dingwall, C. Compartmentalization of beta-secretase (Asp2) into low-buoyant density, noncaveolar lipid rafts. Curr. Biol. 2001, 11, 1288–1293. [Google Scholar] [CrossRef]
- Vetrivel, K.S.; Cheng, H.; Lin, W.; Sakurai, T.; Li, T.; Nukina, N.; Wong, P.C.; Xu, H.; Thinakaran, G. Association of gamma-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 2004, 279, 44945–44954. [Google Scholar] [CrossRef] [PubMed]
- Urano, Y.; Hayashi, I.; Isoo, N.; Reid, P.C.; Shibasaki, Y.; Noguchi, N.; Tomita, T.; Iwatsubo, T.; Hamakubo, T.; Kodama, T. Association of active gamma-secretase complex with lipid rafts. J. Lipid Res. 2005, 46, 904–912. [Google Scholar] [CrossRef] [PubMed]
- McFarlane, I.; Georgopoulou, N.; Coughlan, C.M.; Gillian, A.M.; Breen, K.C. The role of the protein glycosylation state in the control of cellular transport of the amyloid beta precursor protein. Neuroscience 1999, 90, 15–25. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Urano, Y.; Takahachi, M.; Higashiura, R.; Fujiwara, H.; Funamoto, S.; Imai, S.; Futai, E.; Okuda, M.; Sugimoto, H.; Noguchi, N. Curcumin Derivative GT863 Inhibits Amyloid-Beta Production via Inhibition of Protein N-Glycosylation. Cells 2020, 9, 349. https://doi.org/10.3390/cells9020349
Urano Y, Takahachi M, Higashiura R, Fujiwara H, Funamoto S, Imai S, Futai E, Okuda M, Sugimoto H, Noguchi N. Curcumin Derivative GT863 Inhibits Amyloid-Beta Production via Inhibition of Protein N-Glycosylation. Cells. 2020; 9(2):349. https://doi.org/10.3390/cells9020349
Chicago/Turabian StyleUrano, Yasuomi, Mina Takahachi, Ryo Higashiura, Hitomi Fujiwara, Satoru Funamoto, So Imai, Eugene Futai, Michiaki Okuda, Hachiro Sugimoto, and Noriko Noguchi. 2020. "Curcumin Derivative GT863 Inhibits Amyloid-Beta Production via Inhibition of Protein N-Glycosylation" Cells 9, no. 2: 349. https://doi.org/10.3390/cells9020349
APA StyleUrano, Y., Takahachi, M., Higashiura, R., Fujiwara, H., Funamoto, S., Imai, S., Futai, E., Okuda, M., Sugimoto, H., & Noguchi, N. (2020). Curcumin Derivative GT863 Inhibits Amyloid-Beta Production via Inhibition of Protein N-Glycosylation. Cells, 9(2), 349. https://doi.org/10.3390/cells9020349