Amyloid-β Precursor Protein APP Down-Regulation Alters Actin Cytoskeleton-Interacting Proteins in Endothelial Cells

 , , and
, , and 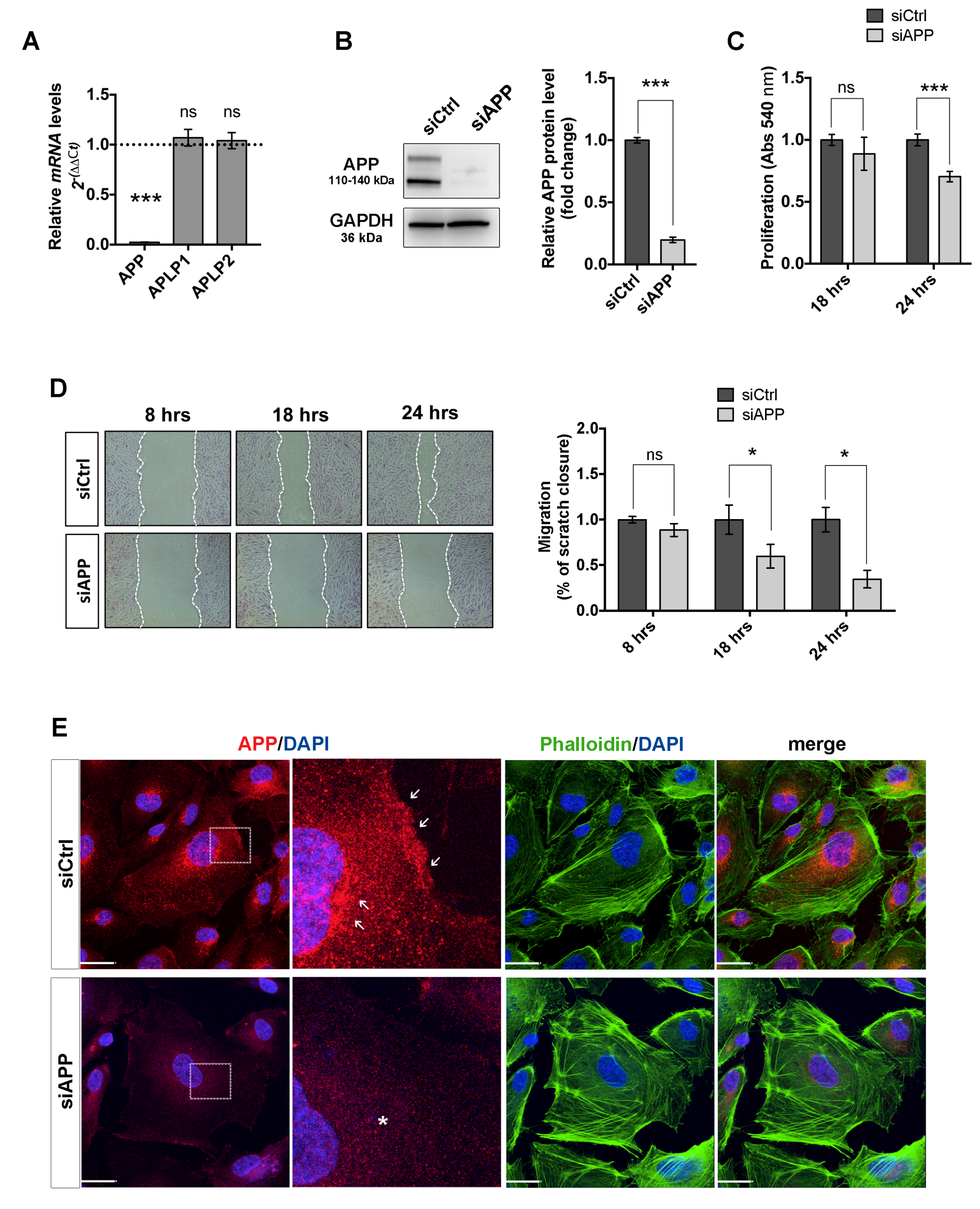{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Small Interfering RNA Transfection
2.3. Real-Time PCR
2.4. Western Blot
2.5. Immunoprecipitation (IP)
2.6. Immunofluorescence Microscopy
2.7. Cell Proliferation Assay
2.8. Wound Healing Scratch Assay
2.9. Cell Adhesion Assay
2.10. In Vitro Permeability Assay
2.11. Tube Formation Assay
2.12. LC-MS/MS Proteomic and Bioinformatic Analysis
2.13. Statistical Analysis
3. Results
3.1. Loss of APP Affects Endothelial Cells Proliferation, Migration and Cytoskeleton Organization In Vitro
3.2. Proteomic Analysis Confirms the Cytoskeleton Organization Defect and Reveals Alteration of Vascular Specific Pathways in APP-Silenced Cells
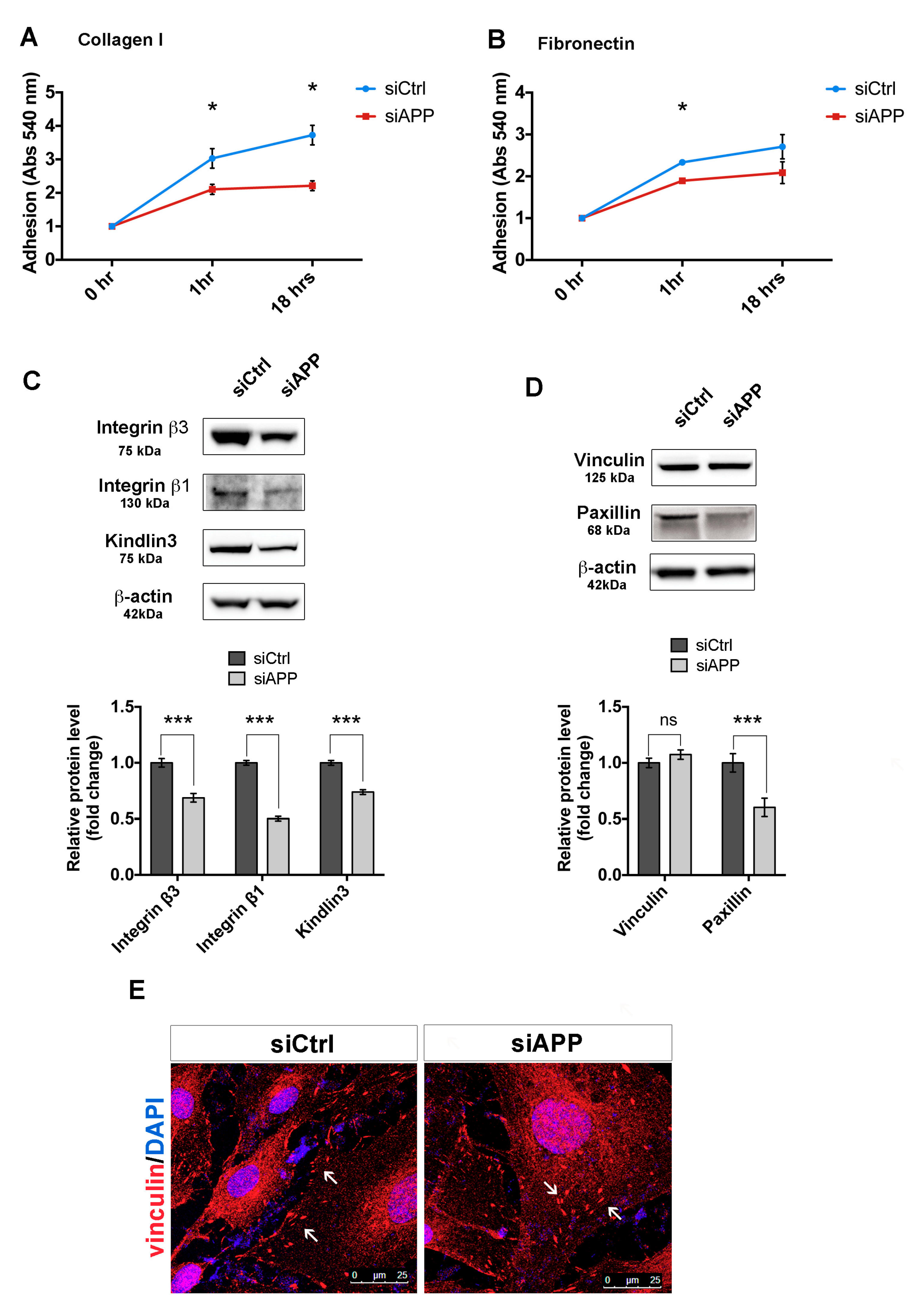
3.3. APP Silencing Affects Endothelial Cells Adhesion and Promotes a Reorganization of Focal Adhesions Complex
3.4. APP Controls Endothelial Barrier Function
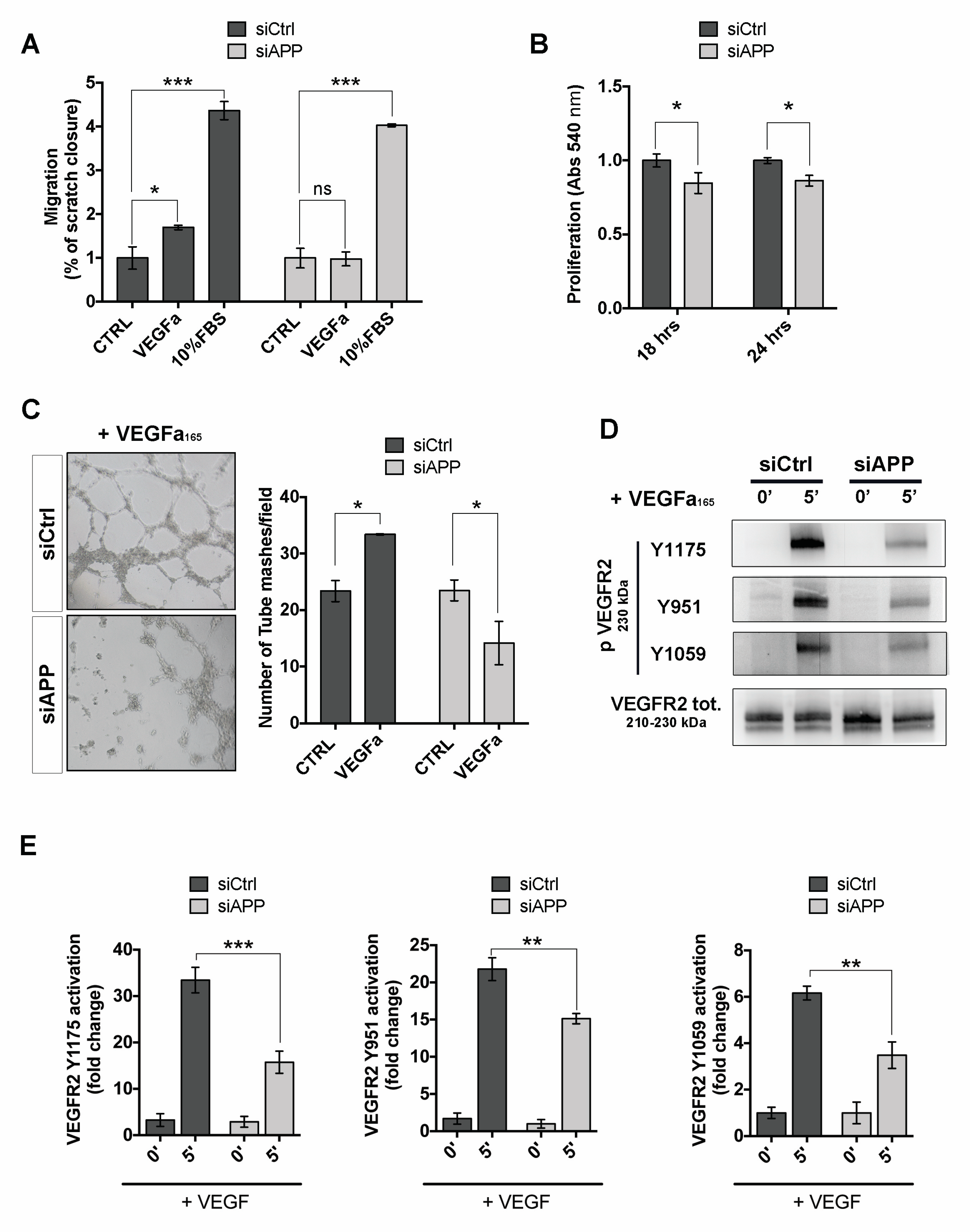
3.5. APP Modulates Endothelial Response to Pro-Angiogenic Stimuli
3.6. APP Modulates Src/FAK Signaling
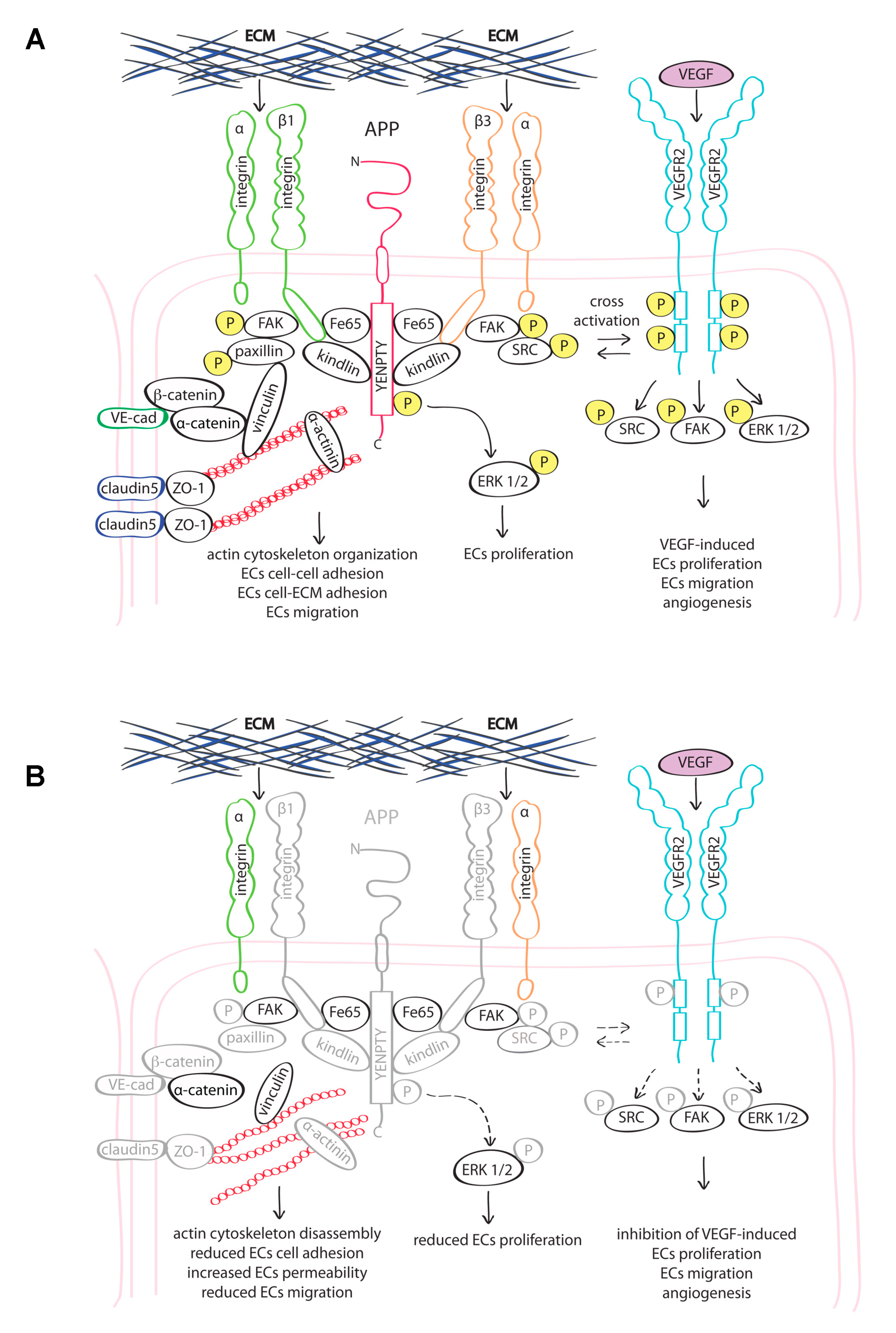
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the beta-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef]
- Corrigan, F.; Pham, C.L.; Vink, R.; Blumbergs, P.C.; Masters, C.L.; van den Heuvel, C.; Cappai, R. The neuroprotective domains of the amyloid precursor protein, in traumatic brain injury, are located in the two growth factor domains. Brain Res. 2011, 1378, 137–143. [Google Scholar] [CrossRef]
- Corrigan, F.; Thornton, E.; Roisman, L.C.; Leonard, A.V.; Vink, R.; Blumbergs, P.C.; van den Heuvel, C.; Cappai, R. The neuroprotective activity of the amyloid precursor protein against traumatic brain injury is mediated via the heparin binding site in residues 96–110. J. Neurochem. 2014, 128, 196–204. [Google Scholar] [CrossRef]
- Itoh, T.; Satou, T.; Nishida, S.; Tsubaki, M.; Hashimoto, S.; Ito, H. Expression of amyloid precursor protein after rat traumatic brain injury. Neurol. Res. 2009, 31, 103–109. [Google Scholar] [CrossRef]
- Nihashi, T.; Inao, S.; Kajita, Y.; Kawai, T.; Sugimoto, T.; Niwa, M.; Kabeya, R.; Hata, N.; Hayashi, S.; Yoshida, J. Expression and distribution of beta amyloid precursor protein and beta amyloid peptide in reactive astrocytes after transient middle cerebral artery occlusion. Acta Neurochir. 2001, 143, 287–295. [Google Scholar] [CrossRef]
- Sayer, R.; Robertson, D.; Balfour, D.J.; Breen, K.C.; Stewart, C.A. The effect of stress on the expression of the amyloid precursor protein in rat brain. Neurosci. Lett. 2008, 431, 197–200. [Google Scholar] [CrossRef]
- Ho, A.; Sudhof, T.C. Binding of F-spondin to amyloid-beta precursor protein: A candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc. Natl. Acad. Sci. USA 2004, 101, 2548–2553. [Google Scholar] [CrossRef]
- Hoe, H.S.; Lee, K.J.; Carney, R.S.; Lee, J.; Markova, A.; Lee, J.Y.; Howell, B.W.; Hyman, B.T.; Pak, D.T.; Bu, G.; et al. Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J. Neurosci. 2009, 29, 7459–7473. [Google Scholar] [CrossRef]
- Reinhard, C.; Borgers, M.; David, G.; De Strooper, B. Soluble amyloid-beta precursor protein binds its cell surface receptor in a cooperative fashion with glypican and syndecan proteoglycans. J. Cell Sci. 2013, 126, 4856–4861. [Google Scholar] [CrossRef]
- Sosa, L.J.; Bergman, J.; Estrada-Bernal, A.; Glorioso, T.J.; Kittelson, J.M.; Pfenninger, K.H. Amyloid precursor protein is an autonomous growth cone adhesion molecule engaged in contact guidance. PLoS ONE 2013, 8, e64521. [Google Scholar] [CrossRef]
- Chen, K.P.; Dou, F. Selective interaction of amyloid precursor protein with different isoforms of neural cell adhesion molecule. J. Mol. Neurosci. 2012, 46, 203–209. [Google Scholar] [CrossRef]
- Young-Pearse, T.L.; Bai, J.; Chang, R.; Zheng, J.B.; LoTurco, J.J.; Selkoe, D.J. A critical function for beta-amyloid precursor protein in neuronal migration revealed by in utero RNA interference. J. Neurosci. 2007, 27, 14459–14469. [Google Scholar] [CrossRef]
- Sosa, L.J.; Caceres, A.; Dupraz, S.; Oksdath, M.; Quiroga, S.; Lorenzo, A. The physiological role of the amyloid precursor protein as an adhesion molecule in the developing nervous system. J. Neurochem. 2017, 143, 11–29. [Google Scholar] [CrossRef]
- Ott, M.O.; Bullock, S.L. A gene trap insertion reveals that amyloid precursor protein expression is a very early event in murine embryogenesis. Dev. Genes Evol. 2001, 211, 355–357. [Google Scholar] [CrossRef]
- Koike, M.A.; Lin, A.J.; Pham, J.; Nguyen, E.; Yeh, J.J.; Rahimian, R.; Tromberg, B.J.; Choi, B.; Green, K.N.; LaFerla, F.M. APP knockout mice experience acute mortality as the result of ischemia. PLoS ONE 2012, 7, e42665. [Google Scholar] [CrossRef]
- Luna, S.; Cameron, D.J.; Ethell, D.W. Amyloid-beta and APP deficiencies cause severe cerebrovascular defects: Important work for an old villain. PLoS ONE 2013, 8, e75052. [Google Scholar] [CrossRef]
- d’Uscio, L.V.; He, T.; Santhanam, A.V.; Katusic, Z.S. Endothelium-specific amyloid precursor protein deficiency causes endothelial dysfunction in cerebral arteries. J. Cereb. Blood Flow Metab. 2018, 38, 1715–1726. [Google Scholar] [CrossRef]
- de la Torre, J. The Vascular Hypothesis of Alzheimer’s Disease: A Key to Preclinical Prediction of Dementia Using Neuroimaging. J. Alzheimers Dis. 2018, 63, 35–52. [Google Scholar] [CrossRef]
- Nannelli, G.; Terzuoli, E.; Giorgio, V.; Donnini, S.; Lupetti, P.; Giachetti, A.; Bernardi, P.; Ziche, M. ALDH2 Activity Reduces Mitochondrial Oxygen Reserve Capacity in Endothelial Cells and Induces Senescence Properties. Oxid. Med. Cell. Longev. 2018, 2018, 9765027. [Google Scholar] [CrossRef]
- Terzuoli, E.; Meini, S.; Cucchi, P.; Catalani, C.; Cialdai, C.; Maggi, C.A.; Giachetti, A.; Ziche, M.; Donnini, S. Antagonism of bradykinin B2 receptor prevents inflammatory responses in human endothelial cells by quenching the NF-kB pathway activation. PLoS ONE 2014, 9, e84358. [Google Scholar] [CrossRef] [PubMed]
- Pezzatini, S.; Morbidelli, L.; Solito, R.; Paccagnini, E.; Boanini, E.; Bigi, A.; Ziche, M. Nanostructured HA crystals up-regulate FGF-2 expression and activity in microvascular endothelium promoting angiogenesis. Bone 2007, 41, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Alghisi, G.C.; Ponsonnet, L.; Ruegg, C. The integrin antagonist cilengitide activates alphaVbeta3, disrupts VE-cadherin localization at cell junctions and enhances permeability in endothelial cells. PLoS ONE 2009, 4, e4449. [Google Scholar] [CrossRef] [PubMed]
- Ciccone, V.; Monti, M.; Antonini, G.; Mattoli, L.; Burico, M.; Marini, F.; Maidecchi, A.; Morbidelli, L. Efficacy of AdipoDren(R) in Reducing Interleukin-1-Induced Lymphatic Endothelial Hyperpermeability. J. Vasc. Res. 2016, 53, 255–268. [Google Scholar] [CrossRef]
- Lin, Y.; Zhou, J.; Bi, D.; Chen, P.; Wang, X.; Liang, S. Sodium-deoxycholate-assisted tryptic digestion and identification of proteolytically resistant proteins. Anal. Biochem. 2008, 377, 259–266. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, T.; Cao, R.; Liu, Z.; Shen, J.; Chen, P.; Wang, X.; Liang, S. Evaluation of the application of sodium deoxycholate to proteomic analysis of rat hippocampal plasma membrane. J. Proteome. Res. 2006, 5, 2547–2553. [Google Scholar] [CrossRef]
- Huang da, W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2009, 37, 1–13. [Google Scholar] [CrossRef]
- Pathan, M.; Keerthikumar, S.; Chisanga, D.; Alessandro, R.; Ang, C.S.; Askenase, P.; Batagov, A.O.; Benito-Martin, A.; Camussi, G.; Clayton, A.; et al. A novel community driven software for functional enrichment analysis of extracellular vesicles data. J. Extracell. Vesicles 2017, 6, 1321455. [Google Scholar] [CrossRef]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef]
- Ringner, M. What is principal component analysis? Nat. Biotechnol. 2008, 26, 303–304. [Google Scholar] [CrossRef] [PubMed]
- Shariati, S.A.; De Strooper, B. Redundancy and divergence in the amyloid precursor protein family. FEBS Lett. 2013, 587, 2036–2045. [Google Scholar] [CrossRef] [PubMed]
- Avraamides, C.J.; Garmy-Susini, B.; Varner, J.A. Integrins in angiogenesis and lymphangiogenesis. Nat. Rev. Cancer 2008, 8, 604–617. [Google Scholar] [CrossRef] [PubMed]
- Khalili, A.A.; Ahmad, M.R. A Review of Cell Adhesion Studies for Biomedical and Biological Applications. Int. J. Mol. Sci. 2015, 16, 18149–18184. [Google Scholar] [CrossRef]
- Schaller, M.D. Paxillin: A focal adhesion-associated adaptor protein. Oncogene 2001, 20, 6459–6472. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F. Endothelial adherens junctions at a glance. J. Cell Sci. 2013, 126, 2545–2549. [Google Scholar] [CrossRef]
- Komarova, Y.A.; Kruse, K.; Mehta, D.; Malik, A.B. Protein Interactions at Endothelial Junctions and Signaling Mechanisms Regulating Endothelial Permeability. Circ. Res. 2017, 120, 179–206. [Google Scholar] [CrossRef]
- Pulous, F.E.; Petrich, B.G. Integrin-dependent regulation of the endothelial barrier. Tissue Barriers 2019, 7, 1685844. [Google Scholar] [CrossRef]
- Puhlmann, M.; Weinreich, D.M.; Farma, J.M.; Carroll, N.M.; Turner, E.M.; Alexander, H.R., Jr. Interleukin-1beta induced vascular permeability is dependent on induction of endothelial tissue factor (TF) activity. J. Transl. Med. 2005, 3, 37. [Google Scholar] [CrossRef]
- Dejana, E.; Orsenigo, F.; Molendini, C.; Baluk, P.; McDonald, D.M. Organization and signaling of endothelial cell-to-cell junctions in various regions of the blood and lymphatic vascular trees. Cell Tissue Res. 2009, 335, 17–25. [Google Scholar] [CrossRef]
- Tornavaca, O.; Chia, M.; Dufton, N.; Almagro, L.O.; Conway, D.E.; Randi, A.M.; Schwartz, M.A.; Matter, K.; Balda, M.S. ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J. Cell Biol. 2015, 208, 821–838. [Google Scholar] [CrossRef] [PubMed]
- Ristori, E.; Donnini, S.; Ziche, M. New Insights Into Blood-Brain Barrier Maintenance: The Homeostatic Role of β-Amyloid Precursor Protein in Cerebral Vasculature. Front. Physiol. 2020, 11, 1056. [Google Scholar] [CrossRef] [PubMed]
- Izawa, Y.; Gu, Y.H.; Osada, T.; Kanazawa, M.; Hawkins, B.T.; Koziol, J.A.; Papayannopoulou, T.; Spatz, M.; Del Zoppo, G.J. beta1-integrin-matrix interactions modulate cerebral microvessel endothelial cell tight junction expression and permeability. J. Cereb. Blood Flow Metab. 2018, 38, 641–658. [Google Scholar] [CrossRef] [PubMed]
- Somanath, P.R.; Malinin, N.L.; Byzova, T.V. Cooperation between integrin alphavbeta3 and VEGFR2 in angiogenesis. Angiogenesis 2009, 12, 177–185. [Google Scholar] [CrossRef]
- Rognoni, E.; Ruppert, R.; Fassler, R. The kindlin family: Functions, signaling properties and implications for human disease. J. Cell Sci. 2016, 129, 17–27. [Google Scholar] [CrossRef]
- Sun, Z.; Costell, M.; Fassler, R. Integrin activation by talin, kindlin and mechanical forces. Nat. Cell Biol. 2019, 21, 25–31. [Google Scholar] [CrossRef]
- Chapuis, J.; Flaig, A.; Grenier-Boley, B.; Eysert, F.; Pottiez, V.; Deloison, G.; Vandeputte, A.; Ayral, A.M.; Mendes, T.; Desai, S.; et al. Genome-wide, high-content siRNA screening identifies the Alzheimer’s genetic risk factor FERMT2 as a major modulator of APP metabolism. Acta Neuropathol. 2017, 133, 955–966. [Google Scholar] [CrossRef]
- Harburger, D.S.; Calderwood, D.A. Integrin signalling at a glance. J. Cell Sci. 2009, 122, 159–163. [Google Scholar] [CrossRef]
- Yamazaki, T.; Koo, E.H.; Selkoe, D.J. Cell surface amyloid beta-protein precursor colocalizes with beta 1 integrins at substrate contact sites in neural cells. J. Neurosci. 1997, 17, 1004–1010. [Google Scholar] [CrossRef]
- Liang, M.; Wang, Y.; Liang, A.; Dong, J.F.; Du, J.; Cheng, J. Impaired integrin beta3 delays endothelial cell regeneration and contributes to arteriovenous graft failure in mice. Arter. Thromb. Vasc. Biol. 2015, 35, 607–615. [Google Scholar] [CrossRef]
- Ren, J.; Avery, J.; Zhao, H.; Schneider, J.G.; Ross, F.P.; Muslin, A.J. Beta3 integrin deficiency promotes cardiac hypertrophy and inflammation. J. Mol. Cell Cardiol. 2007, 42, 367–377. [Google Scholar] [CrossRef] [PubMed]
- Byzova, T.V.; Goldman, C.K.; Pampori, N.; Thomas, K.A.; Bett, A.; Shattil, S.J.; Plow, E.F. A mechanism for modulation of cellular responses to VEGF: Activation of the integrins. Mol. Cell 2000, 6, 851–860. [Google Scholar] [CrossRef]
- Corti, F.; Wang, Y.; Rhodes, J.M.; Atri, D.; Archer-Hartmann, S.; Zhang, J.; Zhuang, Z.W.; Chen, D.; Wang, T.; Wang, Z.; et al. N-terminal syndecan-2 domain selectively enhances 6-O heparan sulfate chains sulfation and promotes VEGFA165-dependent neovascularization. Nat. Commun. 2019, 10, 1562. [Google Scholar] [CrossRef]
- Wang, X.; Freire Valls, A.; Schermann, G.; Shen, Y.; Moya, I.M.; Castro, L.; Urban, S.; Solecki, G.M.; Winkler, F.; Riedemann, L.; et al. YAP/TAZ Orchestrate VEGF Signaling during Developmental Angiogenesis. Dev. Cell 2017, 42, 462–478 e467. [Google Scholar] [CrossRef] [PubMed]
- Kirouac, L.; Rajic, A.J.; Cribbs, D.H.; Padmanabhan, J. Activation of Ras-ERK Signaling and GSK-3 by Amyloid Precursor Protein and Amyloid Beta Facilitates Neurodegeneration in Alzheimer’s Disease. eNeuro 2017, 4. [Google Scholar] [CrossRef]
- Nizzari, M.; Thellung, S.; Corsaro, A.; Villa, V.; Pagano, A.; Porcile, C.; Russo, C.; Florio, T. Neurodegeneration in Alzheimer disease: Role of amyloid precursor protein and presenilin 1 intracellular signaling. J. Toxicol. 2012, 2012, 187297. [Google Scholar] [CrossRef]
- Nizzari, M.; Venezia, V.; Repetto, E.; Caorsi, V.; Magrassi, R.; Gagliani, M.C.; Carlo, P.; Florio, T.; Schettini, G.; Tacchetti, C.; et al. Amyloid precursor protein and Presenilin1 interact with the adaptor GRB2 and modulate ERK 1,2 signaling. J. Biol. Chem. 2007, 282, 13833–13844. [Google Scholar] [CrossRef]
- Venezia, V.; Nizzari, M.; Repetto, E.; Violani, E.; Corsaro, A.; Thellung, S.; Villa, V.; Carlo, P.; Schettini, G.; Florio, T.; et al. Amyloid precursor protein modulates ERK-1 and -2 signaling. Ann. N. Y. Acad. Sci. 2006, 1090, 455–465. [Google Scholar] [CrossRef]
- Kern, A.; Roempp, B.; Prager, K.; Walter, J.; Behl, C. Down-regulation of endogenous amyloid precursor protein processing due to cellular aging. J. Biol. Chem. 2006, 281, 2405–2413. [Google Scholar] [CrossRef]
- Sun, R.; He, T.; Pan, Y.; Katusic, Z.S. Effects of senescence and angiotensin II on expression and processing of amyloid precursor protein in human cerebral microvascular endothelial cells. Aging 2018, 10, 100–114. [Google Scholar] [CrossRef]
- Dawson, G.R.; Seabrook, G.R.; Zheng, H.; Smith, D.W.; Graham, S.; O’Dowd, G.; Bowery, B.J.; Boyce, S.; Trumbauer, M.E.; Chen, H.Y.; et al. Age-related cognitive deficits, impaired long-term potentiation and reduction in synaptic marker density in mice lacking the beta-amyloid precursor protein. Neuroscience 1999, 90, 1–13. [Google Scholar] [CrossRef]
- Senechal, Y.; Kelly, P.H.; Dev, K.K. Amyloid precursor protein knockout mice show age-dependent deficits in passive avoidance learning. Behav. Brain Res. 2008, 186, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Chiocco, M.J.; Lamb, B.T. Spatial and temporal control of age-related APP processing in genomic-based beta-secretase transgenic mice. Neurobiol. Aging 2007, 28, 75–84. [Google Scholar] [CrossRef] [PubMed][Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ristori, E.; Cicaloni, V.; Salvini, L.; Tinti, L.; Tinti, C.; Simons, M.; Corti, F.; Donnini, S.; Ziche, M. Amyloid-β Precursor Protein APP Down-Regulation Alters Actin Cytoskeleton-Interacting Proteins in Endothelial Cells. Cells 2020, 9, 2506. https://doi.org/10.3390/cells9112506
Ristori E, Cicaloni V, Salvini L, Tinti L, Tinti C, Simons M, Corti F, Donnini S, Ziche M. Amyloid-β Precursor Protein APP Down-Regulation Alters Actin Cytoskeleton-Interacting Proteins in Endothelial Cells. Cells. 2020; 9(11):2506. https://doi.org/10.3390/cells9112506
Chicago/Turabian StyleRistori, Emma, Vittoria Cicaloni, Laura Salvini, Laura Tinti, Cristina Tinti, Michael Simons, Federico Corti, Sandra Donnini, and Marina Ziche. 2020. "Amyloid-β Precursor Protein APP Down-Regulation Alters Actin Cytoskeleton-Interacting Proteins in Endothelial Cells" Cells 9, no. 11: 2506. https://doi.org/10.3390/cells9112506
APA StyleRistori, E., Cicaloni, V., Salvini, L., Tinti, L., Tinti, C., Simons, M., Corti, F., Donnini, S., & Ziche, M. (2020). Amyloid-β Precursor Protein APP Down-Regulation Alters Actin Cytoskeleton-Interacting Proteins in Endothelial Cells. Cells, 9(11), 2506. https://doi.org/10.3390/cells9112506