Involvement of NDPK-B in Glucose Metabolism-Mediated Endothelial Damage via Activation of the Hexosamine Biosynthesis Pathway and Suppression of O-GlcNAcase Activity

 ,
,  ,
,  and
and 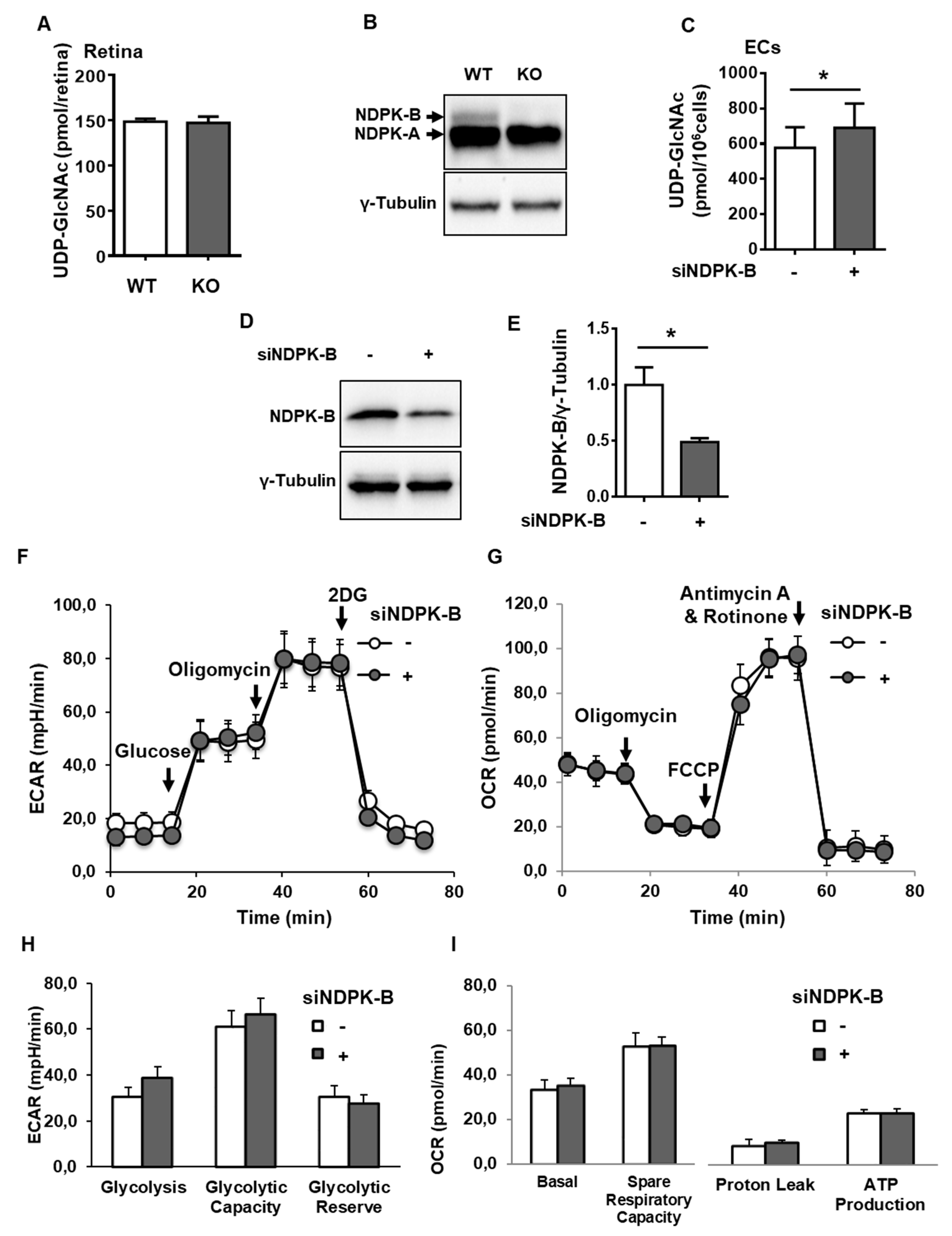{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials and Reagents
2.2. Animals
2.3. Cell Culture
2.4. siRNA-Mediated Gene Silencing
2.5. Determination of Nucleotide Content via UPLC-PDA
2.6. Measurement of Glycolysis and Mitochondrial Respiration
2.7. Protein Extraction and Immunoblotting
2.8. OGA Activity Assay
2.9. Quantitative PCR
2.10. s-WGA Pulldown Assay
2.11. Adenovirus-Mediated Re-Expression of NDPK-B
2.12. NDP Kinase Activity Assay
2.13. Statistical Analysis
3. Results
3.1. Loss of NDPK-B in ECs Causes an Activation of the HBP
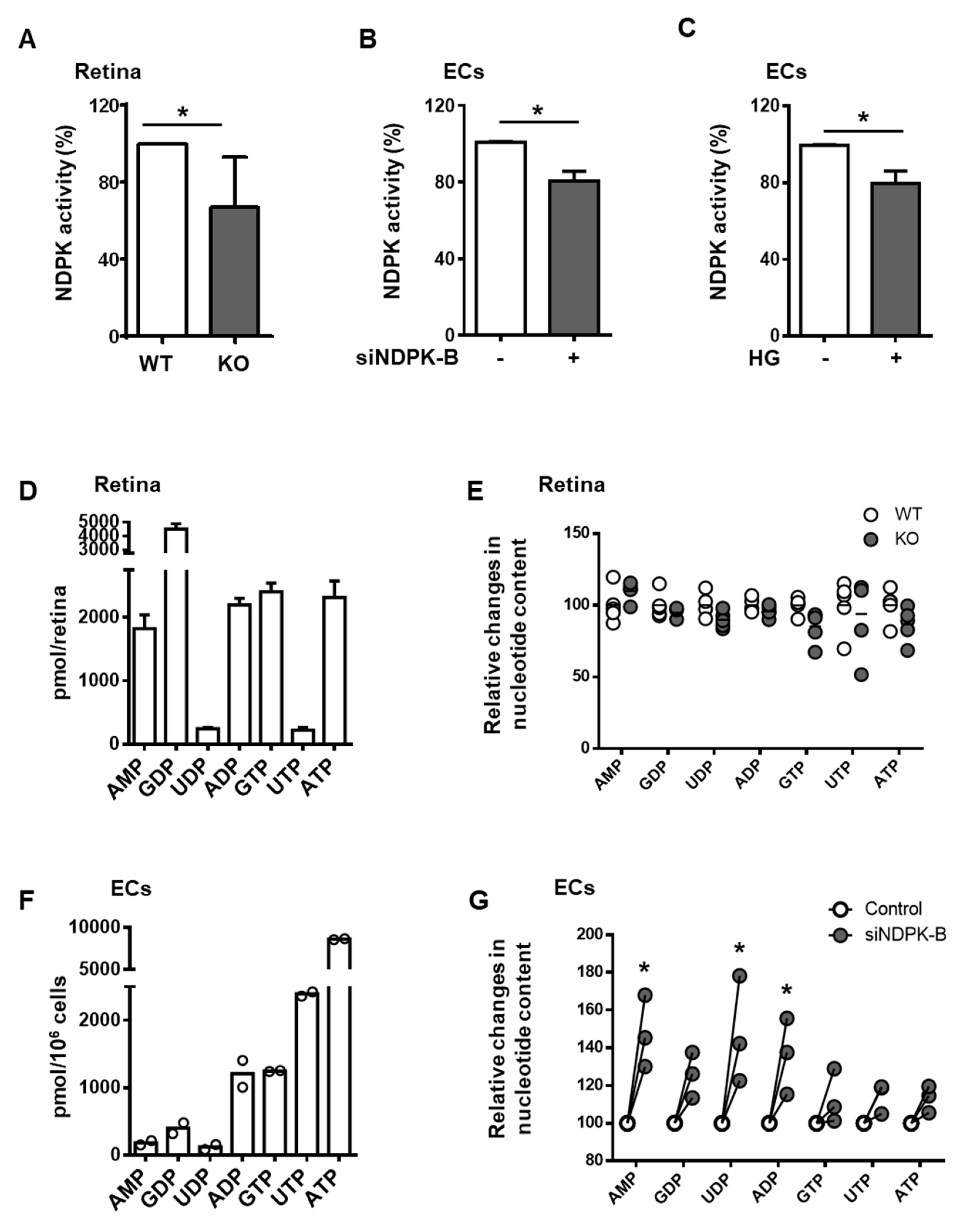
3.2. Loss of NDPK-B in ECs Alters Nucleotide Metabolism without Changing NTP Levels
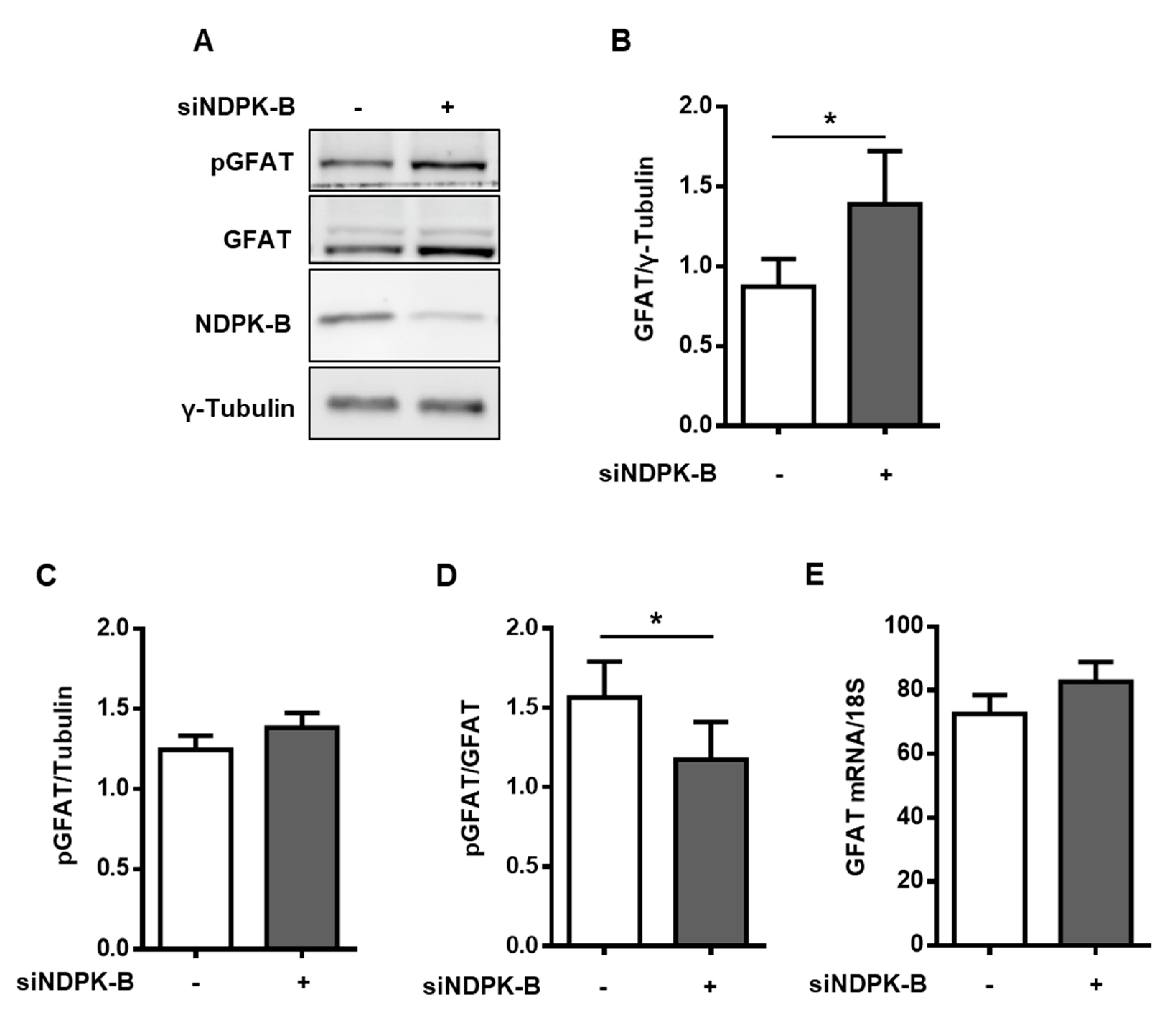
3.3. Loss of NDPK-B in ECs Increases GFAT Content and Activity
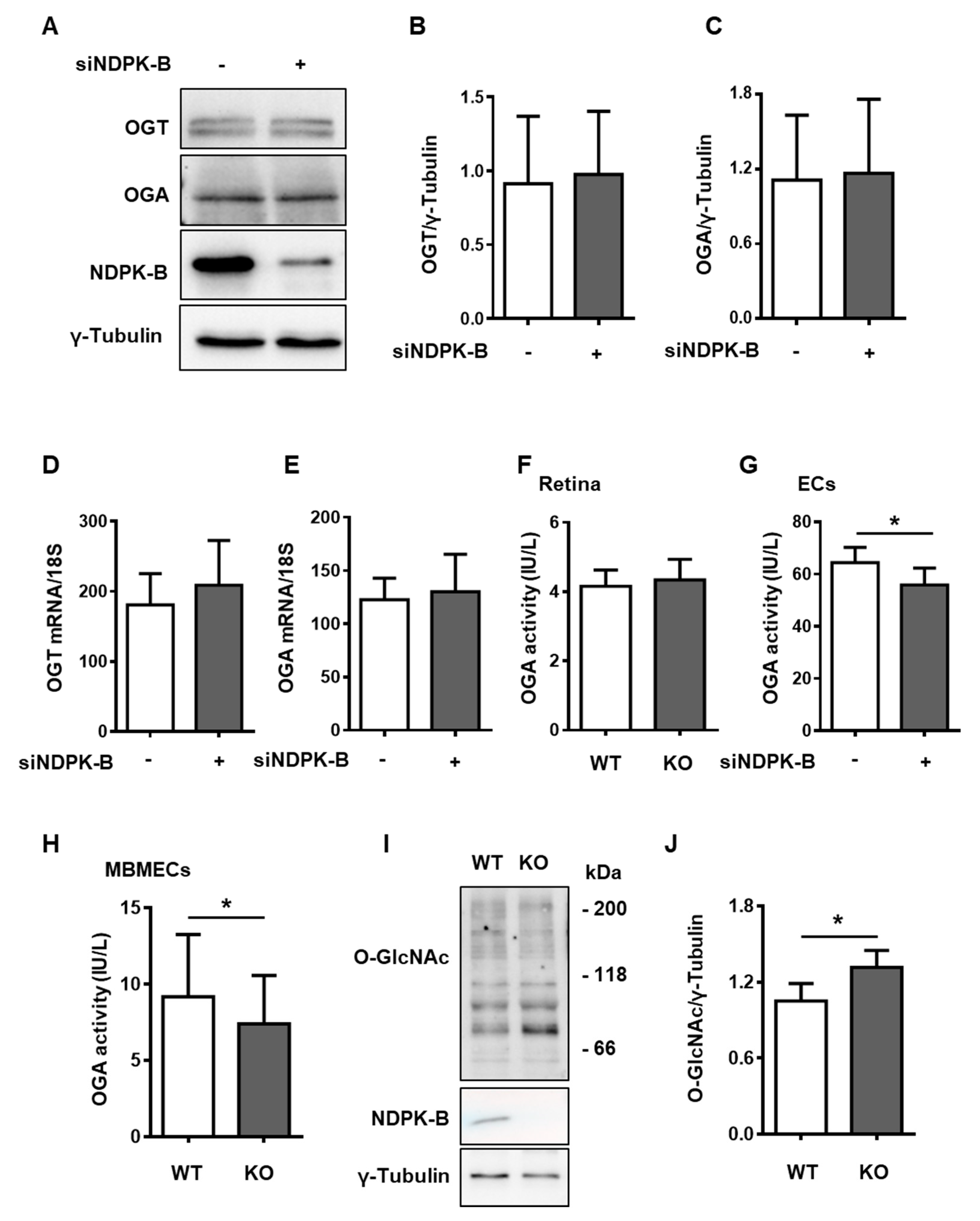
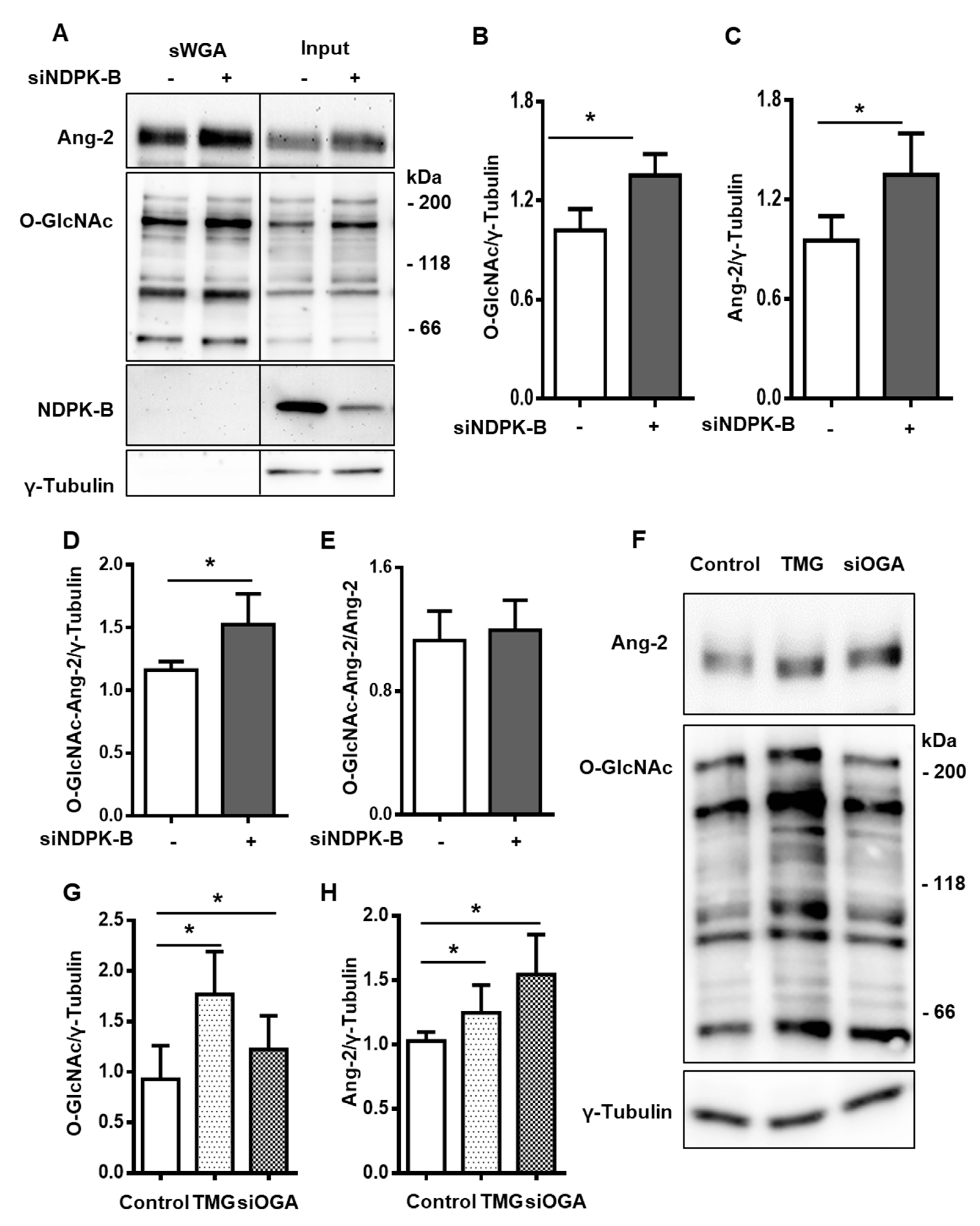
3.4. Reduced OGA Activity Enhances Protein O-GlcNAcylation in NDPK-B Depleted ECs
3.5. Enhanced Protein O-GlcNAcylation Increases Ang-2 Content
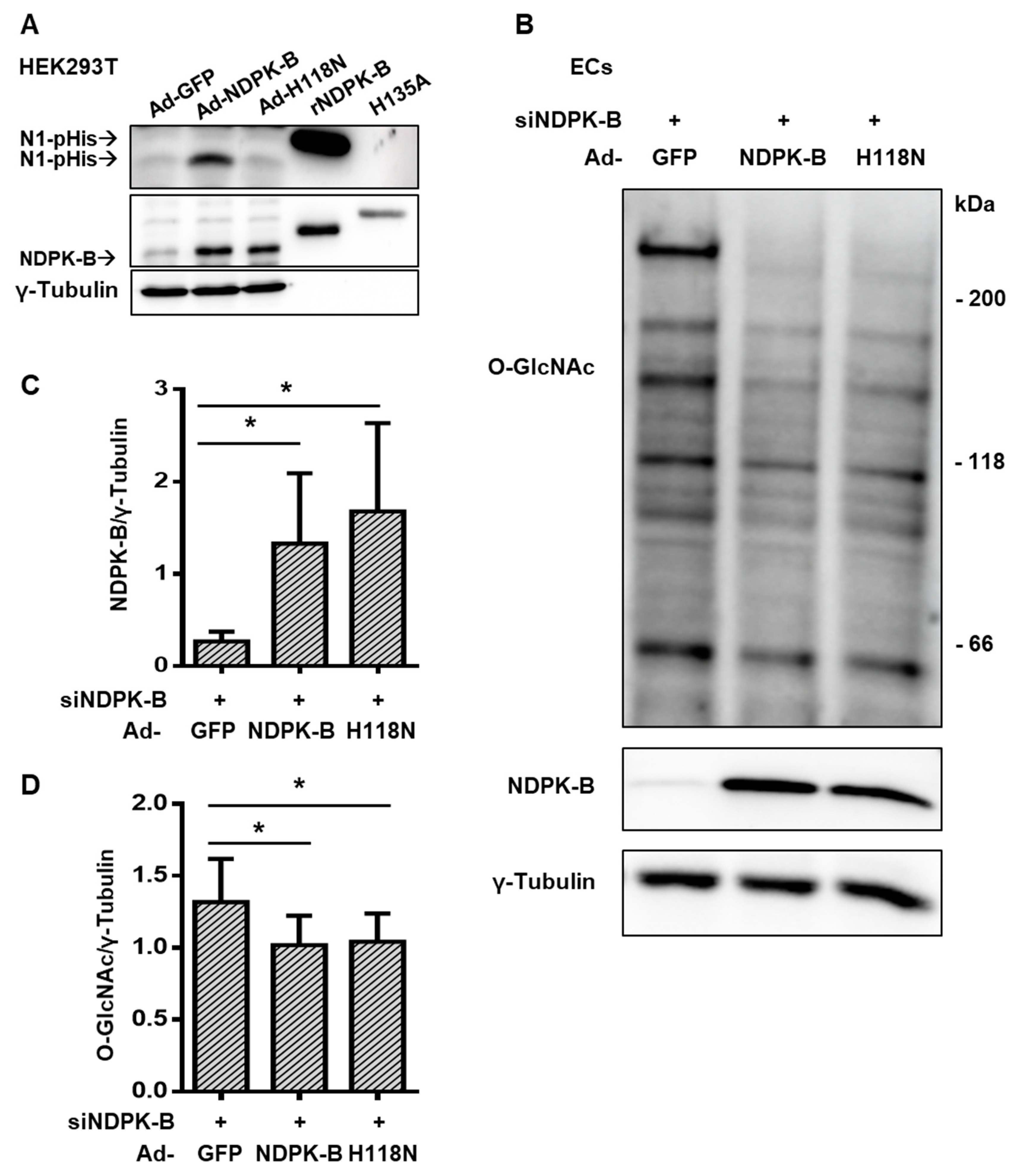
3.6. Regulation of Protein O-GlcNAcylation Is Independent of the Enzymatic NTP/NDP Transphosphorylase and Protein Histidine Activity of NDPK-B Kinase
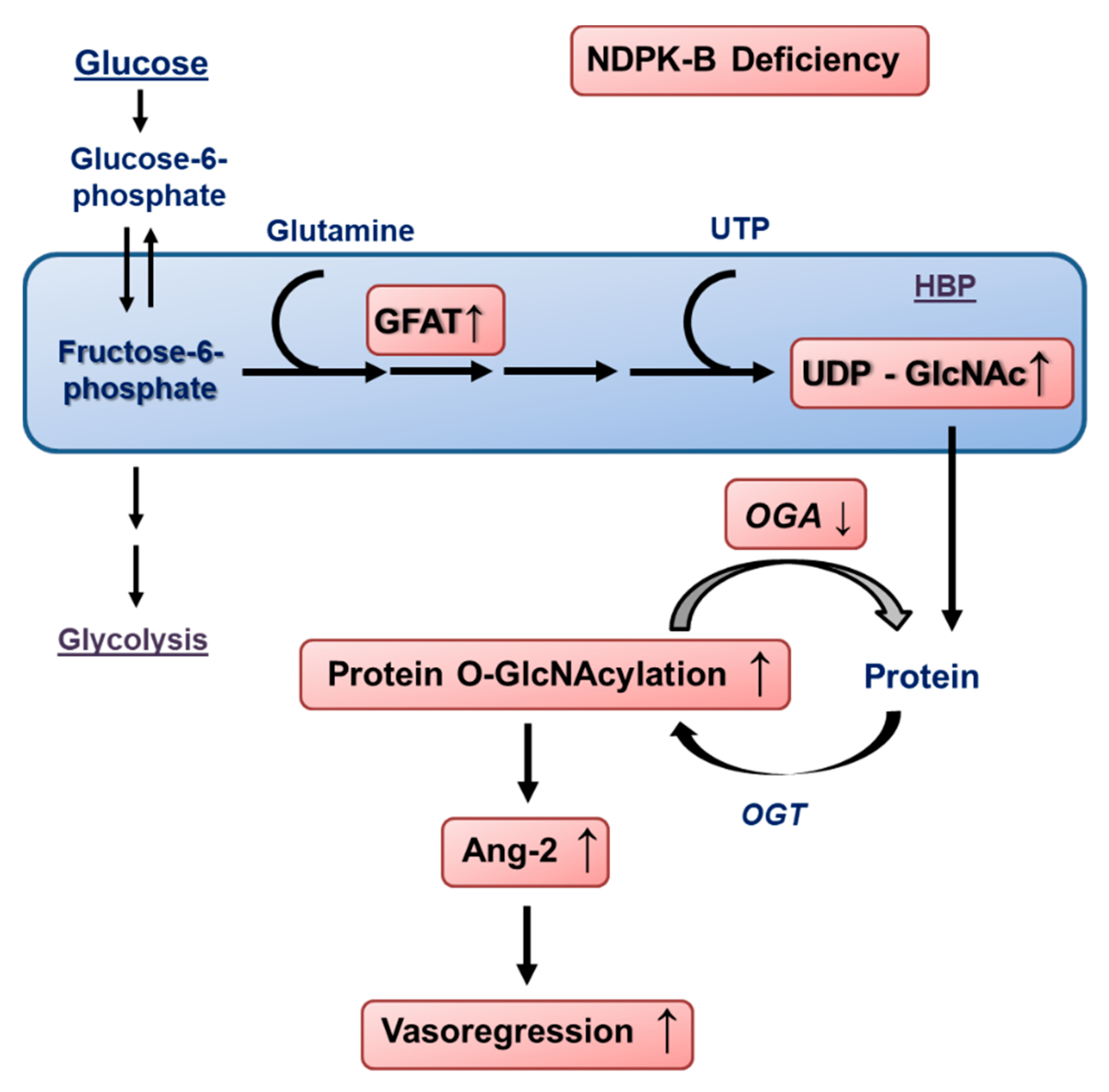
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dorfman, A.; Roseman, S.; Ludowieg, J.; Mayeda, M.; Moses, F.E.; Cifonelli, J.A. The biosynthesis of hyaluronic acid by group A Streptococcus. IV. Role of glucosone as an intermediate in the synthesis of glucosamine. J. Biol. Chem. 1955, 216, 549–552. [Google Scholar]
- Ghosh, S.; Blumenthal, H.J.; Davidson, E.; Roseman, S. Glucosamine metabolism. V. Enzymatic synthesis of glucosamine 6-phosphate. J. Biol. Chem. 1960, 235, 1265–1273. [Google Scholar]
- Marshall, S.; Bacote, V.; Traxinger, R.R. Complete inhibition of glucose-induced desensitization of the glucose transport system by inhibitors of mRNA synthesis. Evidence for rapid turnover of glutamine:fructose-6-phosphate amidotransferase. J. Biol. Chem. 1991, 266, 10155–10161. [Google Scholar]
- Broschat, K.O.; Gorka, C.; Page, J.D.; Martin-Berger, C.L.; Davies, M.S.; Huang Hc, H.C.; Gulve, E.A.; Salsgiver, W.J.; Kasten, T.P. Kinetic characterization of human glutamine-fructose-6-phosphate amidotransferase I: Potent feedback inhibition by glucosamine 6-phosphate. J. Biol. Chem. 2002, 277, 14764–14770. [Google Scholar] [CrossRef] [PubMed]
- Copeland, R.J.; Bullen, J.W.; Hart, G.W. Cross-talk between GlcNAcylation and phosphorylation: Roles in insulin resistance and glucose toxicity. Am. J. Physiol. Endocrinol. Metab. 2008, 295, E17–E28. [Google Scholar] [CrossRef] [PubMed]
- Janetzko, J.; Walker, S. The making of a sweet modification: Structure and function of O-GlcNAc transferase. J. Biol. Chem. 2014, 289, 34424–34432. [Google Scholar] [CrossRef] [PubMed]
- Du, X.L.; Edelstein, D.; Rossetti, L.; Fantus, I.G.; Goldberg, H.; Ziyadeh, F.; Wu, J.; Brownlee, M. Hyperglycemia-induced mitochondrial superoxide overproduction activates the hexosamine pathway and induces plasminogen activator inhibitor-1 expression by increasing Sp1 glycosylation. Proc. Natl. Acad. Sci. USA 2000, 97, 12222–12226. [Google Scholar] [CrossRef] [PubMed]
- Holt, G.D.; Hart, G.W. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J. Biol. Chem. 1986, 261, 8049–8057. [Google Scholar] [PubMed]
- Housley, M.P.; Rodgers, J.T.; Udeshi, N.D.; Kelly, T.J.; Shabanowitz, J.; Hunt, D.F.; Puigserver, P.; Hart, G.W. O-GlcNAc regulates FoxO activation in response to glucose. J. Biol. Chem. 2008, 283, 16283–16292. [Google Scholar] [CrossRef] [PubMed]
- Torres, C.R.; Hart, G.W. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. J. Biol. Chem. 1984, 259, 3308–3317. [Google Scholar] [PubMed]
- Yang, X.; Qian, K. Protein O-GlcNAcylation: Emerging mechanisms and functions. Nat. Rev. Mol. Cell. Biol. 2017, 18, 452–465. [Google Scholar] [CrossRef] [PubMed]
- Yao, D.; Taguchi, T.; Matsumura, T.; Pestell, R.; Edelstein, D.; Giardino, I.; Suske, G.; Rabbani, N.; Thornalley, P.J.; Sarthy, V.P.; et al. High glucose increases angiopoietin-2 transcription in microvascular endothelial cells through methylglyoxal modification of mSin3A. J. Biol. Chem. 2007, 282, 31038–31045. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Feng, Y.; Pfister, F.; Brownlee, M. Diabetic retinopathy: Targeting vasoregression. Diabetes 2011, 60, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Lin, J.; Wagner, P.; Feng, Y.; Vom Hagen, F.; Krzizok, T.; Renner, O.; Breier, G.; Brownlee, M.; Deutsch, U. Angiopoietin-2 causes pericyte dropout in the normal retina: Evidence for involvement in diabetic retinopathy. Diabetes 2004, 53, 1104–1110. [Google Scholar] [CrossRef]
- Pfister, F.; Wang, Y.; Schreiter, K.; vom Hagen, F.; Altvater, K.; Hoffmann, S.; Deutsch, U.; Hammes, H.P.; Feng, Y. Retinal overexpression of angiopoietin-2 mimics diabetic retinopathy and enhances vascular damages in hyperglycemia. Acta Diabetol. 2010, 47, 59–64. [Google Scholar] [CrossRef] [PubMed]
- Gurel, Z.; Sieg, K.M.; Shallow, K.D.; Sorenson, C.M.; Sheibani, N. Retinal O-linked N-acetylglucosamine protein modifications: Implications for postnatal retinal vascularization and the pathogenesis of diabetic retinopathy. Mol. Vis. 2013, 19, 1047–1059. [Google Scholar] [PubMed]
- Morera, S.; Chiadmi, M.; LeBras, G.; Lascu, I.; Janin, J. Mechanism of phosphate transfer by nucleoside diphosphate kinase: X-ray structures of the phosphohistidine intermediate of the enzymes from Drosophila and Dictyostelium. Biochemistry 1995, 34, 11062–11070. [Google Scholar] [CrossRef]
- Tepper, A.D.; Dammann, H.; Bominaar, A.A.; Veron, M. Investigation of the active site and the conformational stability of nucleoside diphosphate kinase by site-directed mutagenesis. J. Biol. Chem. 1994, 269, 32175–32180. [Google Scholar]
- Feng, Y.; Gross, S.; Wolf, N.M.; Butenschon, V.M.; Qiu, Y.; Devraj, K.; Liebner, S.; Kroll, J.; Skolnik, E.Y.; Hammes, H.P.; et al. Nucleoside diphosphate kinase B regulates angiogenesis through modulation of vascular endothelial growth factor receptor type 2 and endothelial adherens junction proteins. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2292–2300. [Google Scholar] [CrossRef]
- Gross, S.; Devraj, K.; Feng, Y.; Macas, J.; Liebner, S.; Wieland, T. Nucleoside diphosphate kinase B regulates angiogenic responses in the endothelium via caveolae formation and c-Src-mediated caveolin-1 phosphorylation. J. Cereb. Blood Flow Metab. 2017, 37, 2471–2484. [Google Scholar] [CrossRef]
- Qiu, Y.; Zhao, D.; Butenschon, V.M.; Bauer, A.T.; Schneider, S.W.; Skolnik, E.Y.; Hammes, H.P.; Wieland, T.; Feng, Y. Nucleoside diphosphate kinase B deficiency causes a diabetes-like vascular pathology via up-regulation of endothelial angiopoietin-2 in the retina. Acta Diabetol. 2016, 53, 81–89. [Google Scholar] [CrossRef]
- Shan, S.; Chatterjee, A.; Qiu, Y.; Hammes, H.P.; Wieland, T.; Feng, Y. O-GlcNAcylation of FoxO1 mediates nucleoside diphosphate kinase B deficiency induced endothelial damage. Sci. Rep. 2018, 8, 10581. [Google Scholar] [CrossRef] [PubMed]
- Di, L.; Srivastava, S.; Zhdanova, O.; Sun, Y.; Li, Z.; Skolnik, E.Y. Nucleoside diphosphate kinase B knock-out mice have impaired activation of the K+ channel KCa3.1, resulting in defective T cell activation. J. Biol. Chem. 2010, 285, 38765–38771. [Google Scholar] [CrossRef] [PubMed]
- Kroll, J.; Epting, D.; Kern, K.; Dietz, C.T.; Feng, Y.; Hammes, H.P.; Wieland, T.; Augustin, H.G. Inhibition of Rho-dependent kinases ROCK I/II activates VEGF-driven retinal neovascularization and sprouting angiogenesis. Am. J. Physiol. Heart Circ. Physiol. 2009, 296, H893–H899. [Google Scholar] [CrossRef] [PubMed]
- Kochanowski, N.; Blanchard, F.; Cacan, R.; Chirat, F.; Guedon, E.; Marc, A.; Goergen, J.L. Intracellular nucleotide and nucleotide sugar contents of cultured CHO cells determined by a fast, sensitive, and high-resolution ion-pair RP-HPLC. Anal. Biochem. 2006, 348, 243–251. [Google Scholar] [CrossRef]
- Krabbendam, I.E.; Honrath, B.; Dilberger, B.; Iannetti, E.F.; Branicky, R.S.; Meyer, T.; Evers, B.; Dekker, F.J.; Koopman, W.J.H.; Beyrath, J.; et al. SK channel-mediated metabolic escape to glycolysis inhibits ferroptosis and supports stress resistance in C. elegans. Cell Death Dis. 2020, 11, 263. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Eshwaran, R.; Huang, H.; Zhao, D.; Schmidt, M.; Wieland, T.; Feng, Y. Role of the Ang2-Tie2 Axis in Vascular Damage Driven by High Glucose or Nucleoside Diphosphate Kinase B Deficiency. Int. J. Mol. Sci. 2020, 21, 3713. [Google Scholar] [CrossRef] [PubMed]
- Hammes, H.P.; Du, X.; Edelstein, D.; Taguchi, T.; Matsumura, T.; Ju, Q.; Lin, J.; Bierhaus, A.; Nawroth, P.; Hannak, D.; et al. Benfotiamine blocks three major pathways of hyperglycemic damage and prevents experimental diabetic retinopathy. Nat. Med. 2003, 9, 294–299. [Google Scholar] [CrossRef]
- Arshavsky, V.Y.; Lamb, T.D.; Pugh, E.N., Jr. G proteins and phototransduction. Annu. Rev. Physiol. 2002, 64, 153–187. [Google Scholar] [CrossRef]
- Eguchi, S.; Oshiro, N.; Miyamoto, T.; Yoshino, K.; Okamoto, S.; Ono, T.; Kikkawa, U.; Yonezawa, K. AMP-activated protein kinase phosphorylates glutamine: Fructose-6-phosphate amidotransferase 1 at Ser243 to modulate its enzymatic activity. Genes Cells 2009, 14, 179–189. [Google Scholar] [CrossRef]
- Zibrova, D.; Vandermoere, F.; Goransson, O.; Peggie, M.; Marino, K.V.; Knierim, A.; Spengler, K.; Weigert, C.; Viollet, B.; Morrice, N.A.; et al. GFAT1 phosphorylation by AMPK promotes VEGF-induced angiogenesis. Biochem. J. 2017, 474, 983–1001. [Google Scholar] [CrossRef] [PubMed]
- Fuhs, S.R.; Hunter, T. pHisphorylation: The emergence of histidine phosphorylation as a reversible regulatory modification. Curr. Opin. Cell. Biol. 2017, 45, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Potente, M.; Urbich, C.; Sasaki, K.; Hofmann, W.K.; Heeschen, C.; Aicher, A.; Kollipara, R.; DePinho, R.A.; Zeiher, A.M.; Dimmeler, S. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Investig. 2005, 115, 2382–2392. [Google Scholar] [CrossRef]
- Ruan, H.B.; Nie, Y.; Yang, X. Regulation of protein degradation by O-GlcNAcylation: Crosstalk with ubiquitination. Mol. Cell Proteom. 2013, 12, 3489–3497. [Google Scholar] [CrossRef] [PubMed]
- Qiu, Y.; Huang, H.; Chatterjee, A.; Teuma, L.D.; Baumann, F.S.; Hammes, H.-P.; Wieland, T.; Feng, Y.J.N. Mediation of FoxO1 in Activated Neuroglia Deficient for Nucleoside Diphosphate Kinase B during Vascular Degeneration. Neuroglia 2018, 1, 280–291. [Google Scholar] [CrossRef]
- Potente, M.; Carmeliet, P. The Link Between Angiogenesis and Endothelial Metabolism. Annu. Rev. Physiol. 2017, 79, 43–66. [Google Scholar] [CrossRef]
- Hawkins, M.; Angelov, I.; Liu, R.; Barzilai, N.; Rossetti, L. The tissue concentration of UDP-N-acetylglucosamine modulates the stimulatory effect of insulin on skeletal muscle glucose uptake. J. Biol. Chem. 1997, 272, 4889–4895. [Google Scholar] [CrossRef]
- Abu-Taha, I.H.; Heijman, J.; Hippe, H.J.; Wolf, N.M.; El-Armouche, A.; Nikolaev, V.O.; Schafer, M.; Wurtz, C.M.; Neef, S.; Voigt, N.; et al. Nucleoside Diphosphate Kinase-C Suppresses cAMP Formation in Human Heart Failure. Circulation 2017, 135, 881–897. [Google Scholar] [CrossRef]
- Chen, C.W.; Wang, H.L.; Huang, C.W.; Huang, C.Y.; Lim, W.K.; Tu, I.C.; Koorapati, A.; Hsieh, S.T.; Kan, H.W.; Tzeng, S.R.; et al. Two separate functions of NME3 critical for cell survival underlie a neurodegenerative disorder. Proc. Natl. Acad. Sci. USA 2019, 116, 566–574. [Google Scholar] [CrossRef]
- Hippe, H.J.; Abu-Taha, I.; Wolf, N.M.; Katus, H.A.; Wieland, T. Through scaffolding and catalytic actions nucleoside diphosphate kinase B differentially regulates basal and beta-adrenoceptor-stimulated cAMP synthesis. Cell Signal. 2011, 23, 579–585. [Google Scholar] [CrossRef]
- Hippe, H.J.; Wolf, N.M.; Abu-Taha, I.; Mehringer, R.; Just, S.; Lutz, S.; Niroomand, F.; Postel, E.H.; Katus, H.A.; Rottbauer, W.; et al. The interaction of nucleoside diphosphate kinase B with Gbetagamma dimers controls heterotrimeric G protein function. Proc. Natl. Acad. Sci. USA 2009, 106, 16269–16274. [Google Scholar] [CrossRef] [PubMed]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chatterjee, A.; Eshwaran, R.; Poschet, G.; Lomada, S.; Halawa, M.; Wilhelm, K.; Schmidt, M.; Hammes, H.-P.; Wieland, T.; Feng, Y. Involvement of NDPK-B in Glucose Metabolism-Mediated Endothelial Damage via Activation of the Hexosamine Biosynthesis Pathway and Suppression of O-GlcNAcase Activity. Cells 2020, 9, 2324. https://doi.org/10.3390/cells9102324
Chatterjee A, Eshwaran R, Poschet G, Lomada S, Halawa M, Wilhelm K, Schmidt M, Hammes H-P, Wieland T, Feng Y. Involvement of NDPK-B in Glucose Metabolism-Mediated Endothelial Damage via Activation of the Hexosamine Biosynthesis Pathway and Suppression of O-GlcNAcase Activity. Cells. 2020; 9(10):2324. https://doi.org/10.3390/cells9102324
Chicago/Turabian StyleChatterjee, Anupriya, Rachana Eshwaran, Gernot Poschet, Santosh Lomada, Mahmoud Halawa, Kerstin Wilhelm, Martina Schmidt, Hans-Peter Hammes, Thomas Wieland, and Yuxi Feng. 2020. "Involvement of NDPK-B in Glucose Metabolism-Mediated Endothelial Damage via Activation of the Hexosamine Biosynthesis Pathway and Suppression of O-GlcNAcase Activity" Cells 9, no. 10: 2324. https://doi.org/10.3390/cells9102324
APA StyleChatterjee, A., Eshwaran, R., Poschet, G., Lomada, S., Halawa, M., Wilhelm, K., Schmidt, M., Hammes, H.-P., Wieland, T., & Feng, Y. (2020). Involvement of NDPK-B in Glucose Metabolism-Mediated Endothelial Damage via Activation of the Hexosamine Biosynthesis Pathway and Suppression of O-GlcNAcase Activity. Cells, 9(10), 2324. https://doi.org/10.3390/cells9102324