Abstract
EGFR and some of the cognate ligands extensively traffic in extracellular vesicles (EVs) from different biogenesis pathways. EGFR belongs to a family of four homologous tyrosine kinase receptors (TKRs). This family are one of the major drivers of cancer and is involved in several of the most frequent malignancies such as non-small cell lung cancer, breast cancer, colorectal cancer and ovarian cancer. The carrier EVs exert crucial biological effects on recipient cells, impacting immunity, pre-metastatic niche preparation, angiogenesis, cancer cell stemness and horizontal oncogene transfer. While EV-mediated EGFR signalling is important to EGFR-driven cancers, little is known about the precise mechanisms by which TKRs incorporated in EVs play their biological role, their stoichiometry and associations to other proteins relevant to cancer pathology and EV biogenesis, and their means of incorporation in the target cell. In addition, it remains unclear whether different subtypes of EVs incorporate different complexes of TKRs with specific functions. A raft of high spatial and temporal resolution methods is emerging that could solve these and other questions regarding the activity of EGFR and its ligands in EVs. More importantly, methods are emerging to block or mitigate EV activity to suppress cancer progression and drug resistance. By highlighting key findings and areas that remain obscure at the intersection of EGFR signalling and EV action, we hope to cross-fertilise the two fields and speed up the application of novel techniques and paradigms to both.
1. Introduction
Extracellular vesicles (EVs) are membranous nanoparticles of a wide range of sizes released from nearly all eukaryotic and prokaryotic cells [1,2,3]. EVs have been identified in a plethora of biological materials including blood, urine, faeces, bile, breast milk, as well as synovial, lacrimal, seminal, ascites and bronchoalveolar lavage fluids [4]. EVs are released naturally in response to cellular stimuli, as well as due to changes in pH, hypoxia, injury and other cellular stressors, and in response to complement activation [5,6,7,8]. While initially thought to be a mechanism of eliminating waste from cells [9], in 1996 Raposo et al. documented that EVs could stimulate adaptive immune responses [10]. From there, EVs became recognised as important mediators of intercellular communication through the bioactive transfer of their protein, lipid and nucleic acid cargoes to recipient cells [11,12,13,14].
Over the last few decades, EVs have been shown to mediate phenotypic alterations in physiological and pathological conditions. Most notably, the role of EVs in cancer development and progression, including their proposed involvement in metastatic growth and chemotherapeutic resistance, has been extensively studied. Along with active efflux mechanisms, EVs have been shown to aid removal of chemotherapeutics from cancer cells [15,16]. Furthermore, EVs have also been shown to be involved in tumour progression and metastasis by mediating epithelial-to-mesenchymal transition (EMT), migration, invasion, angiogenesis, immune modulation and reprogramming healthy cells to a cancerous phenotype in establishing a pre-metastatic niche [17,18].
The Epidermal Growth Factor Receptor (EGFR) family of receptor tyrosine kinases (RTKs) is a major player in a variety of epithelial malignancies, such as breast cancer, non-small cell lung cancer, colorectal, gastric and ovarian cancer, and glioblastoma. This has led to the development of targeted therapeutics, including tyrosine kinase inhibitors (TKIs) and antibodies, such as Herceptin, representing a milestone in cancer therapy [19].
The HER family includes four members—EGFR, Human Epidermal Growth Factor Receptor HER2, HER3 and HER4; their splicing variants; and 11 cognate ligands [20,21,22,23,24,25]. So far, EGFR, HER2 and some ligands have been found to be expressed on EVs, where they enhance the malignant potential of their parental neoplastic cells.
The role of EGFR family proteins on cancer cells is well recognised and has been extensively reviewed (see Maennling et al. [26] for recent updates to the state-of-the-art), highlighting commonalities and specificities in their modes of action and mutational patterns between cancer types [27,28]. As of yet there is no comparable understanding of the roles of this receptor family at the EV level, even though EVs have emerged as a major driver of tumourigenesis and are exploited as biomarkers in liquid biopsies.
Given the biological and clinical relevance of EGFR and its ligands, herein we discuss key findings regarding their expression on EVs and the roles of these proteins in EV signalling and uptake. Additionally, we will shine a spotlight on outstanding questions regarding the relevance of EGFR expression on EVs in different types of cancer, their implications in disease progression, in respective therapeutic approaches and as biomarkers, and discuss what techniques and frameworks can be brought to bear on them.
2. Extracellular Vesicles and Their Biogenesis
The term “extracellular vesicles” encompasses a variety of different vesicle subclasses mainly based on their respective biogenesis and biophysical properties. The most extensively studied subtypes are exosomes and microvesicles (MVs).
Exosomes range in size from roughly 30 nm to 150 nm in diameter [29,30,31]. They are formed within the endolysosomal pathway (Figure 1). First, the inward budding of the cell membrane forms membranous vacuoles called endosomes. Subsequent inward invaginations of endosomal membranes forms intra-luminal vesicles (ILVs) which themselves are early exosomes. Through multiple inward invagination events, endosomes become filled with ILVs and become referred to as multivesicular bodies (MVBs) [32]. MVBs displaying specific surface proteins fuse with lysosomes to degrade ILV contents. These include RAB7A, HSP70-HSP90 organising protein (HOP) complexes and soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) complexes, including vesicle-associated membrane protein 7 (VAMP7), and syntaxin 7 and 8 (STX7/8). Otherwise, MVBs traffic to and fuse with the cell membrane releasing their ILVs, then termed exosomes [33,34].
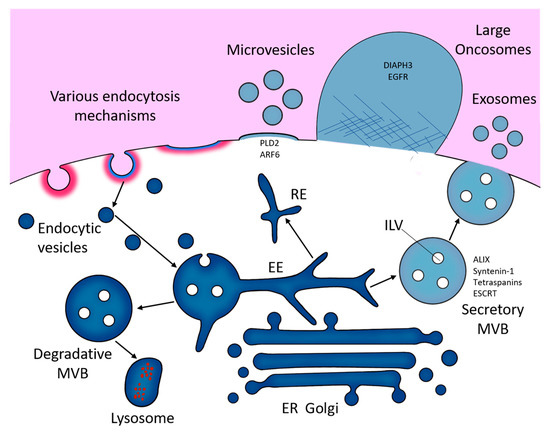
Figure 1.
A diagram of the possible routes of formation of extracellular vesicles (EVs), via the endosomal pathway (exosomes), directly from membrane shedding (microvesicles) and via large-scale, non-apoptotic blebbing (large oncosomes). EE, early endosome; ER, endoplasmic reticulum; RE, recycling endosome.
Exosomal formation in MVBs can occur dependently or independently of proteins called endosomal sorting complexes required for transport (ESCRT-0–III) [35,36]. In ESCRT-dependent exosomal formation ESCRT-0 and -I cluster ubiquitylated membrane-associated proteins and lipids into microdomains on the limiting membrane of MVBs. These microdomains recruit soluble cytosolic proteins and RNAs destined for packing as cargoes into exosomes. ESCRT-II and -III then aid in the exosomal budding and fission [37].
Alternatively, ESCRT-independent formation involves ceramide subdomain mediated curvature of endosomal membranes and cargo sorting into ILVs [38,39]. Similarly, tetraspanin family members CD9, CD63 and CD81, all of which are highly enriched on the surface of exosomes [40,41,42,43], can mediate ILV biogenesis via clustering of these tetraspanins into microdomains with other tetraspanins, transmembrane proteins and cytosolic proteins that bud from the MVB surface [44], or via formation of CD81 rich domains which induce inward budding [45]. Syntenin 1 has also been shown to be involved in targeting of exosome cargo to endosomal membranes for packaging into ILVs [46], and ADP ribosylation factor 6 (ARF6) can aid ILV budding and sorting of epidermal growth factor receptor (EGFR/HER1) into ILVs, then help traffic EGFR to lysosomes for degradation [47].
On the other hand, MVs range between roughly 50 nm to 1300 nm in diameter [29,30,31]. MV formation occurs by direct budding of vesicles from the cell membrane. This process is mediated by ARF6 and RHO family GTPase-dependent rearrangement of the actin cytoskeleton [48,49]. Here, ESCRT-I component tumour susceptibility gene 101 protein (TSG101) traffics to the plasma membrane and interacts with accessory proteins ALG-2- interacting protein X (ALIX) for subsequent cargo sorting and MV release [50]. Furthermore, both neutral and acid sphingomyelinase (N- and A-SMase respectively) modulate downstream activation of receptors that trigger MV release [51,52].
Despite their biogenetic differences, the overlapping size ranges and similar proteomic compositions of exosomes and MVs make them difficult to distinguish from one another. This is particularly important as both exosomes and MVs are released simultaneously from their cells of origin into the same extracellular environment, forming an extensively heterogeneous EV population. Consequently, the selective purification of either exosomes or MVs from culture media or biological fluid remains a persistent issue [53]. Therefore, in non-selective studies, or studies where specific markers of subcellular origin cannot be reliably identified, EVs are classed simply according to their size; small EVs (sEVs), i.e., anything smaller than 200 nm, and large EVs (lEVs), i.e., anything larger than 200 nm [54]. lEVs also include oncosomes, a class of EVs first identified by Dolores Di Vizio and her team in prostate cancer. These oversized EVs range from roughly 0.5–5 μm in diameter and have so far only been detected in cancer cell secretomes [55,56].
3. The EGFR Family
The receptors of the HER family show more than 80% structural homology and are located on the basolateral membrane of epithelial tissues derived from ectoderm and mesoderm [57].
Overall, they are characterised by a ligand-binding extracellular domain (ECD) comprised of two ligand-binding domains (I and III) and two structural cysteine-rich domains (II and IV) [58,59]; a single-helix transmembrane domain, which is essential for receptor interactions and allosteric activation [60,61,62,63,64]; and an intracellular module where the catalytic activity resides.
The intracellular module is comprised of a partially unstructured juxtamembrane domain (JMD) which has regulatory functions and can bind to lipids in the plasma membrane [62,65,66], a tyrosine kinase domain (TKD) and a mostly unstructured c-terminal segment that contains the majority of the phosphorylation sites and plays a regulatory role [67,68] (Figure 2A).

Figure 2.
The EGFR family: (A) Cartoons of EGFR family homodimers showing the extracellular domain in its back-to-back dimer conformation, bound to ligand where applicable, and the intracellular asymmetric tyrosine kinase dimer, joined by the Transmembrane Domain (TMD). The latter leads to phosphorylation of tyrosines on the C-terminal tail (yellow dots) and recruitment of effectors (red, teal, orange and lime green shapes). The red “no entry” symbols indicate the lack of a known soluble ligand for HER2 and the pseudokinase status of HER3. Also shown is a cartoon of a full-length, transmembrane EGFR family (pro)ligand. (B) Molecular model view (above) and cartoon view (below) of the conformational changes involved in ligand-induced dimerisation in the EGFR receptor, adapted from Martin-Fernandez M. et al., (2019).
Of the receptors, only EGFR and HER4 [69,70] are fully signalling competent, i.e., capable of both specific binding ligands and catalysing tyrosine phosphorylation for downstream signalling. HER2 does not have a known soluble ligand [71], while HER3 is considered a pseudokinase, as it can bind ATP but its catalytic activity is very low and does not significantly contribute to signalling [72,73]. In spite of this, the kinase domain of HER3 is indispensable for HER2/HER3 signalling [74,75], acting as the activator partner in an asymmetric kinase heterodimer, in which HER2 phosphorylates the C-terminal tail of HER3 [76]. HER2 and HER3 dimers constitute the strongest signalling unit in the HER family and are often co-expressed [77,78].
Signalling by HER receptors is induced by allosteric changes induced by ligand binding at the ECD, which release a set of intradomain interactions between subdomains II and IV known as the “tether” and enable dimerisation through an interface comprising domains II and IV [70,79,80], which then leads to allosteric kinase activation [81] (Figure 2B). HER2 constitutes an exception to this rule, as its “untethered” extracellular conformation in absence of ligand mimics that of the EGF-activated EGFR, enabling it to form dimers with the other ligand-bound HERs [82,83]. HER2, however appears to be regulated by a different set of inhibitory interactions between its subdomains I and III, mimicking interactions first discovered in invertebrate EGFR orthologues [84], which appear to be relevant for its heterodimerisation with EGFR [85]. Additionally, the control exerted by the “tether” on EGFR signalling was found to be limited [86] and on cell membrane EGFR is reported to be actually quite flexible, sampling a large conformational space [87].
While oligomerisation and clustering of EGFR family receptors have been reported for a long time [88,89,90,91,92], in recent years, multidisciplinary studies have revealed that these structures are functional and that the EGFR signalling pathway includes not just monomers and dimers, but also a variety of multimeric structures, both in the basal and in the ligand-bound state [93,94]. These might reflect different sensitivities to ligand and/or perform specialised signalling functions [95]. Oligomerisation and alternate dimerisation interfaces are also involved in the signalling of the HER2/HER3 signalling unit, both in unperturbed conditions [76] and in response to targeted HER2 TKIs [96], hinting at common mechanisms of regulation for the whole family.
4. EGFR and Its Ligands on EVs
EGFR family members and cognate ligands are expressed by EVs released by neoplastic cells and play a wide variety of roles in enhancing the malignant potential of their parental cells, with a range of effects that covers most if not all of the hallmarks of cancer [97] (Figure 3 and Table 1).
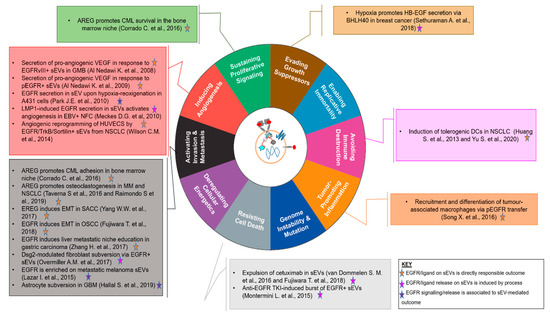
Figure 3.
An EGFR-centric view of the involvement of EVs in the Hallmarks of Cancer [97], summarising the key findings for the role of EGFR+/Ligand+ EVs in the “achievement” of each. The central cartoon schematically depicts a sEV displaying EGFR, a full-length transmembrane EGFR family ligand, a tetraspanin marker and nucleotidic cargo. Findings are denoted by first author and year and colour-coded according to the role played by EGFR and/or its ligands in the biological outcome referenced.

Table 1.
Summary of the effects of EGFR and its ligands when packaged in EVs.
The expression of EGFR was first identified in 2008 at protein and mRNA level in sEVs derived from cultured EGFR-positive cancer cell lines and clinical samples [31,98,99]. In this context, Al-Nedawi et al. found that EGFR and its glioblastoma deletion mutant isoform EGFRvIII could be packaged in EVs, where they could appear in their phosphorylated state, and that they were functional when internalised by recipient cells, increasing downstream MAPK signalling and the expression of oncogenic factors. This process was termed horizontal oncogene transmission [31] and has later been observed also in a variety of other processes involving wt or mutant EGFR, including metastatic niche establishment and immune suppression [100,101,102].
EGFR Ligands on EVs
EGFR can bind to seven cognate ligands, which display different affinities, potency and induction of signalling pathways [113,114,115,116,117]. They are synthesised as high molecular weight transmembrane pro-ligands, which are displayed on the surface of parental cells and then released as soluble fragments in response to various stimuli thanks to the action of ADAM proteases [118], a class of proteins commonly present in the EV cargo [119] (see Figure 2A).
Full-length N-term-out functional EGFR ligands Amphiregulin (AREG), heparin binding EGF (HB-EGF) and transforming growth factor alpha (TGF)-α were first identified in sEVs derived from a panel of breast and colorectal cancer cell lines using fluorescence-activated vesicle sorting (FAVS) [120]. AREG was identified as particularly resistant to degradation when displayed on sEVs and as the strongest inducer of invasiveness among the three ligands in the panel of cell lines, perhaps owing to its high stoichiometry (24 ± 7.6 copies/sEV) [120]. AREG is a low-affinity EGFR ligand which has physiological and pathological roles, including in the progression of various types of cancer [121]. Its pleiotropic effects would explain why it emerged in as a major player in sEV-mediated signalling.
AREG expressed on sEVs derived from Chronic Myeloid Leukaemia (CML) cell lines or patient serum is able to activate EGFR on stromal cells in an in vitro model of a bone marrow (BM) niche. Stromal cells EGFR activation induces via SNAIL pathway the secretion of IL-8 and MMP-9, which promote CML cell survival, as well as expression of Annexin 2 which promotes CML cell adhesion and homing to the niche. The significance of its role in niche regulation is highlighted by the fact that AREG expression on CML sEVs is correlated to aggressiveness and chemoresistance in patients [103].
The role of EGFR signalling, and in particular of EGFR ligands, in osteoclastogenesis is known from breast cancer co-culture models and non-small cell lung cancer (NSCLC) models, where EGFR inhibition with TKIs was shown to prevent osteoclast differentiation [122,123,124]. Recent studies have highlighted a key role of EVs in this pathogenesis mechanism, both in bone metastatic NSCLC, where increased plasma levels of AREG are correlated with poor prognosis [105], and in multiple myeloma [104], a highly osteoclastogenic disease where AREG is a known tumour growth factor [125]. In both cases, AREG displayed on EVs is able to induce the in vitro differentiation of pre-osteoclasts, with reduction in OPG levels, increase in RANKL expression and secretion of osteolytic enzymes such as MMP-9 and TRAP [104,105].
Besides inducing pro-tumoural changes in bystander stromal cells, EGFR ligands can also induce cancer progression by acting on their own parental cells. Salivary Adenoid Cystic Carcinoma (SACC) is a type of head and neck cancer that presents a poor prognosis and a penchant for metastasising to the lungs. In this tumour type, expression of low-affinity, high-efficiency EGFR ligand epiregulin (EREG) [126] is associated with poor outcomes and induces EMT, with consequent increases in angiogenic signalling [106]. While previous studies had already highlighted the role of EREG in the induction of SACC metastasis to the lung via the activation of EMT pathways involving the EGFR-Snail/Slug axis [127], a recent study demonstrates that the vehicle for EREG transport to target cells is constituted by sEVs, and that these are responsible for morphological changes in bystander endothelial cells and the induction of angiogenesis [106].
The secretion of EVs carrying EGFR ligands can occur through stimulation. Cationic lipids induce the release of EGF+ EVs from melanoma cell lines [128], while breast cancer cells are known to respond to hypoxia by secreting increased numbers of sEVs, a mechanism that favours the secretion of EGFR ligands such as HB-EGF [129]. Studies using murine models of breast cancer have identified BHLHE40, a transcriptional target of HIF-1α, which is itself a transcriptional regulator, as an inducer of several cytokines and growth factors in response to hypoxia. BHLHE40 acts as a transcriptional activator of HB-EGF by binding to its promoter and preventing HDAC1/2 from repressing its transcription. Upregulated HB-EGF is then packaged on upregulated sEVs, which induce survival and migration in recipient cells. The clinical relevance of this mechanism seems supported by the positive correlation between HB-EGF and BHLHE40 expression in TGA datasets [107].
However, why would cancer cells invest in releasing membrane-bound vesicles, when they could just secrete soluble ligands? One explanation is that EV-incorporated transmembrane ligands seem to be much more efficient in terms of concentration and length of induced signalling than recombinant soluble ligands in inducing downstream effects in recipient cells [130].
Signalling by full-length, transmembrane ligands to receptors expressed in trans on neighbouring cells, a process called juxtacrine signalling, has been identified since the early 1990s as an independent function for many EGFR ligands, including TGF-α [131], EGF [132], HB-EGF [133], and AREG [134].
More recently, recognising the importance of this mode of signalling, Higginbotham et al. [120] suggested using the term ExTRAcrine (Exosomal Targeted Receptor Activation) for the signalling by transmembrane EGFR ligands displayed on sEVs, a mode which straddles the divide between endocrine for the communication range and juxtacrine, for the binding mode, which is key in determining signalling properties. Owing to their small size, EVs act as a single-point, directional delivery mechanism for the activation of EGFR family receptors, one which concentrates a high density of ligands in a small space. Time-resolved fluorescence experiments have shown not only that point-stimulation of EGFR can be propagated to the entire cell [135], especially if the recipient cell overexpresses EGFR or a local increase in receptor density is created [136], and that multivalency amplifies EGFR signalling, but also that clustering of EGF-displaying particles can lead to even higher signalling amplification [137]. It might also be a matter of fine-tuning the extent of clustering on a spatial level: other investigations have shown that small-scale clusters of EGFR lead to higher phosphorylation and signalling than large-scale clustering [138].
Thus, it seems that there is a clear advantage in being able to deliver a mitogenic signal in well-packaged quanta instead of in a continuous trickle.
5. EVs Expressing EGFR Shape the Tumour Microenvironment
The tumour microenvironment (TME) is composed by the tumour cells, stromal cells including fibroblasts, blood and lymph vessels, immune cells, the extracellular matrix (ECM), cytokines, RNAs and small organelles such as EVs. The interplay of tumour cells with their TME influences tumourigenesis, progression and metastasis, drug resistance and has an impact on patient prognosis [139].
Among the soluble factors, EVs play major roles in the complex tumour–TME relationship as, depending on their parental cells, they can support intercellular communication, contributing to the acquisition of functional features by TME components, such as immune cells and endothelial cells, leading to immune suppression and angiogenesis, to mention only some of the effects detailed below.
EVs have different ways of shaping their microenvironment and distant sites depending on their type of interaction with the recipient cells. The factors and mechanisms that mediate EV cargo transfer as well as those that determine how EVs “choose” their target cells are still yet to be fully understood. However, these processes seem to depend on the cells that the EVs originate from, the phenotype and origin of recipient cells and the subsequent effects elicited by the EVs on their recipients [140,141].
Additionally, it remains unclear which moieties on the cell membrane may promote or inhibit EV docking based on their expression level, protein structure or other features like proximity. Another important question is if EVs travelling as single vesicle or in droves may have different effects during their interaction with target cells. However, ultimately the general mechanisms of uptake and intercellular trafficking of different EV subpopulations, such that EV cargoes can evoke their intended effects, are likely to be shared [141]. Major mechanisms of EV uptake include phagocytosis, macro- and micropinocytosis, clathrin mediated endocytosis (CME) and clathrin-independent endocytosis, all of which and more have been reviewed elsewhere [141,142,143].
With these outstanding questions in mind, in the following sections we will expose the shaping activity of EVs in an EGFR context.
5.1. Proangiogenic Signalling
Since the very beginning of this investigation field, the induction of angiogenesis downstream of EV uptake by bystander cells has been a major concern.
Early on, the uptake of EGFRvIII sEVs by human glioblastoma cell line U373 has been demonstrated to increase their secretion of the proangiogenic factor VEGF [31], which then induces angiogenic responses in neighbouring endothelial cells. Proangiogenic effects have been demonstrated also for activated EGFR (pEGFR) displayed on sEVs originating from other cancer cell lines, including A431, A549 and DLD-1, which, in turn, can induce the activation of MAPK and Akt signalling and initiate an autocrine VEGF signalling loop in recipient endothelial cells. The ability of sEVs to transfer pEGFR from parental cancer cells to endothelial cells confers them an advantage in terms of elicitation of proangiogenic responses and tumour growth, which can be reversed in both cell cultures and mice xenografts by inhibiting the uptake of sEVs with Annexin V [108].
Subsequent proteomics studies have confirmed that hypoxia stimulates EGFR-overexpressing A431 cells to secrete both soluble proangiogenic factors and sEVs loaded with proangiogenic proteins. EGFR itself is increased on the surface of sEVs produced by cells upon a hypoxia-reoxygenation cycle [144], which hints at its role as an indirect inducer of angiogenesis.
Even when EGFR is not the master oncogene, its oncogenic role in the induction of downstream effects through sEVs cannot be overlooked. In EBV-induced nasopharyngeal carcinoma, viral protein LMP1 induces an increase in cellular EGFR expression. Overexpressed EGFR is packaged into sEVs together with LMP1, and its packaging in EVs is increased by LMP1 expression. LMP1+/EGFR+ sEVs are also positive for PI3K and, when taken up by bystander HUVEC cells, they increase the activation of both Erk and Akt downstream pathways, influencing their angiogenic potential [109].
Finally, in human NSCLC cell line A549, EGFR is expressed on sEVs in a trimeric complex with receptors TrkB and sortilin [110], the former of which is another RTK that can cross-talk with EGFR family members in various cellular contexts [145,146,147], and the latter of which has been found to regulate EGFR signalling in lung cancer by promoting its internalisation [148] and is necessary for sEV release from the MVB pathway [110]. HUVEC cells exposed to sEVs displaying this triple EGFR/TkrB/sortilin complex acquire enhanced MAPK and Akt pathway signalling, enhanced migratory capabilities and increased secretion of soluble proangiogenic factors. These last findings highlight the importance of the identification of specific signalling complexes to understand the functional heterogeneity of EGFR-containing sEVs, an area of research that is not yet sufficiently explored.
5.2. Epithelial–Mesenchymal Transition (EMT) Program
Apart from conferring tumour cells an advantage in survival by directly inducing proangiogenic pathways, sEVs are a major player in the induction of EMT and in the propagation of the EMT-mediated effects to the microenvironment in terms of angiogenesis induction, niche subversion and invasion [149]. In addition, EGFR signalling is also heavily involved in EMT processes. On one hand, in a physiological context, EGFR is involved in epithelial wound healing, which includes a partial EMT process through a pathway that includes Erk5 and the Slug transcription factor [150]. EGFR signalling and wound healing appear to be strictly associated with sEVs secretion in renal tubule wound healing, where EGFR signalling seems to decrease sEV output, and vice versa [151].
On the other hand, EGFR can induce EMT in several tumourigenesis contexts, either driving it entirely through Ras or Akt signalling and induction of transcriptional activity [152], or acting downstream of EMT-initiating transforming growth factor beta (TGF)-β signalling [153]. EMT is also involved in resistance to EGFR-targeted therapeutics in NSCLC via activation of Axl in response to EGFR inhibition [154]. This makes the study of the convolution between EMT processes and EGFR signalling even more pivotal for the development of effective therapies.
In HSC-3, an Oral Squamous Cell Carcinoma (OSCC) cell line that expresses high levels of EGFR, exposure to nicotine increases the secretion of EGF, which is a major driver of EMT in this cellular context. EGF signalling then induces the enrichment of EGFR protein in HSC-3 sEVs in a way that cannot be countered by anti-EGFR antibody cetuximab [155], a therapeutic agent in clinical use for colorectal tumours and other EGFR-driven clinical entities which binds to the extracellular domain of EGFR and blocks ligand binding and signalling [156]. HSC-3-derived EGFR+ sEVs, in turn, induce EMT markers such as increased vimentin and spindle-like shapes in bystander epithelial cells. This effect is increased by OSCC cell pretreatment with EGFR ligand EGF, which increases the production of EGFR+/CD9+ sEVs even independently of nicotine. Cetuximab, in this case, is able to block sEV internalisation and inhibit sEV-driven EMT in bystander cells [157]. It is worth noting that metastatic sublines of HSC-3 express higher levels of EGFR in their sEVs in association with heat shock proteins even without exposure to stimuli [158].
5.3. Immune Modulation
Immune modulation is a key tactic for tumour cells as it allows them to escape from immune clearance and receive support for growth, dissemination and resistance to therapy from tumour-associated immune cell populations, thereby becoming even more aggressive and deadly (see, for example, in [159,160,161]). Mechanisms allowing tumour cells to avoid immune recognition and escape from immune clearance are continuing to emerge and the different signalling pathways controlling this phenomenon are interconnected with those controlling angiogenesis, proliferation, survival, senescence and metabolism. Communication via vesicles by tumour cells is a key factor in this context, as shown by the numerous studies demonstrating how tumour EVs contribute to generate an immune suppressive state through the induction and support of immune suppressive cells, of which the major populations are represented by T regulatory cells (Tregs) and the myeloid-derived suppressor cells (MDSCs) [162,163,164,165,166]. Their accumulation at tumour sites but also at a systemic level represents a negative prognosis, especially in patients affected by different cancer types in their advanced stages [167,168]. Additionally, MDSCs and Tregs can hinder effective cancer therapy, including immunotherapy with immune checkpoint inhibitors (ICIs) [169,170].
Copious evidence has documented that tumour-derived EVs contribute directly to the generation of suppressive immune cells, and this occurs through different mechanisms including transfer of genetic and protein material, and direct signalling [171,172,173]. In turn, suppressive immune cells can release their own EVs propagating immune suppressive effects at a cellular level [174].
The involvement of EGFR signalling in the induction of pro-tumour immune tolerance has been characterised particularly well for NSCLC, where it has been demonstrated that EGFR+ sEVs can induce differentiation of tolerogenic dendritic cells (DCs), which suppress the anti-cancer immune response [111]. In the case of cells expressing EGFR-del-19 mutant, this effect has been traced back directly to oncogene transmission to infiltrating DCs through sEV uptake. These subverted DCs were then responsible for repressing T cell response against tumoural cells, and resulted in an overall decrease in intratumoural CD8+ T cells [102]. In addition, sEVs displaying EGFR or HER2 have also been found to be involved in the recruitment and differentiation of tumour-associated macrophages (TAMs). In in vitro experiments, monocytes were stimulated to survive and differentiate within a simulated inflammatory microenvironment by sEVs derived from a panel of cancer cell lines from different histotypes. This survival signalling resulted from the activation of MAPK and Ras pathways via the horizontal transfer of phosphorylated EGFR or HER2 [101].
In summary, incorporation of EGFR in EVs appears to have major relevance in EV-mediated immune modulation with wide ranging consequences impacting also current therapeutic approaches in cancer clinical practice. Unravelling the underlying mechanisms will help targeting the coexisting pathways to enhance therapeutic efficacy.
5.4. Tumour Terraforming: Primary Tumour, Pre-Metastatic and Metastatic Niche Education
First advanced in 1899, Paget’s seed and soil hypothesis regarding the non-random distribution of metastases has since been confirmed by a wealth of data regarding the interactions between tumoural cells and the stroma at the site of the metastasis [175].
A series of seminal studies has highlighted that sEVs are able to educate the pre-metastatic niche (PMN), preparing the terrain for the arrival of cancer cells. sEVs are able to influence vascular permeability and the expression of inflammation and ECM proteins at distal sites from the primary tumour [176], and determine the organ tropism and metastatic potential of cancer cells injected in immunodeficient mice [176,177]. Their specific targeting, down to a single cell type in the complex microenvironment of a target organ, and action is due to the selective enrichment of specific integrin isoforms [177] or of cytokines and growth factors specific to microenvironmental components of the target metastatic site, which induce signalling loops favourable to the establishment of cancer cells [178].
PMN education via sEV has been identified also in EGFR-dependent context. EGFR expressed on gastric carcinoma sEVs is transferred to primary liver cells, where it colocalises with E-Cadherin and induces a decrease in the expression of miR-26a and miR-26b. These miRNAs are responsible for the post-transcriptional regulation of hepatocyte growth factor (HGF) mRNA translation through their binding to specific sequences in its 3′UTR, so the net effect of the horizontal transfer of EGFR to liver cells is an increase in HGF secretion in the liver microenvironment, which creates the ideal conditions for the establishment of gastric carcinoma cells, as they express high levels of the HGF receptors c-Met on their plasma membrane [100].
In EGFR-overexpressing HNSCC cell line A431, instead, the desmosomal cadherin desmoglein-2 (Dsg2), which can activate EGFR through a c-Src/Caveolin-1-dependent pathway [179], has been found to be expressed on sEVs and regulate the packaging of both EGFR and c-Src. sEVs from cells overexpressing Dsg2, which contain also increased EGFR, can induce phosphorylation of EGFR and downstream signalling pathways in normal human fibroblasts [112]. This could be a mechanism by which epithelial tumours subvert fibroblasts in the TME into a tumour-supporting role, creating a favourable niche where the tumour can become locally invasive.
Glioblastoma-derived sEVs have been shown to reprogram the main constituent of their microenvironment, astrocytes, to serve in supporting role for tumour growth [180]. This subversion, whereby astrocytes undergo changes in their proteome that favour the secretion of angiogenesis factors and the degradation of basement membranes to aid glioblastoma invasion, is achieved through the activation of MYC and p53 signalling pathways in recipient cells, where EGFR has been identified as one of the predicted upstream regulators of the network [181]. This seems to indicate that EV-displayed EGFR has a pivotal role in this process, but further studies will be required to confirm it.
EGFR is also part of a set of proteins specifically enriched on metastatic melanoma cell line derived sEVs [182], the internalisation of which by bystander cells increases their migratory potential, hinting at a more general role of EGFR in the establishment of metastasis, perhaps by playing a role akin to that of c-Met [176].
It appears that much like c-Met, EGFR has the potential to be a master regulator of the tumour niche, thanks to its capacity for horizontal transfer and its multiple interactions with other RTKs both from its own family and from the wider RTK superfamily (see, for example, in [145,183,184,185,186,187]).
5.5. Drug Resistance and Its Transfer to Bystander Cells
The role of sEVs in drug resistance mechanisms has been a focus of research since the early 2000s, when Shedden et al. noted a correlation between the expression of genes belonging to a “vesicle shedding index”, comprising most sEV markers identified until then, and the ability to resist a panel of drugs in the NCI-60 cell line panel [188]. Since then, sEVs have been implicated in drug resistance and its transfer to other cell types in the TME in various tumours. EVs can perform this function through a variety of mechanisms, including the transfer of ABC transporters and other drug efflux molecules to bystander cells [189].
Owing to its pivotal role in many of the major killers in the oncological field, EGFR has been at the centre of major efforts in pharmaceutical development [190], which have led to the development of two main classes of drugs, tyrosine kinase inhibitors (TKIs), such as erlotinib, afatinib or osimertinib, that target the ATP binding pocket of the TKD and inhibit its activity [191] and can bind reversibly or irreversibly to EGFR, and monoclonal antibodies such as cetuximab and pertuzumab, which target the extracellular domain and are able to block ligand binding [156], induce antibody-dependent cell cytotoxicity [192], or induce internalisation and downregulation of target receptors [193]. While some of these targeted treatments have been highly effective in the clinic on selected patients harbouring sensitising mutations, resistance to therapy happens inevitably, through genetic and non-genetic mechanisms [194,195].
Regarding the involvement of EVs in this context, findings seem controversial. Cetuximab treatment of EGFR-overexpressing A431 cell line induces a decrease of the expression of EGFR and phosphorylated EGFR on sEVs. However, cetuximab itself is found displayed on sEVs, a mechanism that could represent a way for cells to get rid of an inconvenient inhibitor by expelling it to the extracellular environment [196]. A different study, instead, concluded that while cetuximab does not seem to influence the global production of sEVs by A431 cells or the packaging of EGFR in sEVs, treatment with irreversible EGFR TKIs CI-1033 and PF-00299804 caused a “burst” in both sEV production and in the packaging of EGFR, including most phosphorylated species, Erk and pErk in sEVs. This burst was also accompanied by a peak in Caspase-3 cleavage and could be mitigated both by inhibition of Caspase-3 and by inhibition of neutral sphingomyelinase, which attests to their “classical” MVB origin [197]. Yet a third study, conducted on OSCC cell line HSC-3, instead, recorded an increase in EGFR expression in sEVs in response to cetuximab. In this case, cetuximab was also found to be robustly secreted on EGFR+ sEVs [155].
In the case of glioblastomas, targeted EGFR therapies have demonstrated much promise but relatively little success [198]. Alkylating agent temozolomide (TMZ), which, as of today, is one of the standard chemotherapeutic drugs for this histotype, and HSP90 inhibitor geldanamycin (17AAG) have been demonstrated to reduce both the amount of sEVs produced by the parental cells, and more specifically, the expression of both wild type EGFR and EGFRvIII on sEVs [199]. This seems to hint at a role for both drugs in the fight against EGFR-driven microenvironment subversion.
All of these mechanisms could contribute to the changes in sEV secretion and sEV content in response not just to EGFR-targeted therapies, but also to more generic interventions, by using EGFR as a pivotal point to induce microenvironmental changes.
6. EVs As Therapeutic Devices against Tumours
One of the most interesting qualities of EVs is their ability to transfer their cargos to recipient cells. Based on this quality, EVs may present as highly suitable platform for targeted therapeutic delivery. Additionally, EV mediated therapeutic delivery offers numerous advantages: EVs are biocompatible, can be generated from autologous patient cells if required, and have been shown to cross major biological barriers, including the blood-brain barrier [200,201,202]. As such, together these qualities present EVs as a potentially highly fruitful targeted therapeutic delivery platform, thus warranting the substantial attention they have received over the last few decades.
EVs can be engineered to mediate biological functions that are apart from their innate effects. For example, EVs can be loaded with therapeutic RNA or proteins either exogenously, or endogenously in a manner that exploits EV biogenesis machinery [200,201,202,203]. EVs can also be engineered to express specific molecules on their surface that may themselves be therapeutically active, aid in targeting EVs to specific cells, tissues or organs to deliver a therapeutic payload, help EVs fuse with recipient cell membranes or aid endosomal escape following uptake [204].
However, while engineering EVs is necessary to utilise them in the treatment of diseases they may otherwise not have any effect on, EVs do possess innate therapeutic potential in a number of disease contexts. The innate therapeutic benefits of EVs has mainly been explored in connection with EVs derived from cells that express stem cell-like properties, with particular focus on mesenchymal stem cells (MSCs). A recent review has highlighted the significance of MSC-EVs in this context and the multitude of preclinical and clinical studies that are taking place to exploit the innate therapeutic and regenerative benefits of MSC-EVs. In particular, MSC-EVs are being investigated for the treatment of respiratory diseases, including pulmonary hypertension, hepatic diseases including hepatic injury and liver fibrosis, renal diseases such as acute kidney injury, neurological diseases including global cerebral ischemia and traumatic brain injury and spinal cord injury, cardiovascular complications such as myocardial infarction and musculoskeletal disease including arthritis and even bone fractures [205].
Some of the highest profile uses of therapeutic EVs have been in a cancer context. In 2013, Ohno et al. engineered human embryonic kidney 293 (HEK293) cell-derived EVs to contain the tumour supressing miRNA, let-7a. EVs were also engineered to express GE11 peptide on their surfaces, a strong EGFR-targeting ligand. Together these EVs delivered let-7a to EGFR expressing xenograft breast cancer cells in RAG2−/− mice, eliciting a significant (P < 0.05) ~60% reduction in tumour size following weekly administration of GE11+, let-7a+ EVs for four weeks [206]. Their therapeutic action is also due to the fact that these particles are efficiently and rapidly internalised thanks to their ability to act as EGFR ligands [207].
Similarly, in another high profile study, EVs derived from immature dendritic cells were engineered to express lysosome-associated membrane glycoprotein 2b (Lamp2b) fused with iRGD, an integrin α-V-targeting peptide. These EVs were subsequently loaded with the small molecule chemotherapeutic doxorubicin by electroporation and administer to female BALB/c nude mice transplanted with MDA-MB-231 breast cancer cells. iRGD+, doxorubicin+ EVs caused a significant inhibition of tumour growth compared to the control groups (P < 0.01). iRGD+, doxorubicin+ EVs were also significantly less cytotoxic than free doxorubicin (P < 0.05) [208].
Moreover, unlike the previous two examples, another study exploited the use of a bioactive protein for the treatment of B cell lymphoma and melanoma. K562 multiple myeloma cells were transduced with lentiviral human membrane TRAIL, a protein that binds to death receptors on cancer cells, inducing selective apoptosis. Transduced cells produced TRAIL+ EVs and these latter were administered to mice transplanted with SUDHL4 B cell lymphoma cells and INT12 melanoma cells every 48 h. Subsequently, a significant growth inhibition of SUDHL4 (68%) and INT12 (51%) was observed following four intratumour injections [209]. Each of these examples exploits a different EV cargo for the treatment of various cancers, highlighting the utility and flexibility of EVs as therapeutic devices in a cancer context and others. An interesting aspect of EVs as natural therapeutic delivery vehicles lies in their versatile packaging potential: in case of EGFR targeting, EVs could be engineered to express EGFR ligands, and simultaneously, the same EVs could be loaded with different drugs or molecules to potentiate drug activity. For this reason, the use of EVs in the treatment of cancer has been extensively researched. Further significant contributions to this area have been highlighted in a recent review [210].
7. Exploitation of Circulating EVs As a “Liquid Biopsy” in Tumour Care
In many disease contexts, research is being undertaken to advance diagnostic tools to both complement and replace current diagnostic techniques that may be invasive, painful or otherwise time-consuming. One such avenue of exploration is through noninvasive sampling of bodily fluids for biomarkers of disease known as liquid biopsy. Liquid biopsy can provide real-time feedback on patient condition and response to cellular and molecular therapies [211]. Examples of biomarkers may include circulating cells or DNA [211,212]; however, EVs have also been regarded as biomarkers due to them being potentially released by diseased cells.
Cancer is one such context in which the use of EVs as biomarkers has been rigorously pursued for numerous reasons. First, evidence for the use of EVs in early-stage detection and diagnosis of multiple cancers has been extensively explored [213]. EVs may also help identify cancer subtypes to select the correct treatment [214,215] and assist in determining disease prognosis [216]. Furthermore, while tumour biopsies are often utilised in the diagnostic and monitoring process, non-specific findings during immunohistochemical analysis can elicit false-positive or false-negative diagnoses, resulting in less effective treatment [217].
In recent years, a novel analytical tool name ExoScreen that can be used to profile surface proteins on EVs has been developed. This tool can be used to analyse EVs in patient blood and serum without the need for purification. The system has been tested in the context of colorectal cancer and could be used to aid diagnosis. ExoScreen utilises streptavidin-coated “donor” beads that captures a biotinylated antibody specific to a marker of interest on an EV surface. “Donor” conjugates are also incubated with fluorescent “acceptor” beads conjugated to a second antibody that also binds to an epitope on the EV surface. The streptavidin-coated beads are then excited with a 680 nm laser resulting in the release of singlet oxygen. In turn, this excites the fluorescent acceptor beads that fluoresce, but only when the beads are within 200 nm of the donor beads that have bound to the analyte of interest. The complexity of ExoScreen prevents against false positive detection, however also prevents detection of particles larger than 200 nm such as some microvesicles or apoptotic bodies [218].
EGFR on EVs in Pre-Clinical Studies and in the Clinic
Ever since the discovery that sEVs expressing EGFRvIII from glioblastoma patients can cross the blood brain barrier and be quantified in the serum [219], the EGFR family has garnered notable attention within the EV field, bolstered by its importance as cellular biomarkers.
In pancreatic carcinoma EGFR overexpression is measured in ~30% of cases and cytoplasmic overexpression of EGFR is considered a marker of poor prognosis in the ductal adenocarcinoma subtype [220]. Small studies have evidenced that pancreatic carcinoma cells produce a higher amount of sEVs than their normal counterparts, and that the sEVs display EGFR protein. In addition, while EGFR enrichment on sEVs from pancreatic carcinoma cell lines is still debated [221], EGFR was found to be enriched on sEVs derived from mouse xenografts or from the serum of pancreatic carcinoma patients [222].
A small study using ExoProfile, a novel immunocapture chip, instead identified a significant difference in EGFR expression on plasma-derived sEVs between patients with ovarian carcinoma and normal controls; however, the study only involved a small number (n = 15) of patients, so larger studies will be required to confirm the validity of this putative biomarker [223].
In colorectal carcinoma, where EGFR is overexpressed by a variable but high percentage of patients and EGFR targeted therapy is a mainstay of treatment [224], but EGFR expression levels do not necessarily correlate with therapy efficacy, perhaps owing to inconsistencies in immunohistochemical methods [225], sEV-expressed EGFR was identified as part of an A33+ colorectal carcinoma-specific proteomics signature [226]; however, no specific claims as to its applicability in the clinic were made.
Glioblastoma is another cancer where EGFR signalling plays a pivotal role in tumorigenesis, via overexpression and the expression of truncation mutants [198]. Due to the difficulty to access the main tumour, sEVs have been at the centre of much attention in glioblastoma research [227].
Using a microfluidic detection chip, magnetic nanoparticle labelling and NMR detection on circulating glioblastoma sEVs, Shao et al. identified a diagnostic signature comprising EGFR, EGFRvIII, PDPN and IDH1 R132H which displays a 90% combined accuracy in distinguishing tumour sEVs from control sEVs. Combining this expression signature and a quantification of vesicle numbers, the authors then profiled the circulating sEVs from patients before and after treatment with alkylating agent temozolomide, and found that there was a significant difference in the index derived by the combined marker between responder patients and non-responders [199]. This study was also conducted on a small (n = 24) number of patients, thus the metric needs to be validated in a larger longitudinal cohort before coming into wider clinical use.
Other groups have instead focused on the detection of mutant and wild-type EGFR overexpression via sEV-packaged RNA in the cerebrospinal fluid of patients, where the mutational status of the parental tumour is accurately represented [228]. This access route can serve as a less invasive way to access molecular genetics information about the tumour.
In NSCLC, where EGFR overexpression is present in about 50% of patients and has a significant negative prognostic value, while mutations in the TKD which can be targeted with TKIs are present in ~10% of cases [229], the quest to use sEV EGFR as a biomarker has been conducted mainly using antibody array techniques. Multiple members of the EGFR family, including EGFRvIII, which is normally not expressed outside glioblastomas, have been identified in sEVs derived from the plasma of 109 patients [230].
A larger study, comprising 431 NSCLC patients and 150 controls, however failed to validate any of the EGFR family receptors expressed on plasma sEVs as a potential diagnostic marker [231]. Likewise, in a parallel study conducted on plasma sEVs from 276 patients, none of the EGFR family proteins identified was found to be a significant predictor of overall survival (OS) after applying the Bonferroni correction for multiple comparisons [232].
These clinical studies, however, failed to take into account vesicle heterogeneity and the importance that different EGFR family complexes might play in determining biological effects.
In spite of these less than promising results, the quest for sEV biomarkers continues with new and more refined strategies. An interesting new development is EXosome-focused Translational Research for Afatinib (EXTRA), a prospective study to identify novel predictive biomarkers and a resistance marker for the TKI afatinib in NSCLC patients that express mutant EGFR [233]. Its biomarker mining strategy leverages big data to correlate clinical efficacy with multi-omic data, the latter of which will be derived from serially collected peripheral blood-derived samples, including plasma, serum and serum-derived sEVs.
Conceivably by combining many sources of information that can capture vesicle heterogeneity, this study will be able to extract useful information about how therapy reflects in sEV secretion in different circumstances.
8. Perspectives
As shown by the body of research presented so far, EGFR acts as a major cargo of EVs and participates, with co-expressed proteins and small RNA molecules, in EV uptake and reprogramming of bystander cells in the TME, contributing to most if not all of the hallmarks of cancer (see Figure 3).
Systematic understanding of the effects of EGFR signalling within these contexts is fast-growing, however it is hampered by EV heterogeneity that extends beyond vesicle biophysical and biogenetic properties. Despite enormous technical improvements, the difficulty of distinguishing EV subtypes is yet to be solved. EV heterogeneity constitutes a major issue faced by the entire EV community in that it presents a steep challenge to our ability to purify, tag, and track specific EV subpopulations on their journey through the parental cell, and out in the microenvironment (reviewed, among others, in [234,235]).
To cope with resulting heterogeneous descriptions and individual naming of EVs subtypes, the International Society for Extracellular Vesicles (ISEV) has published a “Minimal information for Studies of Extracellular Vesicles-MISEV” 2018 after an extensive survey in the EV scientific community [54].
EV heterogeneity is also exhibited by their differing cargos, which may elicit a diverse array of biological effects on physiological or pathological status of the recipient cell, as demonstrated in these exemplary studies. At the gene level, Willms et al. showed that B16F10 melanoma cells release two subpopulations of EVs that can be distinguished based on flotation density. HD-EXO altered the expression of 1116 genes, whereas LD-EXO altered the expression of only 257 genes in recipient endothelial cells [236].
In this line at the protein level, Xu et al. (2018) revealed that sEVs and lEVs derived from LIM1863 colon cancer cells displayed 354 and 606 distinctly expressed proteins, respectively, with 256 proteins in common and 350 uniquely enriched proteins in lEVs. Comparatively, lEVs were able to elicit invasive action of recipient fibroblasts, which was roughly three times greater than that induced by sEVs. These differences were attributed to differential enrichment of proteins involved in invasion, migration and cell motility among the two EV populations [237].
EV heterogeneity can also cause interesting paradoxes when effects are observed in bulk. Cancer-derived EVs may exhibit opposing effects on immune cells. This can be explained by the existence of EVs exposing different integrin and tetraspanin profiles that correspond to their subsequent immunostimulatory and immunosuppressive activity, thereby modulating these effects by regulating their uptake into specific recipient cells [53].
Given the vast heterogeneity of secreted EVs, the numerous factors that can affect their composition and the plethora of cell sources, so far it has been difficult to clearly attribute any given function to a particular EVs subtype.
In the case of EGFR, this means that when EGFR expression on sEVs is used as a marker of a specific effect, this does not take into account that the same cell type will likely express several EGFR+ subpopulations displaying EGFR complexes with different structure and stoichiometry, as well as distinct sets of post-translational modifications and associated cargo, some of which will not be involved in the process being investigated at all. This is likely to be one of the key reasons why the development of EGFR protein-based biomarkers for liquid biopsies has proven to be so fraught.
This situation is further complicated by EV heterogeneity and the current purification methods that are unable to identify EVs based on their subtype or biogenesis, due to overlapping sizes and continued uncertainty surrounding biogenetic markers [236,238].
So-called “Omics” methods are fundamental to reveal the overall composition of the proteomes of EVs, as well as to guide subsequent targeted studies. However, these techniques often lack the spatial and temporal resolution and single-molecule sensitivity which are needed to capture the complexity of EGFR EV biogenesis and signalling, leaving many cardinal questions unanswered.
9. Outstanding Questions
In the context of EGFR incorporated in EVs, one key question is how many different EGFR family+ subpopulations are secreted by different cancer cells, and under what conditions EGFR and its ligands are enriched in or excluded from EVs. Second, we are also blind to what subtypes of EVs are secreted in response to EGFR signalling and/or are impacted by its inhibition. Immunocapture strategies can do some of the heavy lifting in separating a general EGFR (or ligand)-positive EV subpopulation, but this is likely to be heterogeneous in turn. Furthermore, this first round of detection must be combined with quantitative proteomics and/or FAVS to gain some insight on what other proteins are coexpressed with our chosen target, data which might lead to a future microfluidics-based detection platform for use in pre-clinical and clinical settings.
Differences in the structure and stoichiometry of the EGFR complex, stoichiometry and clustering of ligands, their interactions with other cargo and the presence of other companion molecules will impact the function of EVs in ways we are still not able to quantify, much less control.
Many studies have shown that EGFR+ or ligand+ EVs display various functional activities once taken up by different types of bystander cells. However, it is almost certain that only some, or even only one, of the EGFR+ subpopulations will be involved in each functional activity. The quest to identify which EGFR+ EVs are relevant for pro-tumoural activities is likely to be an arduous one, and will rely not just on ever more elegant proteomics experiments, but also by a much-needed “democratisation” as well as improvements in FAVS techniques [234], by the design and development of better EV-labelling strategies, both for bulk and targeted labelling and especially by the development of single-particle and microfluidic detection strategies and devices.
Enlightenment will be brought by high-resolution, live-cell 3D imaging technologies such as multifocal and multiplane microscopy [239,240], 3D structured illumination microscopy [241], multiangle interference microscopy [242], tilted light sheet [243] and lattice light sheet microscopy [244,245], to shed light on the aspects of EV interaction that are still obscure with target cells including timescales of interaction and internalisation. Lattice-light sheet microscopy in particular seems an extremely promising method, as it can combine with single-molecule detection techniques and record single-molecule information about membrane receptor dynamics over the entire cell surface with high spatial and temporal resolution [246], an ideal feature to investigate EV internalisation dynamics. These methods, however, come with their own challenges of high technical difficulty, the requirement for specialised expertise in microscope set up, data analysis and data management and low diffusion among non-specialists. Although, these very challenges open opportunities for interdisciplinary collaboration between the EV community and the advanced light microscopy and CLEM communities, which hopefully will help “democratise” these techniques in the medium- to long-term.
These high-resolution techniques will contribute unveiling the mechanisms of cargo transfer between EVs and target cells, thereby answering open questions like if EVs behave more like synaptic vesicles, promptly releasing their cargo upon fusion with the plasma membrane, or more like enveloped viruses, travelling within a cell’s endocytic pathway before releasing their contents in the cytoplasm.
New and improved fractionation methods and improved knowledge about specific markers of different biogenesis pathways, combined with the other technical improvements outlined above, will also help clarify whether functions of EVs are segregated by cellular origin or whether functions are independent from biogenesis but rather depend on their protein, lipid and RNA complement.
The acquisition of this knowledge will be a key step in the development of therapeutic agents and strategies to block “subversive” signalling by EVs in cancer with the aim of attenuating the angiogenic, metastatic, immunosuppressive and EMT potential of tumours and mitigating or even reversing drug resistance.
Author Contributions
Conceptualisation, L.C.Z.-D.; Writing—Original Draft Preparation, L.C.Z.-D., S.E.B. and V.H.; Writing—Review and Editing, L.C.Z.-D., S.E.B., V.H. and M.L.M.-F.; Visualisation, L.C.Z.-D. and M.L.M.-F.; Supervision, M.L.M.-F. All authors have read and agreed to the published version of the manuscript.
Funding
S.E.B. was supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC) BB/M011224/1 and Evox Therapeutics Ltd. V.H. was supported by Fondi 5 × 1000 Ministero della Salute 2015 (D/17/1VH).
Acknowledgments
L.C.Z.-D. is grateful to Helen Towrie and Guglielmo Miccolupi for help with the figures and Photoshop troubleshooting. The authors also warmly acknowledge Serenella Pupa and Elda Tagliabue of the Fondazione IRCCS Istituto Nazionale dei Tumori Milano for their advice and critical reading of early drafts of the manuscript.
Conflicts of Interest
S.E.B has equity in Evox Therapeutics Ltd. and holds an ongoing contract with Evox Therapeutics Ltd. from 2017 to present. Evox Therapeutics Ltd. had no influence on the conception or realisation of this review.
References
- Schorey, J.S.; Cheng, Y.; Singh, P.P.; Smith, V.L. Exosomes and other extracellular vesicles in host-pathogen interactions. EMBO Rep. 2015, 16, 24–43. [Google Scholar] [CrossRef] [PubMed]
- Deatherage, B.L.; Cookson, B.T. Membrane vesicle release in bacteria, eukaryotes, and archaea: A conserved yet underappreciated aspect of microbial life. Infect. Immun. 2012, 80, 1948–1957. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.G.; Ding, Y.; Jiang, L. Unconventional protein secretion in plants: A critical assessment. Protoplasma 2016, 253, 31–43. [Google Scholar] [CrossRef] [PubMed]
- Witwer, K.W.; Buzás, E.I.; Bemis, L.T.; Bora, A.; Lässer, C.; Lötvall, J.; Nolte-’t Hoen, E.N.; Piper, M.G.; Sivaraman, S.; Skog, J.; et al. Standardization of sample collection, isolation and analysis methods in extracellular vesicle research. J. Extracell. Vesicles 2013, 2, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Parolini, I.; Federici, C.; Raggi, C.; Lugini, L.; Palleschi, S.; De Milito, A.; Coscia, C.; Iessi, E.; Logozzi, M.; Molinari, A.; et al. Microenvironmental pH is a key factor for exosome traffic in tumor cells. J. Biol. Chem. 2009, 284, 34211–34222. [Google Scholar] [CrossRef]
- Mittelbrunn, M.; Gutiérrez-Vázquez, C.; Villarroya-Beltri, C.; González, S.; Sánchez-Cabo, F.; González, M.Á.; Bernad, A.; Sánchez-Madrid, F. Unidirectional transfer of microRNA-loaded exosomes from T cells to antigen-presenting cells. Nat. Commun. 2011, 2, 1–10. [Google Scholar] [CrossRef]
- Kucharzewska, P.; Belting, M. Emerging roles of extracellular vesicles in the adaptive response of tumour cells to microenvironmental stress. J. Extracell. Vesicles 2013, 2. [Google Scholar] [CrossRef]
- Patton, M.C.; Zubair, H.; Khan, M.A.; Singh, S.; Singh, A.P. Hypoxia alters the release and size distribution of extracellular vesicles in pancreatic cancer cells to support their adaptive survival. J. Cell. Biochem. 2020, 121, 828–839. [Google Scholar] [CrossRef]
- Johnstone, R.M.; Adam, M.; Hammond, J.R.; Orr, L.; Turbide, C. Vesicle formation during reticulocyte maturation. Association of plasma membrane activities with released vesicles (exosomes). J. Biol. Chem. 1987, 262, 9412–9420. [Google Scholar]
- Raposo, G.; Nijman, H.W.; Stoorvogel, W.; Liejendekker, R.; Harding, C.V.; Melief, C.J.; Geuze, H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996, 183, 1161–1172. [Google Scholar] [CrossRef]
- Colombo, M.; Raposo, G.; Théry, C. Biogenesis, secretion, and intercellular interactions of exosomes and other extracellular vesicles. Annu. Rev. Cell Dev. Biol. 2014, 30, 255–289. [Google Scholar] [CrossRef] [PubMed]
- Lo Cicero, A.; Stahl, P.D.; Raposo, G. Extracellular vesicles shuffling intercellular messages: For good or for bad. Curr. Opin. Cell Biol. 2015, 35, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Yáñez-Mó, M.; Siljander, P.R.-M.; Andreu, Z.; Bedina Zavec, A.; Borràs, F.E.; Buzas, E.I.; Buzas, K.; Casal, E.; Cappello, F.; Carvalho, J.; et al. Biological properties of extracellular vesicles and their physiological functions. J. Extracell. Vesicles 2015, 4, 27066. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; EL Andaloussi, S.; Wood, M.J.A. Exosomes and microvesicles: Extracellular vesicles for genetic information transfer and gene therapy. Hum. Mol. Genet. 2012, 21, R125–R134. [Google Scholar] [CrossRef] [PubMed]
- Györffy, B.; Serra, V.; Jürchott, K.; Abdul-Ghani, R.; Garber, M.; Stein, U.; Petersen, I.; Lage, H.; Dietel, M.; Schäfer, R. Prediction of doxorubicin sensitivity in breast tumors based on gene expression profiles of drug-resistant cell lines correlates with patient survival. Oncogene 2005, 24, 7542–7551. [Google Scholar] [CrossRef] [PubMed]
- Muralidharan-Chari, V.; Kohan, H.G.; Asimakopoulos, A.G.; Sudha, T.; Sell, S.; Kannan, K.; Boroujerdi, M.; Davis, P.J.; Mousa, S.A. Microvesicle removal of anticancer drugs contributes to drug resistance in human pancreatic cancer cells. Oncotarget 2016, 7, 50365–50379. [Google Scholar] [CrossRef]
- Conigliaro, A.; Cicchini, C. Exosome-Mediated signaling in epithelial to mesenchymal transition and tumor progression. J. Clin. Med. 2018, 8, 26. [Google Scholar] [CrossRef]
- Li, K.; Chen, Y.; Li, A.; Tan, C.; Liu, X. Exosomes play roles in sequential processes of tumor metastasis. Int. J. Cancer 2019, 144, 1486–1495. [Google Scholar] [CrossRef]
- Seshacharyulu, P.; Ponnusamy, M.P.; Haridas, D.; Jain, M.; Ganti, A.K.; Batra, S.K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. [Google Scholar] [CrossRef]
- Jackson, C.; Browell, D.; Gautrey, H.; Tyson-Capper, A. Clinical significance of HER-2 splice variants in breast cancer progression and drug resistance. Int. J. Cell Biol. 2013, 2013, 973584. [Google Scholar] [CrossRef]
- Abou-Fayçal, C.; Hatat, A.-S.; Gazzeri, S.; Eymin, B. Splice variants of the RTK family: Their role in tumour progression and response to targeted therapy. Int. J. Mol. Sci. 2017, 18, 383. [Google Scholar] [CrossRef] [PubMed]
- Lemmon, M.A.; Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Carpenter, G.; Coffey, R.J. EGF receptor ligands: Recent advances. F1000Research 2016, 5, 2270. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.S.; Coffing, S.L.; Le, A.T.D.; Cameron, E.M.; Williams, E.E.; Andrew, M.; Blommel, E.N.; Hammer, R.P.; Chang, H.; Riese, D.J. Neuregulin isoforms exhibit distinct patterns of ErbB family receptor activation. Oncogene 2002, 21, 8442–8452. [Google Scholar] [CrossRef] [PubMed]
- Kiuchi, T.; Ortiz-Zapater, E.; Monypenny, J.; Matthews, D.R.; Nguyen, L.K.; Barbeau, J.; Coban, O.; Lawler, K.; Burford, B.; Rolfe, D.J.; et al. The ErbB4 CYT2 variant protects EGFR from ligand-induced degradation to enhance cancer cell motility. Sci. Signal. 2014, 7, ra78. [Google Scholar] [CrossRef] [PubMed]
- Maennling, A.E.; Tur, M.K.; Niebert, M.; Klockenbring, T.; Zeppernick, F.; Gattenlöhner, S.; Meinhold-Heerlein, I.; Hussain, A.F. Molecular targeting therapy against EGFR family in breast cancer: Progress and future potentials. Cancers 2019, 11, 1826. [Google Scholar] [CrossRef] [PubMed]
- Vivanco, I.; Robins, H.I.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N.; et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef]
- Lane, A.; Segura-Cabrera, A.; Komurov, K. A comparative survey of functional footprints of EGFR pathway mutations in human cancers. Oncogene 2013, 1–12. [Google Scholar] [CrossRef]
- Pan, B.T.; Teng, K.; Wu, C.; Adam, M.; Johnstone, R.M. Electron microscopic evidence for externalization of the transferrin receptor in vesicular form in sheep reticulocytes. J. Cell Biol. 1985, 101, 942–948. [Google Scholar] [CrossRef]
- Harding, C.; Heuser, J.; Stahl, P. Endocytosis and intracellular processing of transferrin and colloidal gold-transferrin in rat reticulocytes: Demonstration of a pathway for receptor shedding. Eur. J. Cell Biol. 1984, 35, 256–263. [Google Scholar]
- Al-Nedawi, K.; Meehan, B.; Micallef, J.; Lhotak, V.; May, L.; Guha, A.; Rak, J. Intercellular transfer of the oncogenic receptor EGFRvIII by microvesicles derived from tumour cells. Nat. Cell Biol. 2008, 10, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Piper, R.C.; Katzmann, D.J. Biogenesis and function of multivesicular bodies. Annu. Rev. Cell Dev. Biol. 2007, 23, 519–547. [Google Scholar] [CrossRef] [PubMed]
- Luzio, J.P.; Gray, S.R.; Bright, N.A. Endosome-lysosome fusion. Proc. Biochem. Soc. Trans. 2010, 38, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Bucci, C.; Thomsen, P.; Nicoziani, P.; McCarthy, J.; Van Deurs, B. Rab7: A key to lysosome biogenesis. Mol. Biol. Cell 2000, 11, 467–480. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Stuffers, S.; Sem Wegner, C.; Stenmark, H.; Brech, A. Multivesicular endosome biogenesis in the absence of ESCRTs. Traffic 2009, 10, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Tamai, K.; Tanaka, N.; Nakano, T.; Kakazu, E.; Kondo, Y.; Inoue, J.; Shiina, M.; Fukushima, K.; Hoshino, T.; Sano, K.; et al. Exosome secretion of dendritic cells is regulated by Hrs, an ESCRT-0 protein. Biochem. Biophys. Res. Commun. 2010, 399, 384–390. [Google Scholar] [CrossRef]
- Goñi, F.M.; Alonso, A. Effects of ceramide and other simple sphingolipids on membrane lateral structure. Biochim. Biophys. Acta Biomembr. 2009, 1788, 169–177. [Google Scholar] [CrossRef]
- Kajimoto, T.; Okada, T.; Miya, S.; Zhang, L.; Nakamura, S.I. Ongoing activation of sphingosine 1-phosphate receptors mediates maturation of exosomal multivesicular endosomes. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Theos, A.C.; Truschel, S.T.; Tenza, D.; Hurbain, I.; Harper, D.C.; Berson, J.F.; Thomas, P.C.; Raposo, G.; Marks, M.S. A lumenal domain-dependent pathway for sorting to intralumenal vesicles of multivesicular endosomes involved in organelle morphogenesis. Dev. Cell 2006, 10, 343–354. [Google Scholar] [CrossRef]
- van Niel, G.; Charrin, S.; Simoes, S.; Romao, M.; Rochin, L.; Saftig, P.; Marks, M.S.; Rubinstein, E.; Raposo, G. The Tetraspanin CD63 regulates ESCRT-independent and -dependent endosomal sorting during melanogenesis. Dev. Cell 2011, 21, 708–721. [Google Scholar] [CrossRef] [PubMed]
- Buschow, S.I.; Nolte-’t Hoen, E.N.M.; van Niel, G.; Pols, M.S.; ten Broeke, T.; Lauwen, M.; Ossendorp, F.; Melief, C.J.M.; Raposo, G.; Wubbolts, R.; et al. MHC II In dendritic cells is targeted to lysosomes or t cell-induced exosomes via distinct multivesicular body pathways. Traffic 2009, 10, 1528–1542. [Google Scholar] [CrossRef] [PubMed]
- Chairoungdua, A.; Smith, D.L.; Pochard, P.; Hull, M.; Caplan, M.J. Exosome release of β-catenin: A novel mechanism that antagonizes Wnt signaling. J. Cell Biol. 2010, 190, 1079–1091. [Google Scholar] [CrossRef] [PubMed]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef]
- Zimmerman, B.; Kelly, B.; McMillan, B.J.; Seegar, T.C.M.; Dror, R.O.; Kruse, A.C.; Blacklow, S.C. Crystal structure of a full-length human tetraspanin reveals a cholesterol-binding pocket. Cell 2016, 167, 1041–1051.e11. [Google Scholar] [CrossRef]
- Baietti, M.F.; Zhang, Z.; Mortier, E.; Melchior, A.; Degeest, G.; Geeraerts, A.; Ivarsson, Y.; Depoortere, F.; Coomans, C.; Vermeiren, E.; et al. Syndecan-syntenin-ALIX regulates the biogenesis of exosomes. Nat. Cell Biol. 2012, 14, 677–685. [Google Scholar] [CrossRef]
- Ghossoub, R.; Lembo, F.; Rubio, A.; Gaillard, C.B.; Bouchet, J.; Vitale, N.; Slavík, J.; Machala, M.; Zimmermann, P. Syntenin-ALIX exosome biogenesis and budding into multivesicular bodies are controlled by ARF6 and PLD2. Nat. Commun. 2014, 5, 1–12. [Google Scholar] [CrossRef]
- Muralidharan-Chari, V.; Clancy, J.; Plou, C.; Romao, M.; Chavrier, P.; Raposo, G.; D’Souza-Schorey, C. ARF6-regulated shedding of tumor cell-derived plasma membrane microvesicles. Curr. Biol. CB 2009, 19, 1875–1885. [Google Scholar] [CrossRef]
- Li, B.; Antonyak, M.A.; Zhang, J.; Cerione, R.A. RhoA triggers a specific signaling pathway that generates transforming microvesicles in cancer cells. Oncogene 2012, 31, 4740–4749. [Google Scholar] [CrossRef]
- Nabhan, J.F.; Hu, R.; Oh, R.S.; Cohen, S.N.; Lu, Q. Formation and release of arrestin domain-containing protein 1-mediated microvesicles (ARMMs) at plasma membrane by recruitment of TSG101 protein. Proc. Natl. Acad. Sci. USA 2012, 109, 4146–4151. [Google Scholar] [CrossRef]
- Trajkovic, K.; Hsu, C.; Chiantia, S.; Rajendran, L.; Wenzel, D.; Wieland, F.; Schwille, P.; Brügger, B.; Simons, M. Ceramide triggers budding of exosome vesicles into multivesicular endosomes. Science 2008, 319, 1244–1247. [Google Scholar] [CrossRef] [PubMed]
- Bianco, F.; Perrotta, C.; Novellino, L.; Francolini, M.; Riganti, L.; Menna, E.; Saglietti, L.; Schuchman, E.H.; Furlan, R.; Clementi, E.; et al. Acid sphingomyelinase activity triggers microparticle release from glial cells. EMBO J. 2009, 28, 1043–1054. [Google Scholar] [CrossRef] [PubMed]
- Willms, E.; Cabañas, C.; Mäger, I.; Wood, M.J.A.; Vader, P. Extracellular vesicle heterogeneity: Subpopulations, isolation techniques, and diverse functions in cancer progression. Front. Immunol. 2018, 9, 738. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed]
- Di Vizio, D.; Kim, J.; Hager, M.H.; Morello, M.; Yang, W.; Lafargue, C.J.; True, L.D.; Rubin, M.A.; Adam, R.M.; Beroukhim, R.; et al. Oncosome formation in prostate cancer: Association with a region of frequent chromosomal deletion in metastatic disease. Cancer Res. 2009, 69, 5601–5609. [Google Scholar] [CrossRef]
- Kim, J.; Morley, S.; Le, M.; Bedoret, D.; Umetsu, D.T.; Di Vizio, D.; Freeman, M.R. Enhanced shedding of extracellular vesicles from amoeboid prostate cancer cells: Potential effects on the tumor microenvironment. Cancer Biol. Ther. 2014, 15, 409–418. [Google Scholar] [CrossRef]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; et al. Crystal structure of the complex of human epidermal growth factor and receptor extracellular domains. Cell 2002, 110, 775–787. [Google Scholar] [CrossRef]
- Garrett, T.P.J.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.-J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; et al. Crystal structure of a truncated epidermal growth factor receptor extracellular domain bound to transforming growth factor alpha. Cell 2002, 110, 763–773. [Google Scholar] [CrossRef]
- Fleishman, S.J.; Schlessinger, J.; Ben-Tal, N. A putative molecular-activation switch in the transmembrane domain of erbB2. Proc. Natl. Acad. Sci. USA 2002, 99, 15937–15940. [Google Scholar] [CrossRef]
- Lelimousin, M.; Limongelli, V.; Sansom, M.S.P. Conformational changes in the epidermal growth factor receptor: Role of the transmembrane domain investigated by coarse-grained metadynamics free energy calculations. J. Am. Chem. Soc. 2016, 138, 10611–10622. [Google Scholar] [CrossRef] [PubMed]
- Bragin, P.E.; Mineev, K.S.; Bocharova, O.V.; Volynsky, P.E.; Bocharov, E.V.; Arseniev, A.S. HER2 transmembrane domain dimerization coupled with self-association of membrane-embedded cytoplasmic juxtamembrane regions. J. Mol. Biol. 2016, 428, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Bocharov, E.V.; Lesovoy, D.M.; Pavlov, K.V.; Pustovalova, Y.E.; Bocharova, O.V.; Arseniev, A.S. Alternative packing of EGFR transmembrane domain suggests that protein-lipid interactions underlie signal conduction across membrane. Biochim. Biophys. Acta 2016, 1858, 1254–1261. [Google Scholar] [CrossRef] [PubMed]
- Hedger, G.; Shorthouse, D.; Koldsø, H.; Sansom, M.S.P. Free energy landscape of lipid interactions with regulatory binding sites on the transmembrane domain of the EGF receptor. J. Phys. Chem. B 2016, 120, 8154–8163. [Google Scholar] [CrossRef] [PubMed]
- Abd Halim, K.B.; Koldsø, H.; Sansom, M.S.P. Interactions of the EGFR juxtamembrane domain with PIP2-containing lipid bilayers: Insights from multiscale molecular dynamics simulations. Biochim. Biophys. Acta BBA Gen. Subj. 2015, 1850, 1017–1025. [Google Scholar] [CrossRef]
- Hedger, G.; Sansom, M.S.P.; Koldsø, H. The juxtamembrane regions of human receptor tyrosine kinases exhibit conserved interaction sites with anionic lipids. Sci. Rep. 2015, 5, 9198. [Google Scholar] [CrossRef]
- Margolis, B.L.; Lax, I.; Kris, R.; Dombalagian, M.; Honegger, A.M.; Howk, R.; Givol, D.; Ullrich, A.; Schlessinger, J. All autophosphorylation sites of epidermal growth factor (EGF) receptor and HER2/neu are located in their carboxyl-terminal tails. Identification of a novel site in EGF receptor. J. Biol. Chem. 1989, 264, 10667–10671. [Google Scholar]
- Kovacs, E.; Das, R.; Wang, Q.; Collier, T.S.; Cantor, A.; Huang, Y.; Wong, K.; Mirza, A.; Barros, T.; Grob, P.; et al. Analysis of the role of the C-terminal tail in the regulation of the epidermal growth factor receptor. Mol. Cell. Biol. 2015, 35, 3083–3102. [Google Scholar] [CrossRef]
- Qiu, C.; Tarrant, M.K.; Choi, S.H.; Sathyamurthy, A.; Bose, R.; Pal, A.; Bornmann, W.G.; Lemmon, M.A.; Cole, P.A.; Leahy, D.J. Mechanism of activation and inhibition of the HER4/ErbB4 Kinase. Structure 2010, 16, 460–467. [Google Scholar] [CrossRef]
- Bouyain, S.; Longo, P.A.; Li, S.; Ferguson, K.M.; Leahy, D.J. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. USA 2005, 102, 15024–15029. [Google Scholar] [CrossRef]
- Moasser, M.M. The oncogene HER2: Its signaling and transforming functions and its role in human cancer pathogenesis. Oncogene 2007, 26, 6469–6487. [Google Scholar] [CrossRef] [PubMed]
- Jura, N.; Shan, Y.; Cao, X.; Shaw, D.E.; Kuriyan, J. Structural analysis of the catalytically inactive kinase domain of the human EGF receptor 3. Proc. Natl. Acad. Sci. USA 2009, 106, 21608–21613. [Google Scholar] [CrossRef] [PubMed]
- Shi, F.; Telesco, S.E.; Liu, Y.; Radhakrishnan, R.; Lemmon, M.A. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 7692–7697. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.-K.; Cai, X.; Yuan, B.; Huang, Z.; Fan, X.; Deng, H.; Zhang, N.; An, Z. HER3 intracellular domains play a crucial role in HER3/HER2 dimerization and activation of downstream signaling pathways. Protein Cell 2012, 3, 781–789. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Choi, B.-K.; Fan, X.; Deng, H.; Zhang, N.; An, Z. ERBB3 (HER3) is a key sensor in the regulation of ERBB-mediated signaling in both low and high ERBB2 (HER2) expressing cancer cells. Cancer Med. 2012, 1, 28–38. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Park, E.; Kani, K.; Landgraf, R. Functional isolation of activated and unilaterally phosphorylated heterodimers of ERBB2 and ERBB3 as scaffolds in ligand-dependent signaling. Proc. Natl. Acad. Sci. USA 2012, 2–7. [Google Scholar] [CrossRef]
- Tzahar, E.; Waterman, H.; Chen, X.; Levkowitz, G.; Karunagaran, D.; Lavi, S.; Ratzkin, B.J.; Yarden, Y. A hierarchical network of interreceptor interactions determines signal transduction by Neu differentiation factor/neuregulin and epidermal growth factor. Mol. Cell. Biol. 1996, 16, 5276–5287. [Google Scholar] [CrossRef]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F.; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar] [CrossRef]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF activates its receptor by removing interactions that autoinhibit ectodomain dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar] [CrossRef]
- Cho, H.-S.; Leahy, D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science 2002, 297, 1330–1333. [Google Scholar] [CrossRef]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar] [CrossRef] [PubMed]
- Cho, H.; Mason, K.; Ramyar, K.X.; Stanley, A.M. Structure of the extracellular region of HER2 alone and in complex with the Herceptin Fab. Nature 2003, 421. [Google Scholar] [CrossRef] [PubMed]
- Garrett, T.P.J.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Kofler, M.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; et al. The crystal structure of a truncated ErbB2 ectodomain reveals an active conformation, poised to interact with other ErbB receptors. Mol. Cell 2003, 11, 495–505. [Google Scholar] [CrossRef]
- Alvarado, D.; Klein, D.E.; Lemmon, M.A. ErbB2 resembles an autoinhibited invertebrate epidermal growth factor receptor. Nature 2009, 461, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Arkhipov, A.; Shan, Y.; Kim, E.T.; Dror, R.O.; Shaw, D.E. Her2 activation mechanism reflects evolutionary preservation of asymmetric ectodomain dimers in the human EGFR family. eLife 2013, 2, e00708. [Google Scholar] [CrossRef] [PubMed]
- Mattoon, D.; Klein, P.; Lemmon, M.A.; Lax, I.; Schlessinger, J. The tethered configuration of the EGF receptor extracellular domain exerts only a limited control of receptor function. Proc. Natl. Acad. Sci. USA 2004, 101, 923–928. [Google Scholar] [CrossRef]
- Kaplan, M.; Narasimhan, S.; de Heus, C.; Mance, D.; van Doorn, S.; Houben, K.; Popov-Čeleketić, D.; Damman, R.; Katrukha, E.A.; Jain, P.; et al. EGFR dynamics change during activation in native membranes as revealed by NMR. Cell 2016, 167, 1241–1251.e11. [Google Scholar] [CrossRef]
- Kani, K.; Warren, C.M.; Kaddis, C.S.; Loo, J.A.; Landgraf, R. Oligomers of ERBB3 have two distinct interfaces that differ in their sensitivity to disruption by heregulin. J. Biol. Chem. 2005, 280, 8238–8247. [Google Scholar] [CrossRef]
- Saffarian, S.; Li, Y.; Elson, E.L.; Pike, L.J. Oligomerization of the EGF receptor investigated by live cell fluorescence intensity distribution analysis. Biophys. J. 2007, 93, 1021–1031. [Google Scholar] [CrossRef]
- Szabó, A.; Szöllosi, J.; Nagy, P. Coclustering of ErbB1 and ErbB2 revealed by FRET-sensitized acceptor bleaching. Biophys. J. 2010, 99, 105–114. [Google Scholar] [CrossRef]
- Clayton, A.H.A.; Tavarnesi, M.L.; Johns, T.G. Unligated epidermal growth factor receptor forms higher order oligomers within microclusters on A431 cells that are sensitive to tyrosine kinase inhibitor binding. Biochemistry 2007, 46, 4589–4597. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, R.; Müller, P.; Hildenbrand, G.; Hausmann, M.; Cremer, C. Analysis of Her2/neu membrane protein clusters in different types of breast cancer cells using localization microscopy. J. Microsc. 2011, 242, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Needham, S.R.; Roberts, S.K.; Arkhipov, A.; Mysore, V.P.; Tynan, C.J.; Zanetti-Domingues, L.C.; Kim, E.T.; Losasso, V.; Korovesis, D.; Hirsch, M.; et al. EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat. Commun. 2016, 7, 13307. [Google Scholar] [CrossRef] [PubMed]
- Zanetti-Domingues, L.C.; Korovesis, D.; Needham, S.R.; Tynan, C.J.; Sagawa, S.; Roberts, S.K.; Kuzmanic, A.; Ortiz-Zapater, E.; Jain, P.; Roovers, R.C.; et al. The architecture of EGFR’s basal complexes reveals autoinhibition mechanisms in dimers and oligomers. Nat. Commun. 2018, 9, 4325. [Google Scholar] [CrossRef]
- Krall, J.A.; Beyer, E.M.; MacBeath, G. High- and low-affinity epidermal growth factor receptor-ligand interactions activate distinct signaling pathways. PLoS ONE 2011, 6, e15945. [Google Scholar] [CrossRef]
- Claus, J.; Patel, G.; Autore, F.; Colomba, A.; Weitsman, G.; Soliman, T.N.; Roberts, S.; Zanetti-Domingues, L.C.; Hirsch, M.; Collu, F.; et al. Inhibitor-induced HER2-HER3 heterodimerisation promotes proliferation through a novel dimer interface. eLife 2018, 7, e32271. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Sanderson, M.P.; Keller, S.; Alonso, A.; Riedle, S.; Dempsey, P.J.; Altevogt, P. Generation of novel, secreted Epidermal Growth Factor Receptor (EGFR/ErbB1) isoforms via metalloprotease-dependent ectodomain shedding and exosome secretion. J. Cell. Biochem. 2008, 103, 1783–1797. [Google Scholar] [CrossRef]
- Skog, J.; Würdinger, T.; van Rijn, S.; Meijer, D.H.; Gainche, L.; Curry, W.T.; Carter, B.S.; Krichevsky, A.M.; Breakefield, X.O. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat. Cell Biol. 2008, 10, 1470–1476. [Google Scholar] [CrossRef]
- Zhang, H.; Deng, T.; Liu, R.; Bai, M.; Zhou, L.; Wang, X.; Li, S.; Wang, X.; Yang, H.; Li, J.; et al. Exosome-Delivered EGFR regulates liver microenvironment to promote gastric cancer liver metastasis. Nat. Commun. 2017, 8. [Google Scholar] [CrossRef]
- Song, X.; Ding, Y.; Liu, G.; Yang, X.; Zhao, R.; Zhang, Y.; Zhao, X.; Anderson, G.J.; Nie, G. Cancer cell-derived exosomes induce mitogen-activated protein kinase-dependent monocyte survival by transport of functional receptor tyrosine kinases. J. Biol. Chem. 2016, 291, 8453–8464. [Google Scholar] [CrossRef]
- Yu, S.; Sha, H.; Qin, X.; Chen, Y.; Li, X.; Shi, M.; Feng, J. EGFR E746-A750 deletion in lung cancer represses antitumor immunity through the exosome-mediated inhibition of dendritic cells. Oncogene 2020, 39, 2643–2657. [Google Scholar] [CrossRef] [PubMed]
- Corrado, C.; Saieva, L.; Raimondo, S.; Santoro, A.; De Leo, G.; Alessandro, R. Chronic myelogenous leukaemia exosomes modulate bone marrow microenvironment through activation of epidermal growth factor receptor. J. Cell. Mol. Med. 2016, 20, 1829–1839. [Google Scholar] [CrossRef] [PubMed]
- Raimondo, S.; Saieva, L.; Vicario, E.; Pucci, M.; Toscani, D.; Manno, M.; Raccosta, S.; Giuliani, N.; Alessandro, R. Multiple myeloma-derived exosomes are enriched of amphiregulin (AREG) and activate the epidermal growth factor pathway in the bone microenvironment leading to osteoclastogenesis. J. Hematol. Oncol. 2019, 12. [Google Scholar] [CrossRef] [PubMed]
- Taverna, S.; Pucci, M.; Giallombardo, M.; Di Bella, M.A.; Santarpia, M.; Reclusa, P.; Gil-Bazo, I.; Rolfo, C.; Alessandro, R. Amphiregulin contained in NSCLC-exosomes induces osteoclast differentiation through the activation of EGFR pathway. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef]
- Yang, W.-W.; Yang, L.-Q.; Zhao, F.; Chen, C.-W.; Xu, L.-H.; Fu, J.; Li, S.-L.; Ge, X.-Y. Epiregulin promotes lung metastasis of salivary adenoid cystic carcinoma. Theranostics 2017, 7, 3700–3714. [Google Scholar] [CrossRef]
- Sethuraman, A.; Brown, M.; Krutilina, R.; Wu, Z.-H.; Seagroves, T.N.; Pfeffer, L.M.; Fan, M. BHLHE40 confers a pro-survival and pro-metastatic phenotype to breast cancer cells by modulating HBEGF secretion. Breast Cancer Res. BCR 2018, 20, 117. [Google Scholar] [CrossRef]
- Al-Nedawi, K.; Meehan, B.; Kerbel, R.S.; Allison, A.C.; Rak, J. Endothelial expression of autocrine VEGF upon the uptake of tumor-derived microvesicles containing oncogenic EGFR. Proc. Natl. Acad. Sci. USA 2009, 106, 3794–3799. [Google Scholar] [CrossRef]
- Meckes, D.G.; Shair, K.H.Y.; Marquitz, A.R.; Kung, C.-P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef]
- Wilson, C.M.; Naves, T.; Vincent, F.; Melloni, B.; Bonnaud, F.; Lalloue, F.; Jauberteau, M.-O. Sortilin mediates the release and transfer of exosomes in concert with two tyrosine kinase receptors. J. Cell Sci. 2014, 127, 3983–3997. [Google Scholar] [CrossRef]
- Huang, S.; Li, Y.; Zhang, J.; Rong, J.; Ye, S. Epidermal growth factor receptor-containing exosomes induce tumor-specific regulatory T cells. Cancer Investig. 2013, 31, 330–335. [Google Scholar] [CrossRef] [PubMed]
- Overmiller, A.M.; Pierluissi, J.A.; Wermuth, P.J.; Sauma, S.; Martinez-Outschoorn, U.; Tuluc, M.; Luginbuhl, A.; Curry, J.; Harshyne, L.A.; Wahl, J.K.; et al. Desmoglein 2 modulates extracellular vesicle release from squamous cell carcinoma keratinocytes. FASEB J. 2017, 31, 3412–3424. [Google Scholar] [CrossRef] [PubMed]
- Jones, J.; Akita, R.; Sliwkowski, M. Binding specificities and affinities of egf domains for ErbB receptors. FEBS Lett. 1999, 447, 227–231. [Google Scholar] [CrossRef]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.J.; Lerdrup, M.; Grøvdal, L.; Willumsen, B.M.; van Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar] [CrossRef] [PubMed]
- Wilson, K.J.; Mill, C.; Lambert, S.; Buchman, J.; Wilson, T.R.; Hernandez-Gordillo, V.; Gallo, R.M.; Ades, L.M.C.; Settleman, J.; Riese, D.J. EGFR ligands exhibit functional differences in models of paracrine and autocrine signaling. Growth Factors 2012, 30, 107–116. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, S.L.J.; Mac, A.S.W.; Henriksen, L.; van Deurs, B.; Grøvdal, L.M. EGFR signaling patterns are regulated by its different ligands. Growth Factors 2014, 32, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Macdonald-Obermann, J.L.; Pike, L.J. Different epidermal growth factor (EGF) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J. Biol. Chem. 2014, 289, 26178–26188. [Google Scholar] [CrossRef] [PubMed]
- Blobel, C.P.; Carpenter, G.; Freeman, M. The role of protease activity in ErbB biology. Exp. Cell Res. 2009, 315, 671–682. [Google Scholar] [CrossRef]
- Shimoda, M. Extracellular vesicle-associated MMPs: A modulator of the tissue microenvironment. Adv. Clin. Chem. 2019, 88, 35–66. [Google Scholar] [CrossRef]
- Higginbotham, J.N.; Demory Beckler, M.; Gephart, J.D.; Franklin, J.L.; Bogatcheva, G.; Kremers, G.-J.; Piston, D.W.; Ayers, G.D.; McConnell, R.E.; Tyska, M.J.; et al. Amphiregulin exosomes increase cancer cell invasion. Curr. Biol. CB 2011, 21, 779–786. [Google Scholar] [CrossRef]
- Busser, B.; Sancey, L.; Brambilla, E.; Coll, J.-L.; Hurbin, A. The multiple roles of amphiregulin in human cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2011, 1816, 119–131. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Jia, X.; Xiao, G.; Kang, Y.; Partridge, N.C.; Qin, L. EGF-like ligands stimulate osteoclastogenesis by regulating expression of osteoclast regulatory factors by osteoblasts—Implications for osteolytic bone metastases. J. Biol. Chem. 2007, 282, 26656–26664. [Google Scholar] [CrossRef] [PubMed]
- Furugaki, K.; Moriya, Y.; Iwai, T.; Yorozu, K.; Yanagisawa, M.; Kondoh, K.; Fujimoto-Ohuchi, K.; Mori, K. Erlotinib inhibits osteolytic bone invasion of human non-small-cell lung cancer cell line NCI-H292. Clin. Exp. Metastasis 2011, 28, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Mercatali, L.; La Manna, F.; Miserocchi, G.; Liverani, C.; De Vita, A.; Spadazzi, C.; Bongiovanni, A.; Recine, F.; Amadori, D.; Ghetti, M.; et al. Tumor-Stroma crosstalk in bone tissue: The osteoclastogenic potential of a breast cancer cell line in a co-culture system and the role of EGFR inhibition. Int. J. Mol. Sci. 2017, 18, 1655. [Google Scholar] [CrossRef]
- Mahtouk, K.; Hose, D.; Rème, T.; De Vos, J.; Jourdan, M.; Moreaux, J.; Fiol, G.; Raab, M.; Jourdan, E.; Grau, V.; et al. Expression of EGF-family receptors and amphiregulin in multiple myeloma. Amphiregulin is a growth factor for myeloma cells. Oncogene 2005, 24, 3512–3524. [Google Scholar] [CrossRef]
- Riese, D.J.; Cullum, R.L. Epiregulin: Roles in normal physiology and cancer. Semin. Cell Dev. Biol. 2014, 28, 49–56. [Google Scholar] [CrossRef]
- Liu, S.; Ye, D.; Xu, D.; Liao, Y.; Zhang, L.; Liu, L.; Yu, W.; Wang, Y.; He, Y.; Hu, J.; et al. Autocrine epiregulin activates EGFR pathway for lung metastasis via EMT in salivary adenoid cystic carcinoma. Oncotarget 2016, 7, 25251–25263. [Google Scholar] [CrossRef]
- Emam, S.E.; Ando, H.; Lila, A.S.A.; Shimizu, T.; Okuhira, K.; Ishima, Y.; Mahdy, M.A.; Ghazy, F.S.; Sagawa, I.; Ishida, T. Liposome co-incubation with cancer cells secreted exosomes (extracellular vesicles) with different proteins expressions and different uptake pathways. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef]
- King, H.W.; Michael, M.Z.; Gleadle, J.M. Hypoxic enhancement of exosome release by breast cancer cells. BMC Cancer 2012, 12, 421. [Google Scholar] [CrossRef]
- Zhang, Q.; Higginbotham, J.N.; Jeppesen, D.K.; Yang, Y.-P.; Li, W.; McKinley, E.T.; Graves-Deal, R.; Ping, J.; Britain, C.M.; Dorsett, K.A.; et al. Transfer of functional cargo in exomeres. Cell Rep. 2019, 27, 940–954. [Google Scholar] [CrossRef]
- Wong, S.T.; Winchell, L.F.; McCune, B.K.; Earp, H.S.; Teixidó, J.; Massagué, J.; Herman, B.; Lee, D.C. The TGF-alpha precursor expressed on the cell surface binds to the EGF receptor on adjacent cells, leading to signal transduction. Cell 1989, 56, 495–506. [Google Scholar] [CrossRef]
- Dobashi, Y.; Stern, D.F. Membrane-anchored forms of EGF stimulate focus formation and intercellular communication. Oncogene 1991, 6, 1151–1159. [Google Scholar] [PubMed]
- Higashiyama, S.; Iwamoto, R.; Goishi, K.; Raab, G.; Taniguchi, N.; Klagsbrun, M.; Mekada, E. The membrane protein CD9/DRAP 27 potentiates the juxtacrine growth factor activity of the membrane-anchored heparin-binding EGF-like growth factor. J. Cell Biol. 1995, 128, 929–938. [Google Scholar] [CrossRef] [PubMed]
- Willmarth, N.E.; Ethier, S.P. Autocrine and juxtacrine effects of Amphiregulin on the proliferative, invasive, and migratory properties of Normal and neoplastic human mammary epithelial cells. J. Biol. Chem. 2006, 281, 37728–37737. [Google Scholar] [CrossRef]
- Verveer, P.J.; Wouters, F.S.; Reynolds, A.R.; Bastiaens, P.I.H. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science 2000, 290, 1567–1570. [Google Scholar] [CrossRef]
- Sawano, A.; Takayama, S.; Matsuda, M.; Miyawaki, A. Lateral propagation of EGF signaling after local stimulation is dependent on receptor density. Dev. Cell 2002, 3, 245–257. [Google Scholar] [CrossRef]
- Zhang, Q.; Reinhard, B.M. Ligand density and nanoparticle clustering cooperate in the multivalent amplification of epidermal growth factor receptor activation. ACS Nano 2018, 10473–10485. [Google Scholar] [CrossRef]
- Stabley, D.; Retterer, S.; Marshall, S.; Salaita, K.; Salaita, K.; Nair, P.M.; Petit, R.S.; Neve, R.M.; Das, D.; Gray, J.W.; et al. Manipulating the lateral diffusion of surface-anchored EGF demonstrates that receptor clustering modulates phosphorylation levels. Integr. Biol. 2013, 5, 659. [Google Scholar] [CrossRef]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R.F. Routes and mechanisms of extracellular vesicle uptake. J. Extracell. Vesicles 2014, 3. [Google Scholar] [CrossRef]
- van Niel, G.; D’Angelo, G.; Raposo, G. Shedding light on the cell biology of extracellular vesicles. Nat. Rev. Mol. Cell Biol. 2018, 19, 213–228. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Théry, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- French, K.C.; Antonyak, M.A.; Cerione, R.A. Extracellular vesicle docking at the cellular port: Extracellular vesicle binding and uptake. Semin. Cell Dev. Biol. 2017, 67, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Park, J.E.; Tan, H.S.; Datta, A.; Lai, R.C.; Zhang, H.; Meng, W.; Lim, S.K.; Sze, S.K. Hypoxic tumor cell modulates its microenvironment to enhance angiogenic and metastatic potential by secretion of proteins and exosomes. Mol. Cell. Proteom. MCP 2010, 9, 1085–1099. [Google Scholar] [CrossRef]
- Qiu, L.; Zhou, C.; Sun, Y.; Di, W.; Scheffler, E.; Healey, S.; Kouttab, N.; Chu, W.; Wan, Y. Crosstalk between EGFR and TrkB enhances ovarian cancer cell migration and proliferation. Int. J. Oncol. 2006, 29, 1003–1011. [Google Scholar] [CrossRef][Green Version]
- de Farias, C.B.; Heinen, T.E.; dos Santos, R.P.; Abujamra, A.L.; Schwartsmann, G.; Roesler, R. BDNF/TrkB signaling protects HT-29 human colon cancer cells from EGFR inhibition. Biochem. Biophys. Res. Commun. 2012, 425, 328–332. [Google Scholar] [CrossRef]
- Puehringer, D.; Orel, N.; Lüningschrör, P.; Subramanian, N.; Herrmann, T.; Chao, M.V.; Sendtner, M. EGF transactivation of Trk receptors regulates the migration of newborn cortical neurons. Nat. Neurosci. 2013, 16, 407–415. [Google Scholar] [CrossRef]
- Al-Akhrass, H.; Naves, T.; Vincent, F.; Magnaudeix, A.; Durand, K.; Bertin, F.; Melloni, B.; Jauberteau, M.-O.; Lalloué, F. Sortilin limits EGFR signaling by promoting its internalization in lung cancer. Nat. Commun. 2017, 8, 1182. [Google Scholar] [CrossRef]
- Blackwell, R.H.; Foreman, K.E.; Gupta, G.N. The role of cancer-derived exosomes in tumorigenicity & epithelial-to-mesenchymal transition. Cancers 2017, 9, 105. [Google Scholar] [CrossRef]
- Arnoux, V.; Nassour, M.; L’Helgoualc’h, A.; Hipskind, R.A.; Savagner, P. Erk5 controls slug expression and keratinocyte activation during wound healing. Mol. Biol. Cell 2008, 19, 4738–4749. [Google Scholar] [CrossRef]
- Zhou, X.; Zhang, W.; Yao, Q.; Zhang, H.; Dong, G.; Zhang, M.; Liu, Y.; Chen, J.-K.; Dong, Z. Exosome production and its regulation of EGFR during wound healing in renal tubular cells. Am. J. Physiol. Ren. Physiol. 2017, 312, F963–F970. [Google Scholar] [CrossRef] [PubMed]
- Clapéron, A.; Guedj, N.; Mergey, M.; Vignjevic, D.; Desbois-Mouthon, C.; Boissan, M.; Saubaméa, B.; Paradis, V.; Housset, C.; Fouassier, L. Loss of EBP50 stimulates EGFR activity to induce EMT phenotypic features in biliary cancer cells. Oncogene 2012, 31, 1376–1388. [Google Scholar] [CrossRef] [PubMed]
- Voon, D.C.-C.; Wang, H.; Koo, J.K.W.; Chai, J.H.; Hor, Y.T.; Tan, T.Z.; Chu, Y.-S.; Mori, S.; Ito, Y. EMT-Induced stemness and tumorigenicity are fueled by the EGFR/Ras pathway. PLoS ONE 2013, 8, e70427. [Google Scholar] [CrossRef] [PubMed]
- Tulchinsky, E.; Demidov, O.; Kriajevska, M.; Barlev, N.A.; Imyanitov, E. EMT: A mechanism for escape from EGFR-targeted therapy in lung cancer. Biochim. Biophys. Acta BBA Rev. Cancer 2019, 1871, 29–39. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.; Okamoto, K.; Calderwood, S.K.; Kozaki, K. Anti-EGFR antibody cetuximab is secreted by oral squamous cell carcinoma and alters EGF-driven mesenchymal transition. Biochem. Biophys. Res. Commun. 2018, 503, 1267–1272. [Google Scholar] [CrossRef]
- Li, S.; Schmitz, K.R.; Jeffrey, P.D.; Wiltzius, J.J.W.; Kussie, P.; Ferguson, K.M. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell 2005, 7, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, T.; Eguchi, T.; Sogawa, C.; Ono, K.; Murakami, J.; Ibaragi, S.; Asaumi, J.; Calderwood, S.K.; Okamoto, K.; Kozaki, K. Carcinogenic epithelial-mesenchymal transition initiated by oral cancer exosomes is inhibited by anti-EGFR antibody cetuximab. Oral Oncol. 2018, 86, 251–257. [Google Scholar] [CrossRef] [PubMed]
- Ono, K.; Eguchi, T.; Sogawa, C.; Calderwood, S.K.; Futagawa, J.; Kasai, T.; Seno, M.; Okamoto, K.; Sasaki, A.; Kozaki, K. HSP-enriched properties of extracellular vesicles involve survival of metastatic oral cancer cells. J. Cell. Biochem. 2018, 119, 7350–7362. [Google Scholar] [CrossRef]
- Khaja, A.S.S.; Toor, S.M.; Salhat, H.E.; Faour, I.; Haq, N.U.; Ali, B.R.; Elkord, E. Preferential accumulation of regulatory T cells with highly immunosuppressive characteristics in breast tumor microenvironment. Oncotarget 2017, 8, 33159–33171. [Google Scholar] [CrossRef]
- Chen, Z.; Hambardzumyan, D. Immune microenvironment in glioblastoma subtypes. Front. Immunol. 2018, 9, 1004. [Google Scholar] [CrossRef]
- Pogge von Strandmann, E.; Reinartz, S.; Wager, U.; Müller, R. Tumor-Host cell interactions in ovarian cancer: Pathways to therapy failure. Trends Cancer 2017, 3, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Peng, P.; Yan, Y.; Keng, S. Exosomes in the ascites of ovarian cancer patients: Origin and effects on anti-tumor immunity. Oncol. Rep. 2011, 25, 749–762. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91. [Google Scholar] [CrossRef] [PubMed]
- Martinez, V.G.; O’Neill, S.; Salimu, J.; Breslin, S.; Clayton, A.; Crown, J.; O’Driscoll, L. Resistance to HER2-targeted anti-cancer drugs is associated with immune evasion in cancer cells and their derived extracellular vesicles. Oncoimmunology 2017, 6, e1362530. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.-E.; Sung, K.J.; Sung, Y.H.; Pack, C.-G.; Jung, M.-K.; Han, B.; et al. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Mu, S.; Wang, Y.; Wang, H.; Cai, L.; Li, W.; Hu, Y. Prognostic role of myeloid-derived suppressor cells in cancers: A systematic review and meta-analysis. BMC Cancer 2018, 18, 1220. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. FoxP3+ T regulatory cells in cancer: Prognostic biomarkers and therapeutic targets. Cancer Lett. 2020, 490, 174–185. [Google Scholar] [CrossRef]
- Weber, R.; Fleming, V.; Hu, X.; Nagibin, V.; Groth, C.; Altevogt, P.; Utikal, J.; Umansky, V. Myeloid-Derived suppressor cells hinder the anti-cancer activity of immune checkpoint inhibitors. Front. Immunol. 2018, 9, 1310. [Google Scholar] [CrossRef]
- Saleh, R.; Elkord, E. Treg-mediated acquired resistance to immune checkpoint inhibitors. Cancer Lett. 2019, 457, 168–179. [Google Scholar] [CrossRef]
- Huber, V.; Vallacchi, V.; Fleming, V.; Hu, X.; Cova, A.; Dugo, M.; Shahaj, E.; Sulsenti, R.; Vergani, E.; Filipazzi, P.; et al. Tumor-Derived microRNAs induce myeloid suppressor cells and predict immunotherapy resistance in melanoma. J. Clin. Investig. 2018, 128, 5505–5516. [Google Scholar] [CrossRef] [PubMed]
- Rojas, C.; Campos-Mora, M.; Cárcamo, I.; Villalón, N.; Elhusseiny, A.; Contreras-Kallens, P.; Refisch, A.; Gálvez-Jirón, F.; Emparán, I.; Montoya-Riveros, A.; et al. T regulatory cells-derived extracellular vesicles and their contribution to the generation of immune tolerance. J. Leukoc. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Fleming, V.; Hu, X.; Weller, C.; Weber, R.; Groth, C.; Riester, Z.; Hüser, L.; Sun, Q.; Nagibin, V.; Kirschning, C.; et al. Melanoma extracellular vesicles generate immunosuppressive myeloid cells by upregulating PD-L1 via TLR4 signaling. Cancer Res. 2019, 79, 4715–4728. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wu, J.; Tian, J.; Wang, S. Role of T cell-derived exosomes in immunoregulation. Immunol. Res. 2018, 66, 313–322. [Google Scholar] [CrossRef]
- Langley, R.R.; Fidler, I.J. The seed and soil hypothesis revisited—The role of tumor-stroma interactions in metastasis to different organs. Int. J. Cancer 2011, 128, 2527–2535. [Google Scholar] [CrossRef]
- Peinado, H.; Alečković, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; García-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef]
- Hoshino, A.; Costa-Silva, B.; Shen, T.L.; Rodrigues, G.; Hashimoto, A.; Tesic Mark, M.; Molina, H.; Kohsaka, S.; Di Giannatale, A.; Ceder, S.; et al. Tumour exosome integrins determine organotropic metastasis. Nature 2015, 527, 329–335. [Google Scholar] [CrossRef]
- Costa-Silva, B.; Aiello, N.M.; Ocean, A.J.; Singh, S.; Zhang, H.; Thakur, B.K.; Becker, A.; Hoshino, A.; Mark, M.T.; Molina, H.; et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat. Cell Biol. 2015, 17, 816–826. [Google Scholar] [CrossRef]
- Overmiller, A.M.; McGuinn, K.P.; Roberts, B.J.; Cooper, F.; Brennan-Crispi, D.M.; Deguchi, T.; Peltonen, S.; Wahl, J.K.; Mahoney, M.G.; Mahoney, M.G. c-Src/Cav1-dependent activation of the EGFR by Dsg2. Oncotarget 2016, 7, 37536–37555. [Google Scholar] [CrossRef]
- Matias, D.; Balça-Silva, J.; da Graça, G.C.; Wanjiru, C.M.; Macharia, L.W.; Nascimento, C.P.; Roque, N.R.; Coelho-Aguiar, J.M.; Pereira, C.M.; Dos Santos, M.F.; et al. Microglia/Astrocytes-Glioblastoma Crosstalk: Crucial molecular mechanisms and microenvironmental factors. Front. Cell. Neurosci. 2018, 12, 235. [Google Scholar] [CrossRef]
- Hallal, S.; Mallawaaratchy, D.M.; Wei, H.; Ebrahimkhani, S.; Stringer, B.W.; Day, B.W.; Boyd, A.W.; Guillemin, G.J.; Buckland, M.E.; Kaufman, K.L. Extracellular vesicles released by glioblastoma cells stimulate normal astrocytes to acquire a tumor-supportive phenotype via p53 and MYC signaling pathways. Mol. Neurobiol. 2019, 56, 4566–4581. [Google Scholar] [CrossRef] [PubMed]
- Lazar, I.; Clement, E.; Ducoux-Petit, M.; Denat, L.; Soldan, V.; Dauvillier, S.; Balor, S.; Burlet-Schiltz, O.; Larue, L.; Muller, C.; et al. Proteome characterization of melanoma exosomes reveals a specific signature for metastatic cell lines. Pigment Cell Melanoma Res. 2015, 28, 464–475. [Google Scholar] [CrossRef] [PubMed]
- Greenall, S.A.; Johns, T.G. EGFRvIII: The promiscuous mutation. Cell Death Discov. 2016, 2, 16049. [Google Scholar] [CrossRef] [PubMed]
- Paul, I.; Bhattacharya, S.; Chatterjee, A.; Ghosh, M.K. Current understanding on EGFR and Wnt/β-catenin signaling in Glioma and their possible crosstalk. Genes Cancer 2013, 4, 427–446. [Google Scholar] [CrossRef] [PubMed]
- Soung, Y.H.; Clifford, J.L.; Chung, J. Crosstalk between integrin and receptor tyrosine kinase signaling in breast carcinoma progression. BMB Rep. 2010, 43, 311–318. [Google Scholar] [CrossRef]
- Meyer, A.S.; Miller, M.A.; Gertler, F.B.; Lauffenburger, D.A. The receptor AXL diversifies EGFR signaling and limits the response to EGFR-targeted inhibitors in triple-negative Breast cancer cells. Sci. Signal. 2013, 6, ra66. [Google Scholar] [CrossRef]
- Karamouzis, M.V.; Konstantinopoulos, P.A.; Papavassiliou, A.G. Targeting MET as a strategy to overcome crosstalk-related resistance to EGFR inhibitors. Lancet Oncol. 2009, 10, 709–717. [Google Scholar] [CrossRef]
- Shedden, K.; Xie, X.T.; Chandaroy, P.; Chang, Y.T.; Rosania, G.R. Expulsion of small molecules in vesicles shed by cancer cells: Association with gene expression and chemosensitivity profiles. Cancer Res. 2003, 63, 4331–4337. [Google Scholar]
- Safaei, R.; Larson, B.J.; Cheng, T.C.; Gibson, M.A.; Otani, S.; Naerdemann, W.; Howell, S.B. Abnormal lysosomal trafficking and enhanced exosomal export of cisplatin in drug-resistant human ovarian carcinoma cells. Mol. Cancer Ther. 2005, 4, 1595–1604. [Google Scholar] [CrossRef]
- Mahipal, A.; Kothari, N.; Gupta, S. Epidermal growth factor receptor inhibitors: Coming of age. Cancer Control J. Moffitt Cancer Cent. 2014, 21, 74–79. [Google Scholar] [CrossRef]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Kurai, J.; Chikumi, H.; Hashimoto, K.; Yamaguchi, K.; Yamasaki, A.; Sako, T.; Touge, H.; Makino, H.; Takata, M.; Miyata, M.; et al. Antibody-Dependent cellular cytotoxicity mediated by cetuximab against lung cancer cell lines. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2007, 13, 1552–1561. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Lahiji, A.; Patel, S.; Franklin, M.; Jimenez, X.; Hicklin, D.J.; Kang, X. Monoclonal antibody cetuximab binds to and down-regulates constitutively activated epidermal growth factor receptor vIII on the cell surface. Anticancer Res. 2007, 27, 3355–3366. [Google Scholar] [PubMed]
- Nagano, T.; Tachihara, M.; Nishimura, Y. Mechanism of resistance to epidermal growth factor receptor-tyrosine kinase inhibitors and a potential treatment strategy. Cells 2018, 7, 212. [Google Scholar] [CrossRef]
- Kruser, T.J.; Wheeler, D.L. Mechanisms of resistance to HER family targeting antibodies. Exp. Cell Res. 2010, 316, 1083–1100. [Google Scholar] [CrossRef]
- van Dommelen, S.M.; van der Meel, R.; van Solinge, W.W.; Coimbra, M.; Vader, P.; Schiffelers, R.M. Cetuximab treatment alters the content of extracellular vesicles released from tumor cells. Nanomedicine 2016, 11, 881–890. [Google Scholar] [CrossRef]
- Montermini, L.; Meehan, B.; Garnier, D.; Lee, W.J.; Lee, T.H.; Guha, A.; Al-Nedawi, K.; Rak, J. Inhibition of oncogenic epidermal growth factor receptor kinase triggers release of exosome-like extracellular vesicles and impacts their phosphoprotein and DNA content. J. Biol. Chem. 2015, 290, 24534–24546. [Google Scholar] [CrossRef]
- Westphal, M.; Maire, C.L.; Lamszus, K. EGFR as a target for glioblastoma treatment: An unfulfilled promise. CNS Drugs 2017, 31, 723–735. [Google Scholar] [CrossRef]
- Shao, H.; Chung, J.; Balaj, L.; Charest, A.; Bigner, D.D.; Carter, B.S.; Hochberg, F.H.; Breakefield, X.O.; Weissleder, R.; Lee, H. Protein typing of circulating microvesicles allows real-time monitoring of glioblastoma therapy. Nat. Med. 2012, 18, 1835–1840. [Google Scholar] [CrossRef]
- Alvarez-Erviti, L.; Seow, Y.; Yin, H.; Betts, C.; Lakhal, S.; Wood, M.J.A. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat. Biotechnol. 2011, 29, 341–345. [Google Scholar] [CrossRef]
- El Andaloussi, S.; Lakhal, S.; Mäger, I.; Wood, M.J.A. Exosomes for targeted siRNA delivery across biological barriers. Adv. Drug Deliv. Rev. 2013, 65, 391–397. [Google Scholar] [CrossRef] [PubMed]
- Rufino-Ramos, D.; Albuquerque, P.R.; Carmona, V.; Perfeito, R.; Nobre, R.J.; Pereira de Almeida, L. Extracellular vesicles: Novel promising delivery systems for therapy of brain diseases. J. Control. Release 2017, 262, 247–258. [Google Scholar] [CrossRef] [PubMed]
- Kamerkar, S.; Lebleu, V.S.; Sugimoto, H.; Yang, S.; Ruivo, C.F.; Melo, S.A.; Lee, J.J.; Kalluri, R. Exosomes facilitate therapeutic targeting of oncogenic KRAS in pancreatic cancer. Nature 2017, 546, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, J.; Kadungure, T.; Beyene, J.; Zhang, H.; Lu, Q. ARMMs as a versatile platform for intracellular delivery of macromolecules. Nat. Commun. 2018, 9, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wiklander, O.P.B.; Brennan, M.Á.; Lötvall, J.; Breakefield, X.O.; El Andaloussi, S. Advances in therapeutic applications of extracellular vesicles. Sci. Transl. Med. 2019, 11, eaav8521. [Google Scholar] [CrossRef] [PubMed]
- Ohno, S.I.; Takanashi, M.; Sudo, K.; Ueda, S.; Ishikawa, A.; Matsuyama, N.; Fujita, K.; Mizutani, T.; Ohgi, T.; Ochiya, T.; et al. Systemically injected exosomes targeted to EGFR deliver antitumor microrna to breast cancer cells. Mol. Ther. 2013, 21, 185–191. [Google Scholar] [CrossRef]
- Mickler, F.M.; Möckl, L.; Ruthardt, N.; Ogris, M.; Wagner, E.; Bräuchle, C. Tuning nanoparticle uptake: Live-Cell imaging reveals two distinct endocytosis mechanisms mediated by natural and artificial EGFR targeting ligand. Nano Lett. 2012, 3417–3423. [Google Scholar] [CrossRef]
- Tian, Y.; Li, S.; Song, J.; Ji, T.; Zhu, M.; Anderson, G.J.; Wei, J.; Nie, G. A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383–2390. [Google Scholar] [CrossRef]
- Rivoltini, L.; Chiodoni, C.; Squarcina, P.; Tortoreto, M.; Villa, A.; Vergani, B.; Bürdek, M.; Botti, L.; Arioli, I.; Cova, A.; et al. TNF-related apoptosis-inducing ligand (trail)-armed exosomes deliver proapoptotic signals to tumor site. Clin. Cancer Res. 2016, 22, 3499–3512. [Google Scholar] [CrossRef]
- Xu, R.; Rai, A.; Chen, M.; Suwakulsiri, W.; Greening, D.W.; Simpson, R.J. Extracellular vesicles in cancer—Implications for future improvements in cancer care. Nat. Rev. Clin. Oncol. 2018, 15, 617–638. [Google Scholar] [CrossRef]
- Riva, F.; Dronov, O.I.; Khomenko, D.I.; Huguet, F.; Louvet, C.; Mariani, P.; Stern, M.H.; Lantz, O.; Proudhon, C.; Pierga, J.Y.; et al. Clinical applications of circulating tumor DNA and circulating tumor cells in pancreatic cancer. Mol. Oncol. 2016, 10, 481–493. [Google Scholar] [CrossRef] [PubMed]
- Cristofanilli, M.; Budd, G.T.; Ellis, M.J.; Stopeck, A.; Matera, J.; Miller, M.C.; Reuben, J.M.; Doyle, G.V.; Allard, W.J.; Terstappen, L.W.M.M.; et al. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N. Engl. J. Med. 2004, 351, 781–791. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.; Jiang, F.; Ma, Y.; Wang, J.; Li, H.; Zhang, J. Isolation and detection technologies of extracellular vesicles and application on cancer diagnostic. Dose Response 2019, 17, 1559325819891004. [Google Scholar] [CrossRef] [PubMed]
- Rontogianni, S.; Synadaki, E.; Li, B.; Liefaard, M.C.; Lips, E.H.; Wesseling, J.; Wu, W.; Altelaar, M. Proteomic profiling of extracellular vesicles allows for human breast cancer subtyping. Commun. Biol. 2019, 2. [Google Scholar] [CrossRef] [PubMed]
- Lane, R.; Simon, T.; Vintu, M.; Solkin, B.; Koch, B.; Stewart, N.; Benstead-Hume, G.; Pearl, F.M.G.; Critchley, G.; Stebbing, J.; et al. Cell-Derived extracellular vesicles can be used as a biomarker reservoir for glioblastoma tumor subtyping. Commun. Biol. 2019, 2, 1–12. [Google Scholar] [CrossRef]
- Verma, M.; Lam, T.K.; Hebert, E.; Divi, R.L. Extracellular vesicles: Potential applications in cancer diagnosis, prognosis, and epidemiology. BMC Clin. Pathol. 2015, 15, 6. [Google Scholar] [CrossRef]
- Zaha, D.C. Significance of immunohistochemistry in breast cancer. World J. Clin. Oncol. 2014, 5, 382–392. [Google Scholar] [CrossRef]
- Yoshioka, Y.; Kosaka, N.; Konishi, Y.; Ohta, H.; Okamoto, H.; Sonoda, H.; Nonaka, R.; Yamamoto, H.; Ishii, H.; Mori, M.; et al. Ultra-Sensitive liquid biopsy of circulating extracellular vesicles using ExoScreen. Nat. Commun. 2014, 5, 3591. [Google Scholar] [CrossRef]
- Graner, M.W.; Alzate, O.; Dechkovskaia, A.M.; Keene, J.D.; Sampson, J.H.; Mitchell, D.A.; Bigner, D.D. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009, 23, 1541–1557. [Google Scholar] [CrossRef]
- Ueda, S.; Ogata, S.; Tsuda, H.; Kawarabayashi, N.; Kimura, M.; Sugiura, Y.; Tamai, S.; Matsubara, O.; Hatsuse, K.; Mochizuki, H. The correlation between cytoplasmic overexpression of epidermal growth factor receptor and tumor aggressiveness: Poor prognosis in patients with pancreatic ductal adenocarcinoma. Pancreas 2004, 29, e1. [Google Scholar] [CrossRef]
- Adamczyk, K.A.; Klein-Scory, S.; Tehrani, M.M.; Warnken, U.; Schmiegel, W.; Schnölzer, M.; Schwarte-Waldhoff, I. Characterization of soluble and exosomal forms of the EGFR released from pancreatic cancer cells. Life Sci. 2011, 89, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Kharmate, G.; Hosseini-Beheshti, E.; Caradec, J.; Chin, M.Y.; Tomlinson Guns, E.S. Epidermal growth factor receptor in prostate cancer derived exosomes. PLoS ONE 2016, 11, e0154967. [Google Scholar] [CrossRef]
- Zhang, P.; Zhou, X.; Zeng, Y. Multiplexed immunophenotyping of circulating exosomes on nano-engineered ExoProfile chip towards early diagnosis of cancer. Chem. Sci. 2019, 10, 5495–5504. [Google Scholar] [CrossRef] [PubMed]
- Krasinskas, A.M. EGFR signaling in colorectal carcinoma. Pathol. Res. Int. 2011, 932932. [Google Scholar] [CrossRef] [PubMed]
- Chung, K.Y.; Shia, J.; Kemeny, N.E.; Shah, M.; Schwartz, G.K.; Tse, A.; Hamilton, A.; Pan, D.; Schrag, D.; Schwartz, L.; et al. Cetuximab shows activity in colorectal cancer patients with tumors that do not express the epidermal growth factor receptor by immunohistochemistry. J. Clin. Oncol. 2005, 23, 1803–1810. [Google Scholar] [CrossRef] [PubMed]
- Mathivanan, S.; Lim, J.W.E.; Tauro, B.J.; Ji, H.; Moritz, R.L.; Simpson, R.J. Proteomics analysis of A33 immunoaffinity-purified exosomes released from the human colon tumor cell line LIM1215 reveals a tissue-specific protein signature. Mol. Cell. Proteom. MCP 2010, 9, 197–208. [Google Scholar] [CrossRef]
- Graner, M.; Redzic, J.; Ung, T. Glioblastoma extracellular vesicles: Reservoirs of potential biomarkers. Pharmacogenom. Pers. Med. 2014, 7, 65. [Google Scholar] [CrossRef]
- Figueroa, J.M.; Skog, J.; Akers, J.; Li, H.; Komotar, R.; Jensen, R.; Ringel, F.; Yang, I.; Kalkanis, S.; Thompson, R.; et al. Detection of wild-type EGFR amplification and EGFRvIII mutation in CSF-derived extracellular vesicles of glioblastoma patients. Neuro Oncol. 2017, 19, 1494–1502. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Jakobsen, K.R.; Paulsen, B.S.; Bæk, R.; Varming, K.; Sorensen, B.S.; Jørgensen, M.M. Exosomal proteins as potential diagnostic markers in advanced non-small cell lung carcinoma. J. Extracell. Vesicles 2015, 4. [Google Scholar] [CrossRef]
- Sandfeld-Paulsen, B.; Jakobsen, K.R.; Bæk, R.; Folkersen, B.H.; Rasmussen, T.R.; Meldgaard, P.; Varming, K.; Jørgensen, M.M.; Sorensen, B.S. Exosomal proteins as diagnostic biomarkers in lung cancer. J. Thorac. Oncol. 2016, 11, 1701–1710. [Google Scholar] [CrossRef] [PubMed]
- Sandfeld-Paulsen, B.; Aggerholm-Pedersen, N.; Bæk, R.; Jakobsen, K.R.; Jørgensen, M.M.; Sorensen, B.S. Exosomal proteins as prognostic biomarkers in non-small cell lung cancer. Mol. Oncol. 2016, 10, 1595–1602. [Google Scholar] [CrossRef] [PubMed]
- Okuma, Y.; Morikawa, K.; Tanaka, H.; Yokoyama, T.; Itani, H.; Horiuchi, K.; Nakagawa, H.; Takahashi, N.; Bessho, A.; Soejima, K.; et al. Prospective exosome-focused translational research for afatinib study of non-small cell lung cancer patients expressing EGFR (EXTRA study). Thorac. Cancer 2019, 10, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Russell, A.E.; Sneider, A.; Witwer, K.W.; Bergese, P.; Bhattacharyya, S.N.; Cocks, A.; Cocucci, E.; Erdbrügger, U.; Falcon-Perez, J.M.; Freeman, D.W.; et al. Biological membranes in EV biogenesis, stability, uptake, and cargo transfer: An ISEV position paper arising from the ISEV membranes and EVs workshop. J. Extracell. Vesicles 2019, 8. [Google Scholar] [CrossRef]
- Ramirez, M.I.; Amorim, M.G.; Gadelha, C.; Milic, I.; Welsh, J.A.; Freitas, V.M.; Nawaz, M.; Akbar, N.; Couch, Y.; Makin, L.; et al. Technical challenges of working with extracellular vesicles. Nanoscale 2018, 10, 881–906. [Google Scholar] [CrossRef]
- Willms, E.; Johansson, H.J.; Mäger, I.; Lee, Y.; Emelie Blomberg, K.M.; Sadik, M.; Alaarg, A.; Edvard Smith, C.; Lehtiö, J.; El Andaloussi, S.; et al. Cells release subpopulations of exosomes with distinct molecular and biological properties. Sci. Rep. 2016, 6, 22519. [Google Scholar] [CrossRef]
- Xu, R.; Greening, D.W.; Rai, A.; Ji, H.; Simpson, R.J. Highly-Purified exosomes and shed microvesicles isolated from the human colon cancer cell line LIM1863 by sequential centrifugal ultrafiltration are biochemically and functionally distinct. Methods 2015, 87, 11–25. [Google Scholar] [CrossRef]
- Kowal, J.; Arras, G.; Colombo, M.; Jouve, M.; Morath, J.P.; Primdal-Bengtson, B.; Dingli, F.; Loew, D.; Tkach, M.; Théry, C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA 2016, 113, E968–E977. [Google Scholar] [CrossRef]
- Ram, S.; Prabhat, P.; Chao, J.; Ward, E.S.; Ober, R.J. High accuracy 3D quantum dot tracking with multifocal plane microscopy for the study of fast intracellular dynamics in live cells. Biophys. J. 2008, 95, 6025–6043. [Google Scholar] [CrossRef]
- van den Broek, B.; Ashcroft, B.; Oosterkamp, T.H.; van Noort, J. Parallel nanometric 3D tracking of intracellular gold Nanorods using multifocal two-photon microscopy. Nano Lett. 2013, 13, 980–986. [Google Scholar] [CrossRef]
- Fiolka, R.; Shao, L.; Rego, E.H.; Davidson, M.W.; Gustafsson, M.G.L. Time-lapse two-color 3D imaging of live cells with doubled resolution using structured illumination. Proc. Natl. Acad. Sci. USA 2012, 3–7. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Liu, W.; Zhang, Z.; Zheng, C.; Huang, Y.; Cao, R.; Zhu, D.; Xu, L.; Zhang, M.; Zhang, Y.-H.; et al. Multi-color live-cell super-resolution volume imaging with multi-angle interference microscopy. Nat. Commun. 2018, 9, 4818. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, A.-K.; Petrov, P.N.; Lee, M.Y.; Shechtman, Y.; Moerner, W.E. 3D single-molecule super-resolution microscopy with a tilted light sheet. Nat. Commun. 2018, 9, 123. [Google Scholar] [CrossRef]
- Schöneberg, J.; Dambournet, D.; Liu, T.-L.; Forster, R.; Hockemeyer, D.; Betzig, E.; Drubin, D.G. 4D cell biology: Big data image analytics and lattice light-sheet imaging reveal dynamics of clathrin-mediated endocytosis in stem cell–derived intestinal organoids. Mol. Biol. Cell 2018, 29, 2959–2968. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.; Lou, J.; Coelho, S.; Lim, Y.J.; Seidlitz, S.; Nicovich, P.R.; Wunder, C.; Johannes, L.; Gaus, K. Imaging galectin-3 dependent endocytosis with lattice light-sheet microscopy. In Proceedings of the SPIE 10340 International Conference on Biophotonics V, Perth, Australia, 30 April–1 May 2017; p. 103400J. [Google Scholar] [CrossRef]
- Wäldchen, F.; Schlegel, J.; Götz, R.; Luciano, M.; Schnermann, M.; Doose, S.; Sauer, M. Whole-Cell imaging of plasma membrane receptors by 3D lattice light-sheet d STORM. Nat. Commun. 2020, 11, 887. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).