Laron Syndrome Research Paves the Way for New Insights in Oncological Investigation





Abstract
1. The Growth Hormone-Insulin-Like Growth Factor 1 Endocrine Axis
2. The GH-IGF1 Axis and Growth Retardation
3. IGF1: A Validated Cancer Risk Factor
4. Laron Syndrome: A Prototypical Case of Congenital IGF1 Deficiency
5. Laron Syndrome and Cancer Protection: Epidemiological Data
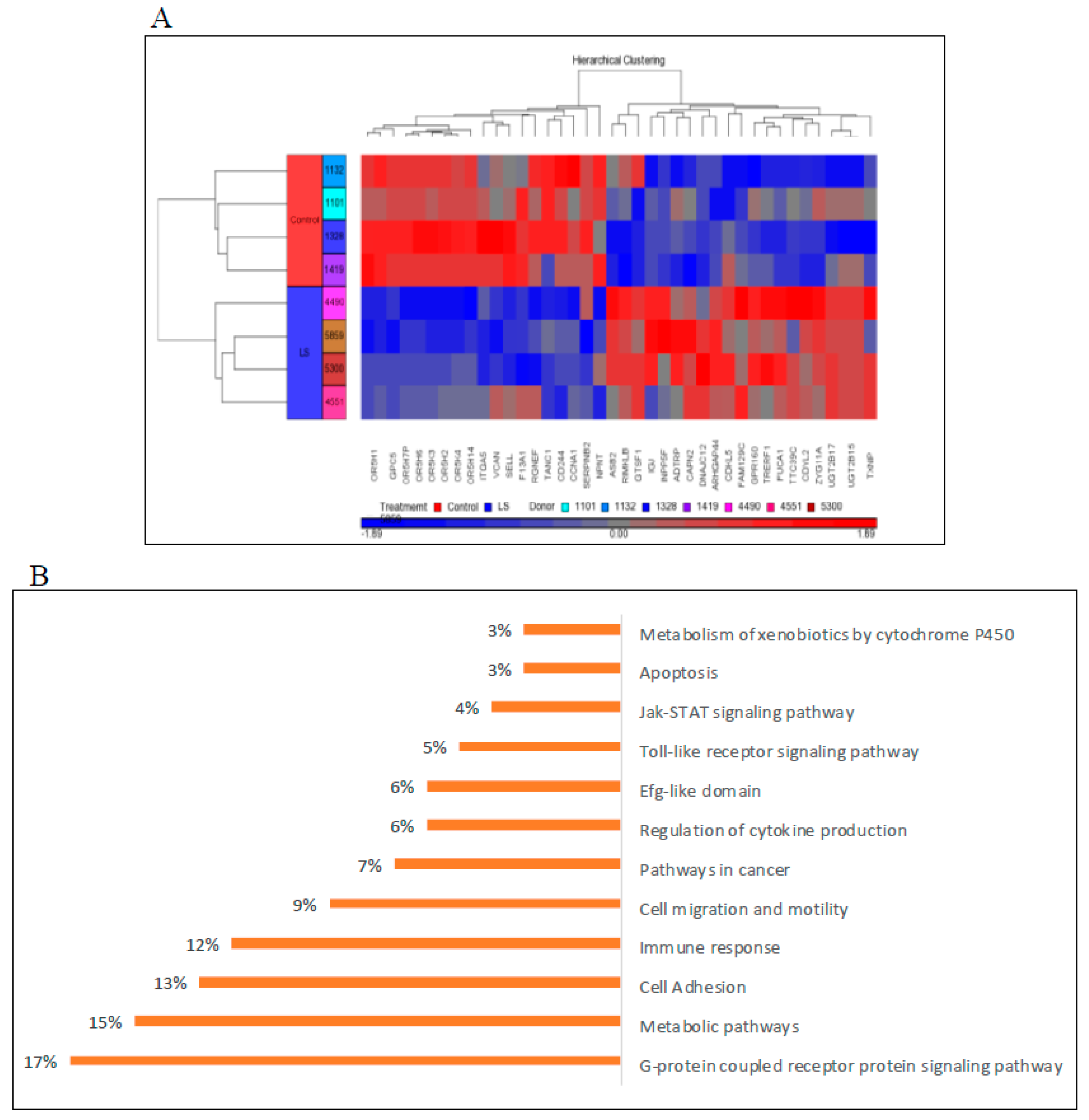
6. Genome-Wide Profiling of Laron Syndrome Patients
7. Bioinformatic Analyses Identify New IGF1 Target Genes and Signaling Pathways
8. LS Cells Display Altered Mitogenic Properties
- (i)
- What are the mitogenic properties of LS-derived cells?
- (ii)
- Are the different phases of the cell cycle altered in LS?
- (iii)
- What is the apoptotic index of LS cells?
- (iv)
- Do LS cells respond differently to oxidative stress?
- (v)
- Is autophagic machinery involved in cancer evasion in LS?
9. Identification of Thioredoxin-Interacting Protein as a New Target for IGF1 Action
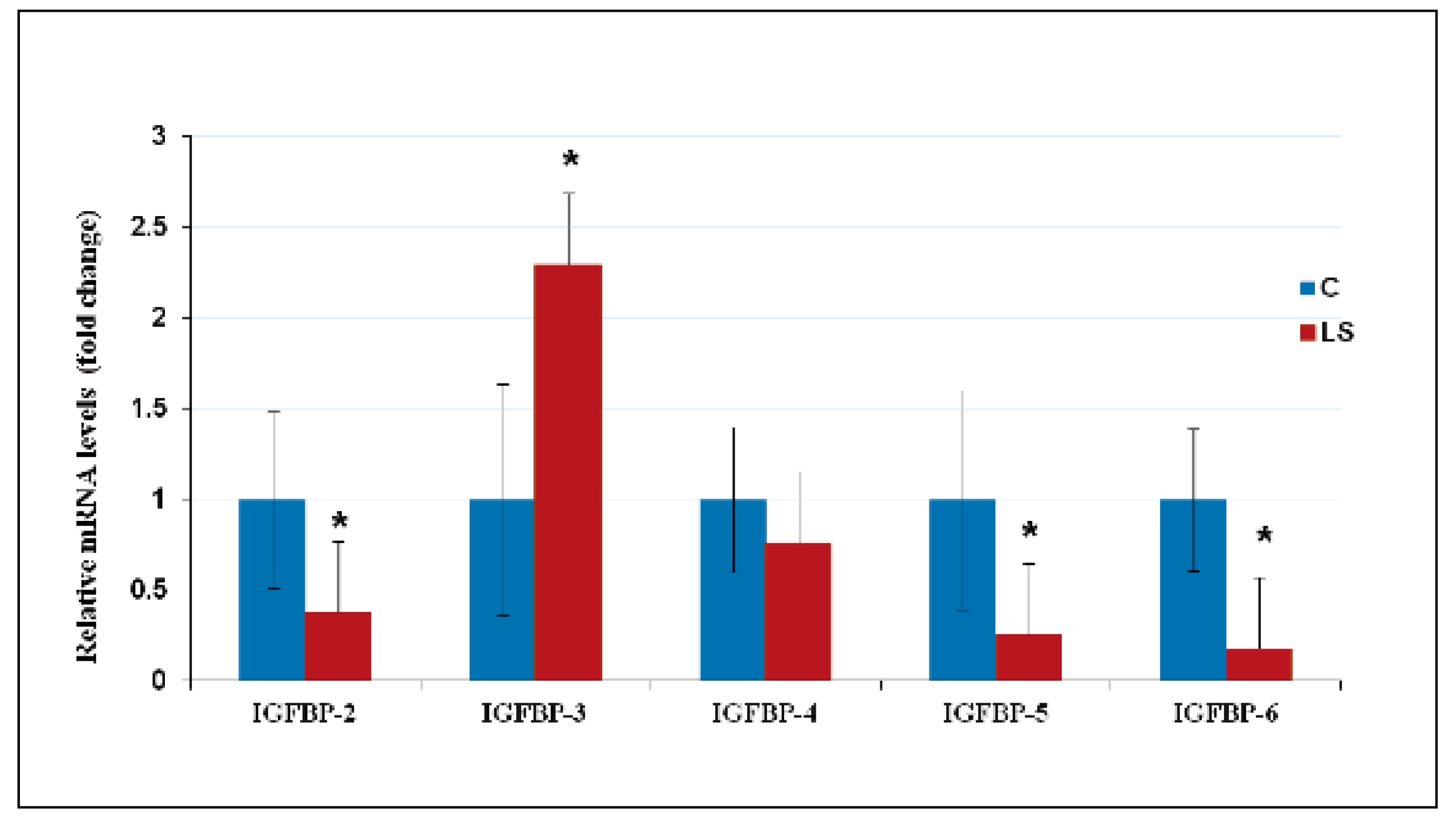
10. Deregulated Expression of IGFBPs in LS Contributes to Cancer Protection
11. Interactions between Tumor Suppressor p53- and IGF1-Signaling Pathways
12. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- LeRoith, D.; Yakar, S. Mechanisms of disease: Metabolic effects of growth hormone and insulin-like growth factor-1. Nat. Clin. Pract. Endocrinol. Metab. 2007, 3, 302–310. [Google Scholar] [CrossRef]
- Yakar, S.; Adamo, M.L. Insulin-like growth factor 1 physiology: Lessons from mouse models. Endocrinol. Metab. Clin. N. Am. 2012, 41, 231–247. [Google Scholar] [CrossRef]
- Salmon, W.D.; Daughaday, W.H. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J. Lab. Clin. Med. 1957, 49, 825–836. [Google Scholar] [PubMed]
- Bentov, I.; Werner, H. Insulin-like growth factors and breast cancer. Curr. Med. Lit.-Breast Cancer 2009, 21, 113–120. [Google Scholar]
- Longo, V.; Finch, C. Evolutionary medicine: From dwarf model systems to healthy centenarians? Science 2003, 299, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Bentov, I.; Werner, H. IGF, IGF receptor and overgrowth syndromes. Pediatr. Endocrinol. Rev. 2004, 1, 352–360. [Google Scholar]
- Baserga, R.; Peruzzi, F.; Reiss, K. The IGF-1 receptor in cancer biology. Int. J. Cancer 2003, 107, 873–877. [Google Scholar] [CrossRef]
- Lu, K.; Campisi, J. Ras proteins are essential and selective for the action of insulin-like growth factor 1 late in the G1 phase of the cell cycle in BALB/c murine fibroblasts. Proc. Natl. Acad. Sci. USA 1992, 89, 3889–3893. [Google Scholar] [CrossRef]
- Moschos, S.J.; Mantzoros, C.S. The role of the IGF system in cancer: From basic to clinical studies and clinical applications. Oncology 2002, 63, 317–332. [Google Scholar] [CrossRef]
- Werner, H. Tumor suppressors govern insulin-like growth factor signaling pathways: Implications in metabolism and cancer. Oncogene 2012, 31, 2703–2714. [Google Scholar] [CrossRef] [PubMed]
- Baserga, R. The contradictions of the insulin-like growth factor 1 receptor. Oncogene 2000, 19, 5574–5581. [Google Scholar] [CrossRef]
- Werner, H.; Bruchim, I. The insulin-like growth factor-I receptor as an oncogene. Arch. Physiol. Biochem. 2009, 115, 58–71. [Google Scholar] [CrossRef]
- Resnicoff, M.; Burgaud, J.-L.; Rotman, H.L.; Abraham, D.; Baserga, R. Correlation between apoptosis, tumorigenesis, and levels of insulin-like growth factor I receptors. Cancer Res. 1995, 55, 3739–3741. [Google Scholar]
- Chitnis, M.M.; Yuen, J.S.P.; Protheroe, A.S.; Pollak, M.; Macaulay, V.M. The type I insulin-like growth factor-I receptor pathway. Clin. Cancer Res. 2008, 14, 6364–6370. [Google Scholar] [CrossRef]
- Werner, H.; Shalita-Chesner, M.; Abramovitch, S.; Idelman, G.; Shaharabani-Gargir, L.; Glaser, T. Regulation of the insulin-like growth factor-I receptor gene by oncogenes and antioncogenes: Implications in human cancer. Mol. Gen. Metab. 2000, 71, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Maor, S.; Reizner, N.; Abramovitch, S.; Werner, H. Transcriptional regulation of the insulin-like growth factor-1 receptor in breast cancer. Mol. Cell. Endocrinol. 2006, 252, 241–246. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Biernacka, K.; Perks, C.M. The neglected insulin: IGF-II, a metabolic regulator with implications for diabetes, obesity, and cancer. Cells 2019, 8, 1207. [Google Scholar] [CrossRef]
- Kessler, S.M.; Haybaeck, J.; Kiemer, A.K. Insulin-like growth factor 2—The oncogene and its accomplices. Curr. Pharm. Des. 2016, 22, 5948–5961. [Google Scholar] [CrossRef]
- Bach, L. Insulin-like growth factor binding proteins—An update. Pediatr. Endocrinol. Rev. 2015, 13, 521–530. [Google Scholar]
- Baxter, R.C. IGF binding proteins in cancer: Mechanistic and clinical insights. Nat. Rev. Cancer 2014, 14, 329–341. [Google Scholar] [CrossRef] [PubMed]
- Klammt, J.; Pfaffle, R.; Werner, H.; Kiess, W. IGF signaling defects as causes of growth failure and IUGR. Trends Endocrinol. Metab. 2008, 19, 197–205. [Google Scholar] [CrossRef]
- Rosenfeld, R.G. The molecular basis of idiophatic short stature. Growth Horm. IGF Res. 2005, 15 (Suppl. A), S3–S5. [Google Scholar] [CrossRef]
- Solomon-Zemler, R.; Basel-Vanagaite, L.; Steier, D.; Yakar, S.; Mel, E.; Phillip, M.; Bazak, L.; Bercovich, D.; Werner, H.; de Vries, L. A novel heterozygous IGF-1 receptor mutation associated with hypoglycemia. Endocr. Connect. 2017, 6, 395–403. [Google Scholar] [CrossRef]
- Cohen, P.; Rogol, A.D.; Deal, C.L.; Saenger, P.; Reiter, E.O.; Ross, J.L.; Chernausek, S.D.; Savage, M.O.; Wit, J.M. 2007 ISS Consensus Workshop participants. Consensus statement on the diagnosis and treatment of children with idiopathis short stature: A summary of the Growth Hormone Research Society, The Lawson Wilkins Pediatric Endocrine Society and the European Society for Pediatric Endocrinology Workshop. J. Clin. Endocrinol. Metab. 2008, 93, 4210–4217. [Google Scholar]
- Woods, K.A.; Camacho-Hubner, C.; Savage, M.O.; Clark, A.J.L. Intrauterine growth retardation and postnatal growth failure associated with deletion of the insulin-like growth factor I gene. N. Engl. J. Med. 1996, 335, 1363–1367. [Google Scholar] [CrossRef]
- Domene, H.; Bengolea, S.V.; Martinez, A.S.; Ropelato, M.G.; Pennisi, P.; Scaglia, P.; Heinrich, J.; Jasper, H. Deficiency of the circulating IGF system associated with inactivation of the acid-labile subunit gene. N. Engl. J. Med. 2004, 350, 570–577. [Google Scholar] [CrossRef]
- Domené, S.; Domené, H.M. Genetic mutations in the GH/IGF axis. Pediatr. Endocrinol. Rev. 2018, 16, 39–62. [Google Scholar]
- Kofoed, E.M.; Hwa, V.; Little, B.; Woods, K.A.; Buckway, C.K.; Tsubaki, J.; Pratt, K.L.; Bezrodni, L.; Jasper, H.; Tepper, A.; et al. Growth hormone insensitivity associated with a STAT5b mutation. N. Engl. J. Med. 2003, 349, 1139–1147. [Google Scholar] [CrossRef] [PubMed]
- Argente, J.; Chowen, J.A.; Perez-Jurada, L.A.; Frystyk, J.; Oxvig, C. One level up: Abnormal proteolytic regulation of IGF activity plays a role in human pathophysiology. EMBO Mol. Med. 2017, 9, 1338–1345. [Google Scholar] [CrossRef]
- Dauber, A.; Muñoz-Calvo, M.T.; Barrios, V.; Domené, H.M.; Kloverpris, S.; Serra-Juhé, C.; Desikan, V.; Pozo, J.; Muzumdar, R.; Martos-Moreno, G.Á.; et al. Mutations in pregnancy-associated plasma protein A2 cause short stature due to low IGF-I availability. EMBO Mol. Med. 2016, 8, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.; Liu, J.-P.; Robertson, E.J.; Efstratiadis, A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell 1993, 75, 73–82. [Google Scholar] [CrossRef]
- Liu, J.-P.; Baker, J.; Perkins, A.S.; Robertson, E.J.; Estratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 1993, 75, 59–72. [Google Scholar] [CrossRef]
- Lupu, F.; Terwilliger, J.D.; Lee, K.; Segre, G.V.; Efstratiadis, A. Roles of growth hormone and insulin-like growth factor 1 in mouse postnatal growth. Dev. Biol. 2001, 229, 141–162. [Google Scholar] [CrossRef]
- Yakar, S.; Liu, J.L.; Stannard, B.; Butler, A.; Accili, D.; Sauer, B.; LeRoith, D. Normal growth and development in the absence of hepatic insulin-like growth factor I. Proc. Natl. Acad. Sci. USA 1999, 96, 7324–7329. [Google Scholar] [CrossRef]
- List, E.O.; Berryman, D.E.; Funk, K.; Jara, A.; Kelder, B.; Wang, F.; Stout, M.B.; Zhi, X.; Sun, L.; White, T.A.; et al. Liver-specific GH receptor gene-disrupted (LiGHRKO) mice have decreased endocrine IGF-I, increased local IGF-I, and altered body size, body composition, and adipokine profiles. Endocrinology 2014, 155, 1793–1805. [Google Scholar] [CrossRef] [PubMed]
- Mancarella, C.; Scotlandi, K. IGF system in sarcomas: A crucial pathway with many unknowns to exploit for therapy. J. Mol. Endocrinol. 2018, 61, T45–T60. [Google Scholar] [CrossRef]
- Hankinson, S.E.; Willett, W.C.; Colditz, G.A.; Hunter, D.J.; Michaud, D.S.; Deroo, B.; Rosner, B.; Speizer, F.E.; Pollak, M. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet 1998, 351, 1393–1396. [Google Scholar] [CrossRef]
- Chan, J.M.; Stampfer, M.J.; Giovannucci, E.; Gann, P.H.; Ma, J.; Wilkinson, P.; Hennekens, C.H.; Pollak, M. Plasma insulin-like growth factor-I and prostate cancer risk: A prospective study. Science 1998, 279, 563–566. [Google Scholar] [CrossRef]
- Ma, J.; Pollack, M.N.; Giovannucci, E.; Chan, J.M.; Tao, Y.; Hennekens, C.H.; Stampfer, M.J. Prospective study of colorectal cancer risk in men and plasma levels of insulin-like growth factor (IGF)-I and IGF-binding protein-3. J. Natl. Cancer Inst. 1999, 91, 620–625. [Google Scholar] [CrossRef]
- Holly, J.M.P.; Gunnell, D.J.; Davey Smith, G. Growth hormone, IGF-I and cancer. Less intervention to avoid cancer? More intervention to prevent cancer? J. Endocrinol. 1999, 162, 321–330. [Google Scholar] [CrossRef][Green Version]
- Cianfarani, S.; Tedeschi, B.; Germani, D.; Prete, S.P.; Rossi, P.; Vernole, P.; Caporossi, D.; Boscherini, B. In vitro effects of growth hormone (GH) and insulin-like growth factor I and II (IGF-I and -II) on chromosome fragility and p53 protein expression in human lymphocytes. Eur. J. Clin. Investig. 1998, 28, 41–47. [Google Scholar] [CrossRef]
- Cohen, S.M.; Ellwein, L.B. Cell proliferation in carcinogenesis. Science 1990, 249, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. Lessons from 50 years of study of Laron syndrome. Endocr. Pract. 2015, 21, 1395–1402. [Google Scholar] [CrossRef]
- Laron, Z. Extensive personal experience. Laron syndrome (primary growth hormone resistance or insensitivity): The personal experience 1958–2003. J. Clin. Endocrinol. Metab. 2004, 89, 1031–1044. [Google Scholar] [CrossRef]
- Laron, Z.; Kopchik, J.J. Laron Syndrome—From Man to Mouse; Springer: Berlin/Heidelberg, Germany, 2011; pp. 1–525. [Google Scholar]
- Werner, H.; Lapkina-Gendler, L.; Laron, Z. Fifty years on: New lessons from Laron syndrome. Isr. Med. Assoc. J. 2017, 19, 6–7. [Google Scholar]
- Laron, Z.; Pertzelan, A.; Mannheimer, S. Genetic pituitary dwarfism with high serum concentration of growth hormone-a new inborn error of metabolism? Isr. J. Med. Sci. 1966, 2, 152–155. [Google Scholar]
- Rosenfeld, R.G.; Rosenbloom, A.L.; Guevara-Aguirre, J. Growth hormone insensitivity due to primary GH receptor deficiency. Endocr. Rev. 1994, 15, 369–390. [Google Scholar] [CrossRef]
- Glick, S.M.; Roth, J.; Yallow, R.S.; Berson, S.A. Immunoassay of human growth hormone in plasma. Nature 1963, 199, 784–788. [Google Scholar] [CrossRef]
- Eshet, R.; Laron, Z.; Brown, M.; Arnon, R. Immunoreactive properties of the plasma hGH from patients with the syndrome of familial dwarfism with high plasma IR-hGH. J. Clin. Endocrinol. Metab. 1973, 37, 819–821. [Google Scholar] [CrossRef]
- Eshet, R.; Laron, Z.; Pertzelan, A.; Arnon, R.; Dintzman, M. Defects of human growth hormone receptors in the liver of two patients with Laron-type dwarfism. Isr. J. Med. Sci. 1984, 20, 8–11. [Google Scholar]
- Godowski, P.J.; Leung, D.W.; Meacham, L.R.; Galgani, J.P.; Hellmiss, R.; Keret, R.; Rotwein, P.S.; Parks, J.S.; Laron, Z.; Wood, W.I. Characterization of the human growth hormone receptor gene and demonstration of a partial gene deletion in two patients with Laron-type dwarfism. Proc. Natl. Acad. Sci. USA 1989, 86, 8083–8087. [Google Scholar] [CrossRef]
- Amselem, S.; Duquesnoy, P.; Attree, O.; Novelli, G.; Bousnina, S.; Postel-Vinay, M.C.; Goossens, M. Laron dwarfism and mutations of the growth hormone-receptor gene. N. Engl. J. Med. 1989, 321, 989–995. [Google Scholar] [CrossRef]
- Shevah, O.; Laron, Z. Genetic analysis of the pedigrees and molecular defects of the GH-receptor gene in the Israeli cohort of patients with Laron syndrome. Pediatr. Endocrinol. Rev. 2006, 3 (Suppl. 3), 489–497. [Google Scholar]
- Rosenbloom, A.L.; Guevara-Aguirre, J. Lessons from the genetics of Laron syndrome. Trends Endocrinol. Metab. 1998, 9, 276–283. [Google Scholar] [CrossRef]
- Goncalves, F.T.; Fridman, C.; Pinto, E.M.; Guevara-Aguirre, J.; Shevah, O.; Rosenbloom, A.L.; Hwa, V.; Cassorla, F.; Rosenfeld, R.G.; Lins, T.S.S.; et al. The E180 splice mutation in the GHR gene causing Laron syndrome: Witness of a Sephardic Jewish exodus from the Iberian Peninsula to the New World? Am. J. Med. Genet. 2014, 164, 1204–1208. [Google Scholar] [CrossRef]
- Zhou, Y.; Xu, B.C.; Maheshwari, H.G.; He, L.; Reed, M.; Lozykowski, M.; Okada, S.; Cataldo, L.; Coschigamo, K.; Wagner, T.E.; et al. A mammalian model for Laron syndrome produced by targeted disruption of the mouse growth hormone receptor/binding protein gene (the Laron mouse). Proc. Natl. Acad. Sci. USA 1997, 94, 13215–13220. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Rosenbloom, A.L.; Vasconez, O.; Martinez, V.; Gargosky, S.E.; Allen, L.; Rosenfeld, R.G. Two year treatment of GH receptor deficiency with recombinant IGF-I in 22 children: Comparison of two dosage levels and to GH treated GH deficiency. J. Clin. Endocrinol. Metab. 1997, 82, 629–633. [Google Scholar]
- Backeljauw, P.F.; Underwood, L.E.; GHIS Collaborative Group. Therapy for 6.5–7.5 years with recombinant IGF-I in children with growth hormone insensitivity syndrome: A clinical research center study. J. Clin. Endocrinol. Metab. 2001, 86, 1504–1510. [Google Scholar]
- Laron, Z.; Klinger, B. Comparison of the growth promoting effects of IGF-I and growth hormone in the early years of life. Acta Paediatr. 2000, 88, 38–41. [Google Scholar] [CrossRef]
- Werner, H.; Lapkina-Gendler, L.; Nagaraj, K.; Sarfstein, R.; Laron, Z. Genome-wide profiling of congenital IGF1 deficient patients: Translational implications in cancer prevention and metabolism. Transl. Med. Rep. 2017, 1, 6657. [Google Scholar] [CrossRef]
- Shevah, O.; Laron, Z. Patients with congenital deficiency of IGF-I seem protected from the development of malignancies: A preliminary report. Growth Horm. IGF Res. 2007, 17, 54–57. [Google Scholar] [CrossRef] [PubMed]
- Steuerman, R.; Shevah, O.; Laron, Z. Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur. J. Endocrinol. 2011, 164, 485–489. [Google Scholar] [CrossRef]
- Guevara-Aguirre, J.; Balasubramanian, P.; Guevara-Aguirre, M.; Wei, M.; Madia, F.; Cheng, C.W.; Hwang, D.; Martin-Montalvo, A.; Saavedra, J.; Ingles, S.; et al. Growth hormone receptor deficiency is associated with a major reduction in pro-aging signaling, cancer, and diabetes in humans. Sci. Transl. Med. 2011, 3, 70ra13. [Google Scholar] [CrossRef]
- Wang, Z.; Prins, G.S.; Coschigano, K.T.; Kopchick, J.J.; Green, J.E.; Ray, V.H.; Hedayat, S.; Christov, K.T.; Unterman, T.G.; Swanson, S.M. Disruption of growth hormone signaling retards early stages of prostate carcinogenesis in the C3(1)/T antigen mouse. Endocrinology 2005, 146, 5188–5196. [Google Scholar] [CrossRef]
- Moore, T.; Carbajal, S.; Beltran, L.; Perkins, S.N.; Yakar, S.; LeRoith, D.; Hursting, S.D.; Digiovanni, J. Reduced susceptibility to two-stage skin carcinogenesis in mice with low circulating insulin-like growth factor-I levels. Cancer Res. 2008, 68, 3680–3688. [Google Scholar] [CrossRef]
- Lapkina-Gendler, L.; Rotem, I.; Pasmanik-Chor, M.; Gurwitz, D.; Sarfstein, R.; Laron, Z.; Werner, H. Identification of signaling pathways associated with cancer protection in Laron syndrome. Endocr. Relat. Cancer 2016, 23, 399–410. [Google Scholar] [CrossRef]
- Morag, A.; Kirchheiner, J.; Rehavi, M.; Gurwitz, D. Human lymphoblastoid cell line panels: Novel tools for assessing shared drug pathways. Pharmacogenomics 2010, 11, 327–340. [Google Scholar] [CrossRef]
- Vidal, A.C.; Tucker, C.; Schildkraut, J.M.; Richardson, R.M.; McPhail, M.; Freedland, S.J.; Hoyo, C.; Grant, D.J. Novel associations of UDP-glucuronosyltransferase 2B gene variants with prostate cancer risk in a multiethnic study. BMC Cancer 2013, 13, 556. [Google Scholar] [CrossRef]
- Zhang, H.; Basit, A.; Busch, D.; Yabut, K.; Bhatt, D.K.; Drozdzik, M.; Ostrowski, M.; Li, A.; Collins, C.; Oswald, S.; et al. Quantitative characterization of UDP-glucuronosyltransferase 2B17 in human liver and intestine and its role in testosterone first-pass metabolism. Biochem. Pharmacol. 2018, 156, 32–42. [Google Scholar] [CrossRef]
- Achlaug, L.; Sarfstein, R.; Nagaraj, K.; Lapkina-Gendler, L.; Bruchim, I.; Dixit, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of ZYG11A as a candidate IGF1-dependent proto-oncogene in endometrial cancer. Oncotarget 2019, 10, 4437–4448. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Lapkina-Gendler, L.; Nagaraj, K.; Laron, Z.; Werner, H. Identification of nephronectin as a new target for IGF1 action. Eur. J. Cancer 2020, 141, 115–127. [Google Scholar] [CrossRef]
- Rosenfeldt, M.T.; Ryan, K.M. The multiple roles of autophagy in cancer. Carcinogenesis 2011, 32, 955–963. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Yoshimori, T. How to interpret LC3 immunoblotting. Autophagy 2007, 3, 542–545. [Google Scholar] [CrossRef]
- Moscat, J.; Diaz-Meco, M.T. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell 2009, 137, 1001–1004. [Google Scholar] [CrossRef]
- Patwari, P.; Higgins, L.J.; Chutkow, W.A.; Yoshioka, J.; Lee, R.T. The interaction of thioredoxin with Txnip: Evidence for formation of a mixed disulfide by disulfide exchange. J. Biol. Chem. 2006, 281, 21884–21891. [Google Scholar] [CrossRef]
- Chen, K.S.; DeLuca, H.F. Isolation and characterization of a novel cDNA from HL-60 cells treated with 1,25-dihydroxyvitamin D-3. Biochim. Biophys. Acta Gene Struct. Expr. 1994, 1219, 26–32. [Google Scholar] [CrossRef]
- Takeuchi, J.; Hirota, K.; Itoh, T.; Shinkura, R.; Kitada, K.; Yodoi, J.; Namba, T.; Fukuda, K. Thioredoxin inhibits tumor necrosis factor- or interleukin-1-induced NF-kappaB activation at a level upstream of NF-kappaB-inducing kinase. Antioxid. Redox Signal. 2000, 2, 83–92. [Google Scholar] [CrossRef]
- Shah, A.; Xia, L.; Goldberg, H.; Lee, K.W.; Quaggin, S.E.; Fantus, I.G. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the NADPH oxidase, Nox4, in mesangial cells. J. Biol. Chem. 2013, 288, 6835–6848. [Google Scholar] [CrossRef]
- Junn, E.; Han, S.H.; Im, J.Y.; Yang, Y.; Cho, E.W.; Um, H.D.; Kim, D.K.; Lee, K.W.; Han, P.L.; Rhee, S.G.; et al. Vitamin D3 up-regulated protein 1 mediates oxidative stress via suppressing the thioredoxin function. J. Immunol. 2000, 164, 6287–6295. [Google Scholar] [CrossRef]
- Nagaraj, K.; Lapkina-Gendler, L.; Sarfstein, R.; Gurwitz, D.; Pasmanik-Chor, M.; Laron, Z.; Yakar, S.; Werner, H. Identification of thioredoxin-interacting protein (TXNIP) as a downstream target for IGF1 action. Proc. Natl. Acad. Sci. USA 2018, 115, 1045–1050. [Google Scholar] [CrossRef]
- Spindel, O.N.; World, C.; Berk, B.C. Thioredoxin interacting protein: Redox dependent and independent regulatory mechanisms. Antioxid. Redox Signal. 2012, 16, 587–596. [Google Scholar] [CrossRef]
- Thielen, L.A.; Chen, J.; Jing, G.; Moukha-Chafiq, O.; Xu, G.; Jo, S.-H.; Grayson, T.B.; Lu, B.; Li, P.; Augelli-Szafran, C.E.; et al. Identification of an anti-diabetic, orally available small molecule that regulates TXNIP expression and glucagon action. Cell Metab. 2020, 32, 353–365. [Google Scholar] [CrossRef]
- Somri, L.; Sarfstein, R.; Lapkina-Gendler, L.; Nagaraj, K.; Laron, Z.; Bach, L.A.; Werner, H. Differential expression of IGFBPs in Laron syndrome-derived lymphoblastoid cell lines: Potential correlation with reduced cancer incidence. Growth Horm. IGF Res. 2018, 39, 6–12. [Google Scholar] [CrossRef]
- Hettmer, S.; Dannecker, L.; Foell, J.; Elmlinger, M.; Dannecker, G. Effects of insulin-like growth factors and insulin-like growth factor binding protein-2 on the in vitro proliferation of peripheral blood mononuclear cells. Hum. Immunol. 2005, 66, 95–103. [Google Scholar] [CrossRef]
- Yasuoka, H.; Yamaguchi, Y.; Feghali-Bostwick, C.A. The pro-fibrotic factor IGFBP-5 induces lung fibroblast and mononuclear cell migration. Am. J. Respir. Cell. Mol. Biol. 2009, 41, 179–188. [Google Scholar] [CrossRef]
- Liso, A.; Castellani, S.; Massenzio, F.; Trotta, R.; Pucciarini, A.; Bigerna, B.; De Luca, P.; Zoppoli, P.; Castiglione, F.; Palumbo, M.C.; et al. Human monocyte-derived dendritic cells exposed to hyperthermia show a distinct gene expression profile and selective upregulation of IGFBP6. Oncotarget 2017, 8, 60826–60840. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Lee, K.; Anzo, M.; Zhang, B.; Zi, X.; Tao, Y.; Shiry, L.; Pollak, M.; Lin, S.; Cohen, P. Insulin-like growth factor-binding protein-3 inhibition of prostate cancer growth involves suppression of angiogenesis. Oncogene 2007, 26, 1811–1819. [Google Scholar] [CrossRef] [PubMed]
- Fielder, P.J.; Guevara-Aguirre, J.; Rosenbloom, A.L.; Carlsson, L.; Hintz, R.L.; Rosenfeld, R.G. Expression of serum insulin-like growth factors, insulin-like growth factor-binding proteins, and the growth hormone-binding protein in heterozygote relatives of Ecuadorian growth hormone receptor deficient patients. J. Clin. Endocrinol. Metab. 1992, 74, 743–750. [Google Scholar] [CrossRef]
- Cotterill, A.M.; Holly, J.M.P.; Taylor, A.M.; Davies, S.C.; Coulson, V.J.; Preece, M.A.; Wass, J.A.H.; Savage, M.O. The insulin-like growth factor (IGF)-binding proteins and IGF bioactivity in Laron-type dwarfism. J. Clin. Endocrinol. Metab. 1992, 74, 56–63. [Google Scholar]
- Kanety, H.; Karasik, A.; Klinger, B.; Silbergeld, A.; Laron, Z. Long-term treatment of Laron type dwarfs with insulin-like growth factor-1 increases serum IGF-binding protein-3 in the absence of growth hormone activity. Acta Endocrinol. 1993, 128, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Laron, Z. IGF-I binding proteins in Laron syndrome. In Laron Syndrome: From Man to Mouse; Laron, Z., Kopchik, J., Eds.; Springer: Berlin/Heidelberg, Germany, 2011. [Google Scholar]
- Werner, H.; Sarfstein, R.; LeRoith, D.; Bruchim, I. Insulin-like growth factor 1 signaling axis meets p53 genome protection pathways. Front. Oncol. 2016, 6, 159. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Kruiswijk, F.; Labuschagne, C.F.; Vousden, K.H. p53 in survival, death and metabolic health: A lifeguard with a licence to kill. Nat. Rev. Mol. Cell Biol. 2015, 16, 393–405. [Google Scholar] [CrossRef]
- Abramovitch, S.; Werner, H. Functional and physical interactions between BRCA1 and p53 in transcriptional regulation of the IGF-IR gene. Horm. Metab. Res. 2003, 35, 758–762. [Google Scholar] [PubMed]
- Attias-Geva, Z.; Bentov, I.; Kidron, D.; Amichay, K.; Sarfstein, R.; Fishman, A.; Bruchim, I.; Werner, H. p53 regulates insulin-like growth factor-I receptor gene expression in uterine serous carcinoma and predicts responsiveness to an insulin-like growth factor-I receptor-directed targeted therapy. Eur. J. Cancer 2012, 48, 1570–1580. [Google Scholar] [CrossRef]
- Werner, H.; Karnieli, E.; Rauscher, F.J., III; LeRoith, D. Wild type and mutant p53 differentially regulate transcription of the insulin-like growth factor I receptor gene. Proc. Natl. Acad. Sci. USA 1996, 93, 8318–8323. [Google Scholar] [CrossRef]
- Nahor, I.; Abramovitch, S.; Engeland, K.; Werner, H. The p53-family members p63 and p73 inhibit insulin-like growth factor-I receptor gene expression in colon cancer cells. Growth Horm. IGF Res. 2005, 15, 388–396. [Google Scholar] [CrossRef]
- Heron-Milhavet, L.; LeRoith, D. Insulin-like growth factor I induces MDM2-dependent degradation of p53 via the p38 MAPK pathway in response to DNA damage. J. Biol. Chem. 2002, 277, 15600–15606. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Girnita, A.; Larsson, O. Mdm2-dependent ubiquitination and degradation of the insulin-like growth factor-I receptor. Proc. Natl. Acad. Sci. USA 2003, 100, 8247–8252. [Google Scholar] [CrossRef] [PubMed]
- Girnita, L.; Girnita, A.; Brodin, B.; Xie, Y.; Nilsson, G.; Dricu, A.; Lundeberg, J.; Wejde, J.; Bartolazzi, A.; Wiman, K.G.; et al. Increased expression of insulin-like growth factor I receptor in malignant cells expressing aberrant p53: Functional impact. Cancer Res. 2000, 60, 5278–5283. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laron Syndrome | First-Degree Relatives | Further Relatives | Sibilings Only | |
|---|---|---|---|---|
| Total number (n) | 230 | 218 | 113 | 86 |
| Malignancies (number of events) | 0 | 18 | 25 | 5 |
| Prevalence of malignancy (%) | 0 | 8.3 | 22.1 | 5.8 |
| p-value (versus LS) | <0.001 | <0.001 | 0.005 |
| Genes up-Regulated in LS | Biological Role |
|---|---|
| UDP glucuronosyltransferase 2 family | Elimination of xenobiotic substances |
| G-protein coupled receptor | Signaling |
| Thioredoxin interacting protein | Metabolic regulation |
| ZYG11A | Cell cycle regulator |
| CAPN2 | Extracellular matrix disassembly |
| Genes down-regulated in LS | Biological role |
| Cyclin A1 | Cell cycle |
| AKT3 | Apoptosis |
| Versican | Extracellular matrix proteoglycan |
| Olfactory receptor | Detection of odor molecules |
| Nephronectin | Cell adhesion |
| Serpin B2 | Apoptosis and proliferation |
| Sp1 | Transcription factor |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Werner, H.; Sarfstein, R.; Nagaraj, K.; Laron, Z. Laron Syndrome Research Paves the Way for New Insights in Oncological Investigation. Cells 2020, 9, 2446. https://doi.org/10.3390/cells9112446
Werner H, Sarfstein R, Nagaraj K, Laron Z. Laron Syndrome Research Paves the Way for New Insights in Oncological Investigation. Cells. 2020; 9(11):2446. https://doi.org/10.3390/cells9112446
Chicago/Turabian StyleWerner, Haim, Rive Sarfstein, Karthik Nagaraj, and Zvi Laron. 2020. "Laron Syndrome Research Paves the Way for New Insights in Oncological Investigation" Cells 9, no. 11: 2446. https://doi.org/10.3390/cells9112446
APA StyleWerner, H., Sarfstein, R., Nagaraj, K., & Laron, Z. (2020). Laron Syndrome Research Paves the Way for New Insights in Oncological Investigation. Cells, 9(11), 2446. https://doi.org/10.3390/cells9112446