Aprotinin Inhibits SARS-CoV-2 Replication

 , ,
, ,  , , , , , and
, , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Drugs
2.2. Cell Culture
2.3. Virus Infection
2.4. Antiviral Assay
2.5. Viability Assay
2.6. Immunostaining for SARS-CoV-2 S Protein
2.7. Caspase 3/7 Activation
2.8. qPCR
2.9. Western Blot
2.10. Sample Preparation for LC–MS
2.11. Targeted Analysis by SPS–MS3
2.12. Data Analysis
2.13. Data Availability
3. Results
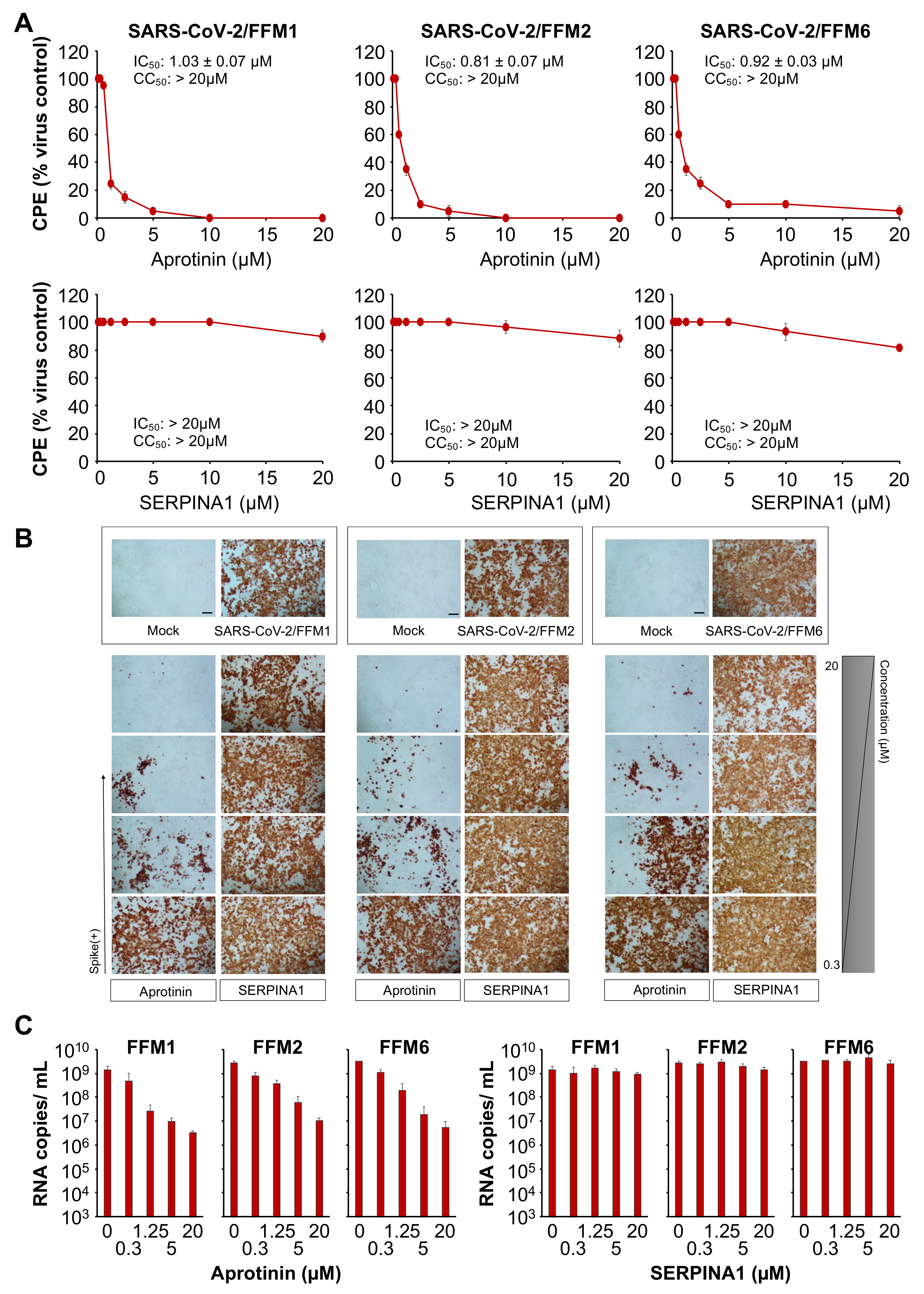
3.1. The Protease Inhibitor Aprotinin Exerts Superior Anti-SARS-CoV-2 Activity Relative to the Endogenous Protease Inhibitor SERPINA1/alpha-1 Antitrypsin
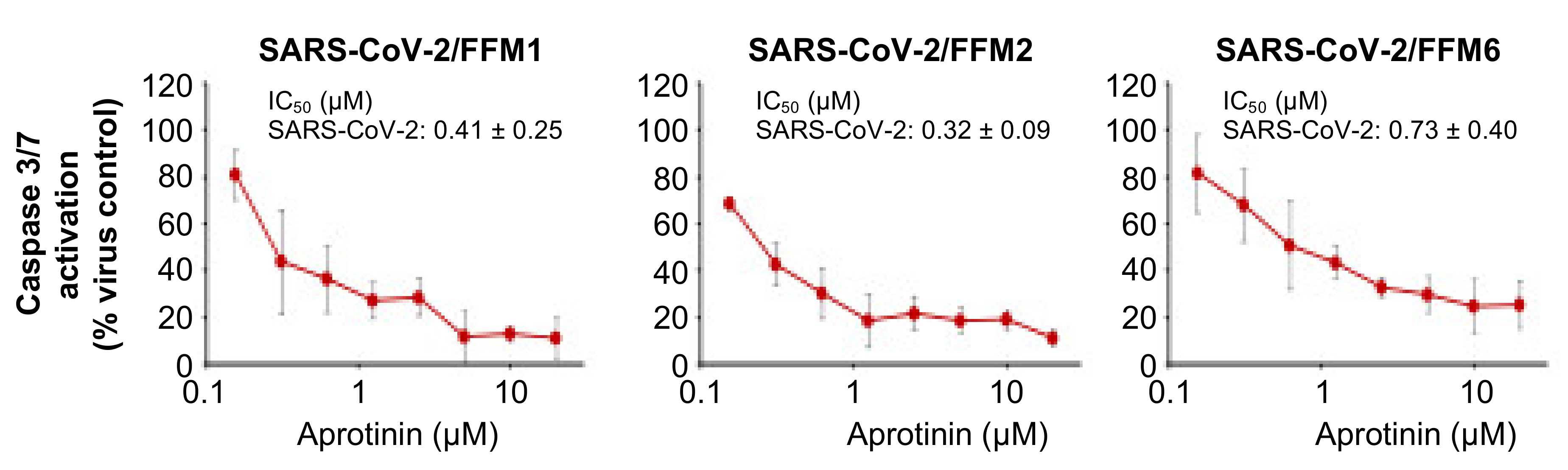
3.2. Quantification of the Antiviral Effects of Aprotinin by Measuring SARS-CoV-2-Induced Caspase 3/7 Activation
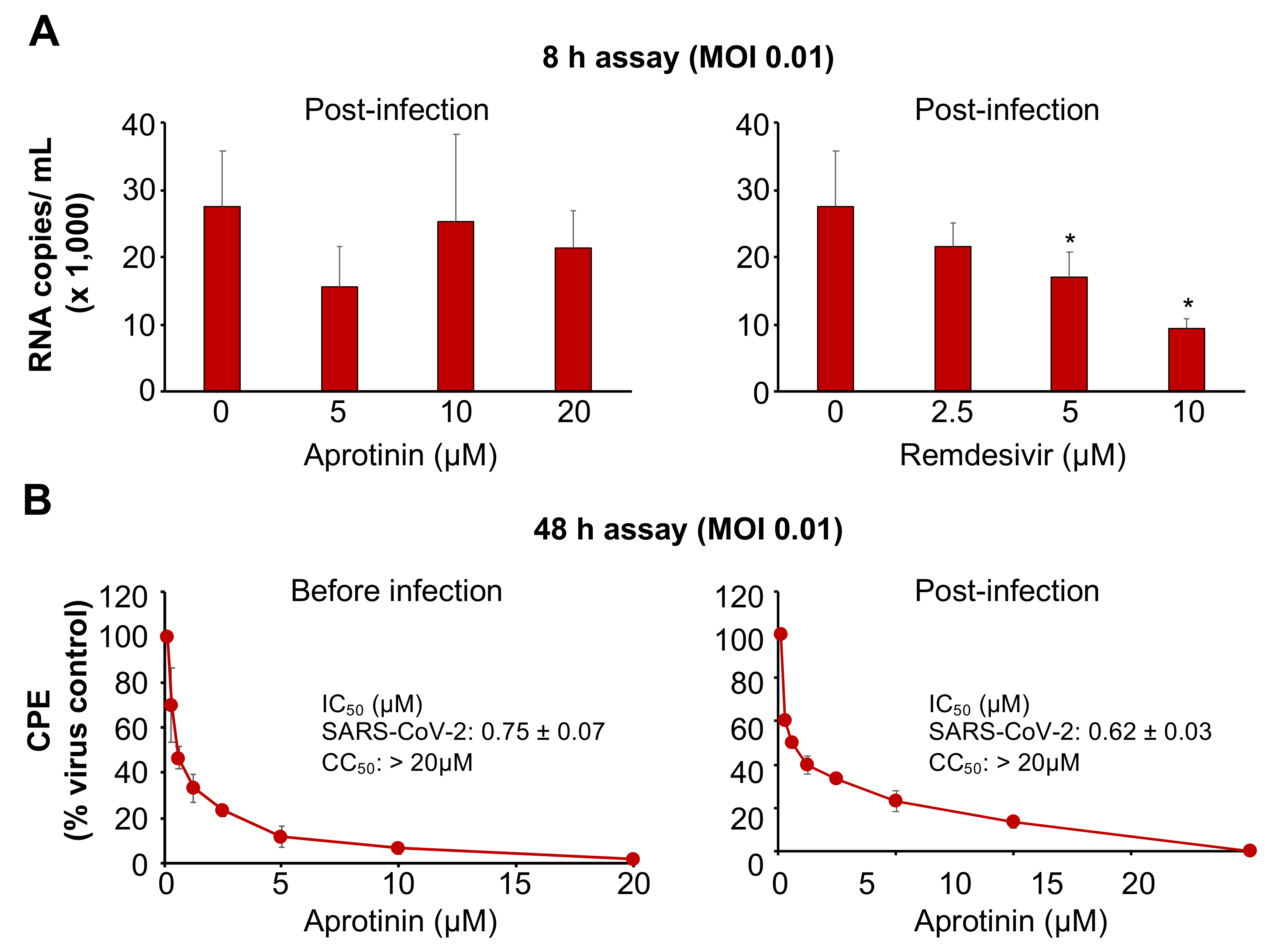
3.3. Aprotinin Inhibits Virus Entry
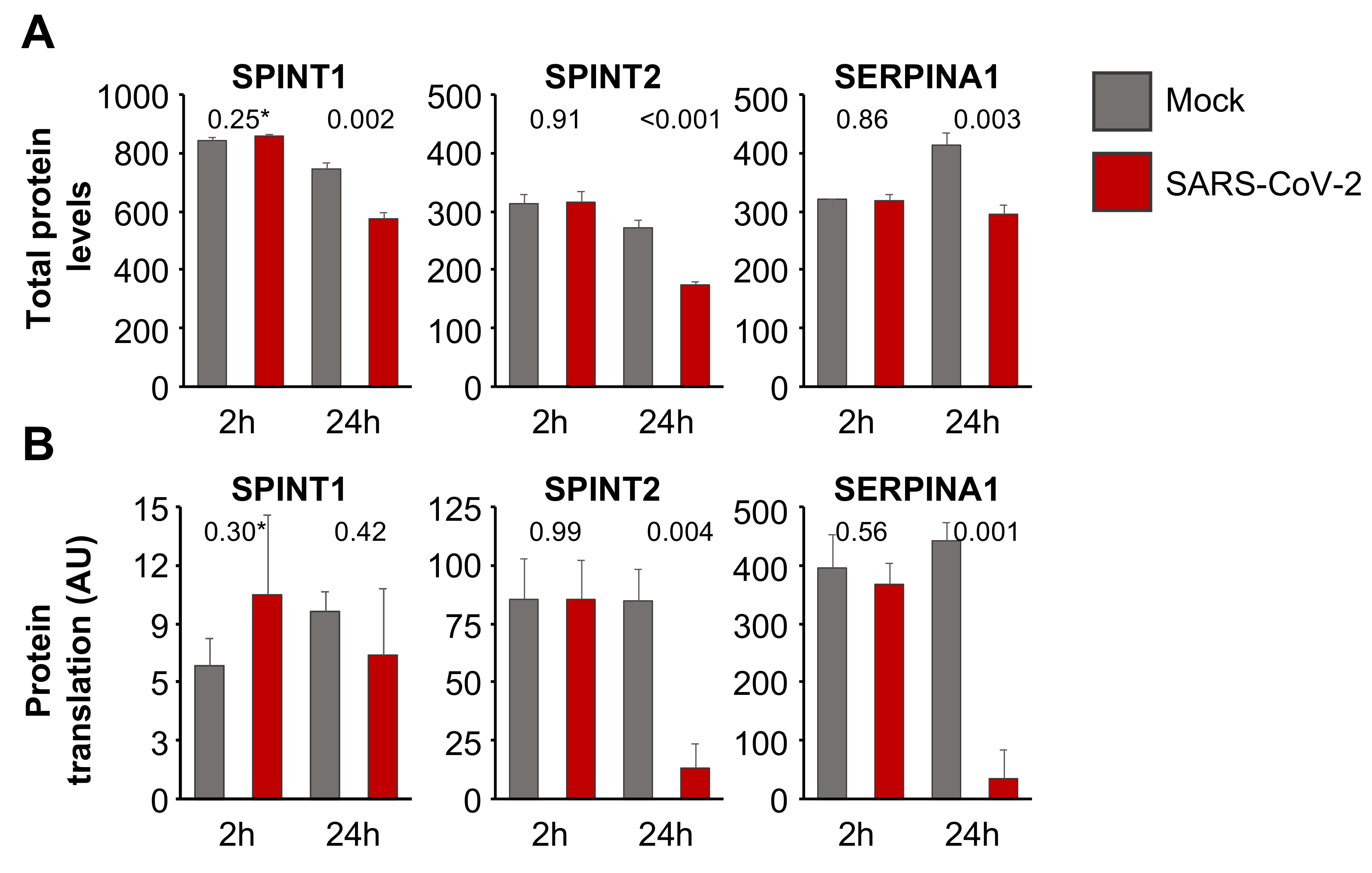
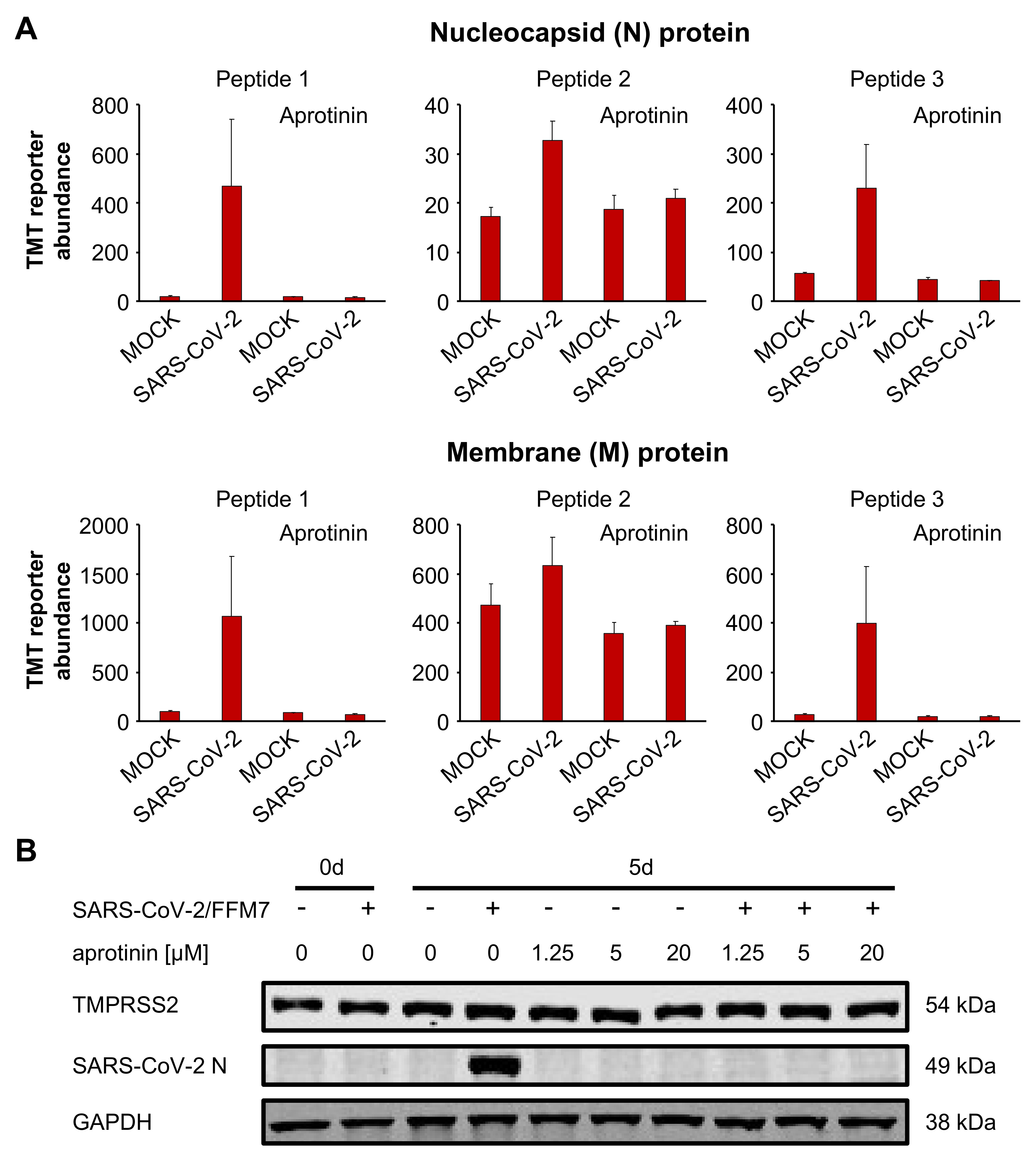
3.4. Aprotinin May Interfere with SARS-CoV-2-Mediated Downregulation of Host Cell Protease Inhibitors
3.5. Aprotinin Exerts Anti-SARS-CoV-2 Activity in Air–Liquid Interface (ALI) Cultures from Primary Bronchial Epithelial Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Chen, N.; Zhou, M.; Dong, X.; Qu, J.; Gong, F.; Han, Y.; Qiu, Y.; Wang, J.; Liu, Y.; Wei, Y.; et al. Epidemiological and clinical characteristics of 99 cases of 2019 novel coronavirus pneumonia in Wuhan, China: A descriptive study. Lancet 2020, 395, 507–513. [Google Scholar] [CrossRef]
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef]
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Infect. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. China Novel Coronavirus Investigating and Research Team. N. Engl. J. Med. 2020, 382, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e8. [Google Scholar] [CrossRef]
- Matsuyama, S.; Nao, N.; Shirato, K.; Kawase, M.; Saito, S.; Takayama, I.; Nagata, N.; Sekizuka, T.; Katoh, H.; Kato, F.; et al. Enhanced isolation of SARS-CoV-2 by TMPRSS2-expressing cells. Proc. Natl. Acad. Sci. USA 2020, 117, 7001–7003. [Google Scholar] [CrossRef]
- Hoffmann, M.; Schroeder, S.; Kleine-Weber, H.; Müller, M.A.; Drosten, C.; Pöhlmann, S. Nafamostat mesylate blocks activation of SARS-CoV-2: New treatment option for COVID-19. Antimicrob. Agents Chemother. 2020, 64, e00754-20. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Kiso, M.; Sakai-Tagawa, Y.; Iwatsuki-Horimoto, K.; Imai, M.; Takeda, M.; Kinoshita, N.; Ohmagari, N.; Gohda, J.; Semba, K.; et al. The Anticoagulant Nafamostat Potently Inhibits SARS-CoV-2 S Protein-Mediated Fusion in a Cell Fusion Assay System and Viral Infection In Vitro in a Cell-Type-Dependent Manner. Viruses 2020, 12, 629. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, S.; Silakari, O. Scaffold morphing of arbidol (umifenovir) in search of multi-targeting therapy halting the interaction of SARS-CoV-2 with ACE2 and other proteases involved in COVID-19. Virus. Res. 2020, 289, 198146. [Google Scholar] [CrossRef] [PubMed]
- Zhirnov, O.P.; Klenk, H.D.; Wright, P.F. Aprotinin and similar protease inhibitors as drugs against influenza. Antiviral Res. 2011, 92, 27–36. [Google Scholar] [CrossRef]
- Shen, L.W.; Mao, H.J.; Wu, Y.L.; Tanaka, Y.; Zhang, W. TMPRSS2: A potential target for treatment of influenza virus and coronavirus infections. Biochimie 2017, 142, 1–10. [Google Scholar] [CrossRef]
- Van Wetering, S.; van der Linden, A.C.; van Sterkenburg, M.A.; de Boer, W.I.; Kuijpers, A.L.; Schalkwijk, J.; Hiemstra, P.S. Regulation of SLPI and elafin release from bronchial epithelial cells by neutrophil defensins. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 278, L51–L58. [Google Scholar] [CrossRef]
- Hoehl, S.; Berger, A.; Kortenbusch, M.; Cinatl, J.; Bojkova, D.; Rabenau, H.; Behrens, P.; Böddinghaus, B.; Götsch, U.; Naujoks, F.; et al. Evidence of SARS-CoV-2 Infection in Returning Travelers from Wuhan, China. N. Engl. J. Med. 2020, 382, 1278–1280. [Google Scholar] [CrossRef]
- Toptan, T.; Hoehl, S.; Westhaus, S.; Bojkova, D.; Berger, A.; Rotter, B.; Hoffmeier, K.; Cinatl, J., Jr.; Ciesek, S.; Widera, M. Optimized qRT-PCR Approach for the Detection of Intra- and Extra-Cellular SARS-CoV-2 RNAs. Int. J. Mol. Sci. 2020, 21, 4396. [Google Scholar] [CrossRef]
- Cinatl, J.; Morgenstern, B.; Bauer, G.; Chandra, P.; Rabenau, H.; Doerr, H.W. Glycyrrhizin, an active component of liquorice roots, and replication of SARS-associated coronavirus. Lancet 2003, 361, 2045–2046. [Google Scholar] [CrossRef]
- Cinatl, J., Jr.; Michaelis, M.; Morgenstern, B.; Doerr, H.W. High-dose hydrocortisone reduces expression of the pro-inflammatory chemokines CXCL8 and CXCL10 in SARS coronavirus-infected intestinal cells. Int. J. Mol. Med. 2005, 15, 323–327. [Google Scholar] [CrossRef]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Onafuye, H.; Pieper, S.; Mulac, D.; Cinatl, J., Jr.; Wass, M.N.; Langer, K.; Michaelis, M. Doxorubicin-loaded human serum albumin nanoparticles overcome transporter-mediated drug resistance in drug-adapted cancer cells. Beilstein J. Nanotechnol. 2019, 10, 1707–1715. [Google Scholar] [CrossRef] [PubMed]
- Cinatl, J.; Cinatl, J.; Weber, B.; Rabenau, H.; Gümbel, H.O.; Chenot, J.F.; Scholz, M.; Encke, A.; Doerr, H.W. In vitro inhibition of human cytomegalovirus replication in human foreskin fibroblasts and endothelial cells by ascorbic acid 2-phosphate. Antiviral Res. 1995, 27, 405–418. [Google Scholar] [CrossRef]
- Klann, K.; Tascher, G.; Münch, C. Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by mTORC1 and eIF2α. Mol. Cell. 2020, 77, 913–925.e4. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
- Solun, B.; Shoenfeld, Y. Inhibition of metalloproteinases in therapy for severe lung injury due to COVID-19. Med. Drug Discov. 2020, 7, 100052. [Google Scholar] [CrossRef]
- Strnad, P.; McElvaney, N.G.; Lomas, D.A. Alpha(1)-Antitrypsin Deficiency. N. Engl. J. Med. 2020, 382, 1443–1455. [Google Scholar] [CrossRef]
- Gettins, P.G. Serpin structure, mechanism, and function. Chem. Rev. 2002, 102, 4751–4804. [Google Scholar] [CrossRef]
- Michaelis, M.; Kleinschmidt, M.C.; Doerr, H.W.; Cinatl, J., Jr. Minocycline inhibits West Nile virus replication and apoptosis in human neuronal cells. J. Antimicrob. Chemother. 2007, 60, 981–986. [Google Scholar] [CrossRef]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell. Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Li, S.; Zhang, Y.; Guan, Z.; Li, H.; Ye, M.; Chen, X.; Shen, J.; Zhou, Y.; Shi, Z.L.; Zhou, P.; et al. SARS-CoV-2 triggers inflammatory responses and cell death through caspase-8 activation. Signal. Transduct. Target. Ther. 2020, 5, 235. [Google Scholar] [CrossRef]
- Esumi, M.; Ishibashi, M.; Yamaguchi, H.; Nakajima, S.; Tai, Y.; Kikuta, S.; Sugitani, M.; Takayama, T.; Tahara, M.; Takeda, M.; et al. Transmembrane serine protease TMPRSS2 activates hepatitis C virus infection. Hepatology 2015, 61, 437–446. [Google Scholar] [CrossRef]
- Straus, M.R.; Kinder, J.T.; Segall, M.; Dutch, R.E.; Whittaker, G.R. SPINT2 inhibits proteases involved in activation of both influenza viruses and metapneumoviruses. Virology 2020, 543, 43–53. [Google Scholar] [CrossRef]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Münch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef]
- Azouz, N.P.; Klingler, A.M.; Rothenberg, M.E. Alpha 1 Antitrypsin is an Inhibitor of the SARS-CoV2–Priming Protease TMPRSS2. bioRxiv 2020. preprint. [Google Scholar] [CrossRef]
- Bestle, D.; Heindl, M.R.; Limburg, H.; Van Lam van, T.; Pilgram, O.; Moulton, H.; Stein, D.A.; Hardes, K.; Eickmann, M.; Dolnik, O.; et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci. Alliance 2020, 3, e202000786. [Google Scholar] [CrossRef]
- Cheng, Y.W.; Chao, T.L.; Li, C.L.; Chiu, M.F.; Kao, H.C.; Wang, S.H.; Pang, Y.H.; Lin, C.H.; Tsai, Y.M.; Lee, W.H.; et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020, 33, 108254. [Google Scholar] [CrossRef]
- Levy, J.H.; Bailey, J.M.; Salmenperä, M. Pharmacokinetics of aprotinin in preoperative cardiac surgical patients. Anesthesiology 1994, 80, 1013–1018. [Google Scholar] [CrossRef]
- Dietrich, W. Reducing thrombin formation during cardiopulmonary bypass: Is there a benefit of the additional anticoagulant action of aprotinin? J. Cardiovasc. Pharmacol. 1996, 27, S50–S57. [Google Scholar] [CrossRef]
- Terrell, M.R.; Walenga, J.M.; Koza, M.J.; Pifarré, R. Efficacy of aprotinin with various anticoagulant agents in cardiopulmonary bypass. Ann. Thorac. Surg. 1996, 62, 506–511. [Google Scholar] [CrossRef]
- Kuitunen, A.; Hiippala, S.; Vahtera, E.; Rasi, V.; Salmenperä, M. The effects of aprotinin and tranexamic acid on thrombin generation and fibrinolytic response after cardiac surgery. Acta Anaesthesiol. Scand. 2005, 49, 1272–1279. [Google Scholar] [CrossRef] [PubMed]
- Sperzel, M.; Huetter, J. Evaluation of aprotinin and tranexamic acid in different in vitro and in vivo models of fibrinolysis, coagulation and thrombus formation. J. Thromb. Haemost. 2007, 5, 2113–2118. [Google Scholar] [CrossRef]
- Marchandot, B.; Sattler, L.; Jesel, L.; Matsushita, K.; Schini-Kerth, V.; Grunebaum, L.; Morel, O. COVID-19 Related Coagulopathy: A Distinct Entity? J. Clin. Med. 2020, 9, 1651. [Google Scholar] [CrossRef]
- Lega, S.; Naviglio, S.; Volpi, S.; Tommasini, A. Recent Insight into SARS-CoV2 Immunopathology and Rationale for Potential Treatment and Preventive Strategies in COVID-19. Vaccines 2020, 8, 224. [Google Scholar] [CrossRef] [PubMed]
- Polycarpou, A.; Howard, M.; Farrar, C.A.; Greenlaw, R.; Fanelli, G.; Wallis, R.; Klavinskis, L.S.; Sacks, S. Rationale for targeting Complement in COVID-19. EMBO Mol. Med. 2020, 12, e202012642. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (µM) | |||
|---|---|---|---|
| FFM1 | FFM2 | FFM6 | |
| CPE formation | 1.03 ± 0.07 | 0.81 ± 0.07 | 0.92 ± 0.03 |
| S levels | 0.79 ± 0.15 | 1.04 ± 0.21 | 1.65 ± 0.30 |
| Caspase 3/7 activation | 0.41 ± 0.25 | 0.32 ± 0.09 | 0.73 ± 0.40 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bojkova, D.; Bechtel, M.; McLaughlin, K.-M.; McGreig, J.E.; Klann, K.; Bellinghausen, C.; Rohde, G.; Jonigk, D.; Braubach, P.; Ciesek, S.; et al. Aprotinin Inhibits SARS-CoV-2 Replication. Cells 2020, 9, 2377. https://doi.org/10.3390/cells9112377
Bojkova D, Bechtel M, McLaughlin K-M, McGreig JE, Klann K, Bellinghausen C, Rohde G, Jonigk D, Braubach P, Ciesek S, et al. Aprotinin Inhibits SARS-CoV-2 Replication. Cells. 2020; 9(11):2377. https://doi.org/10.3390/cells9112377
Chicago/Turabian StyleBojkova, Denisa, Marco Bechtel, Katie-May McLaughlin, Jake E. McGreig, Kevin Klann, Carla Bellinghausen, Gernot Rohde, Danny Jonigk, Peter Braubach, Sandra Ciesek, and et al. 2020. "Aprotinin Inhibits SARS-CoV-2 Replication" Cells 9, no. 11: 2377. https://doi.org/10.3390/cells9112377
APA StyleBojkova, D., Bechtel, M., McLaughlin, K.-M., McGreig, J. E., Klann, K., Bellinghausen, C., Rohde, G., Jonigk, D., Braubach, P., Ciesek, S., Münch, C., Wass, M. N., Michaelis, M., & Cinatl, J., Jr. (2020). Aprotinin Inhibits SARS-CoV-2 Replication. Cells, 9(11), 2377. https://doi.org/10.3390/cells9112377