The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer

 ,
, 
Abstract
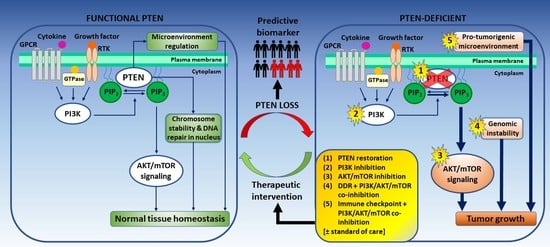
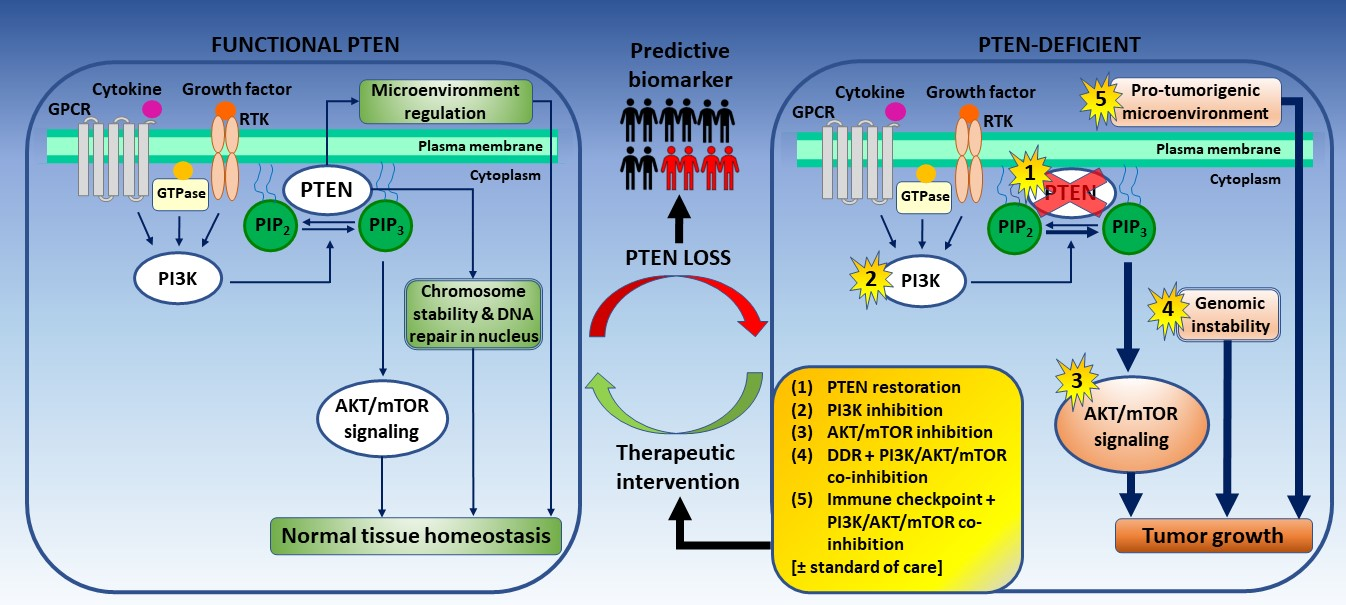{kind=link}
{kind=link}
{kind=link}
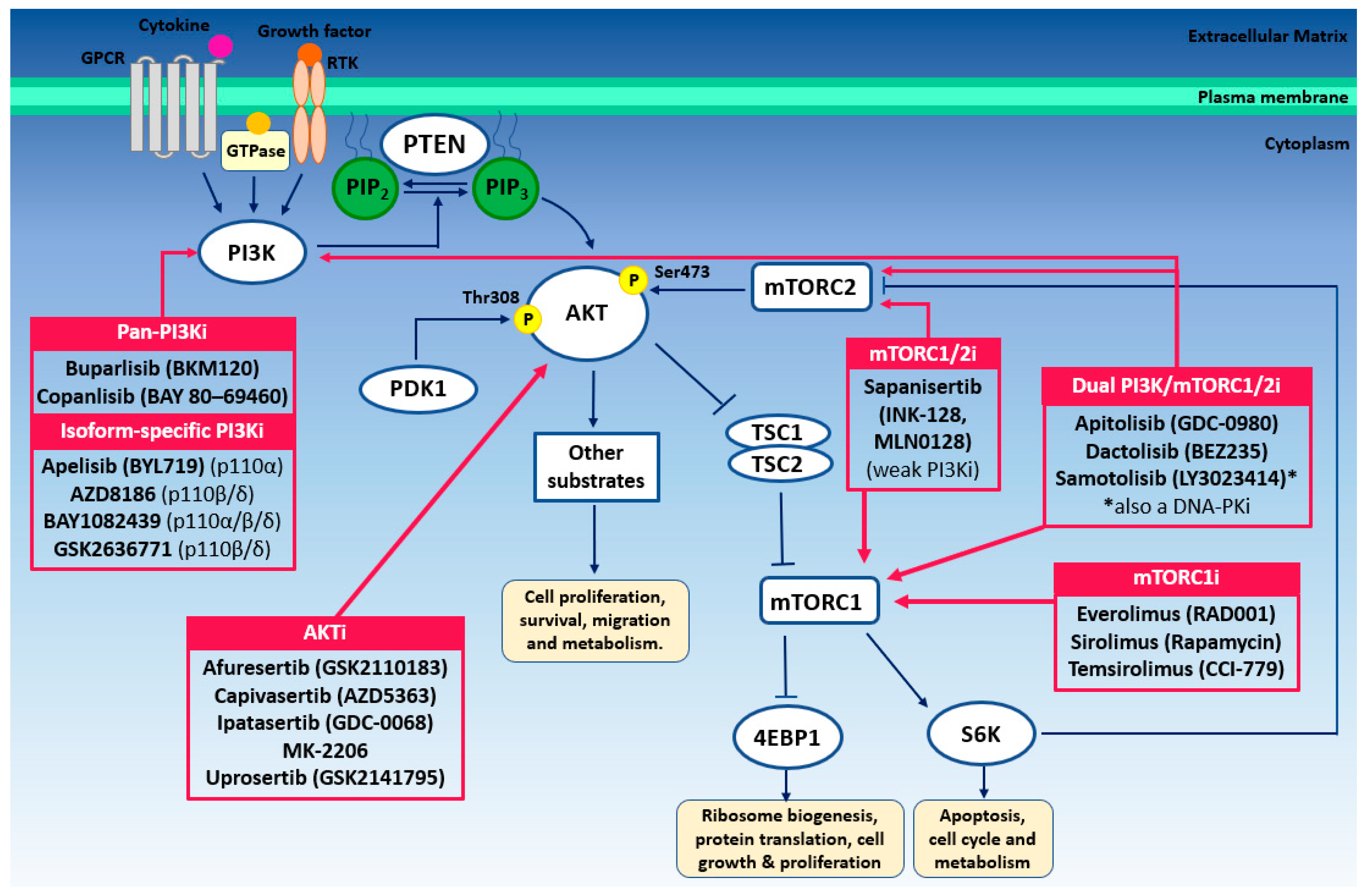{kind=link}
1. Introduction
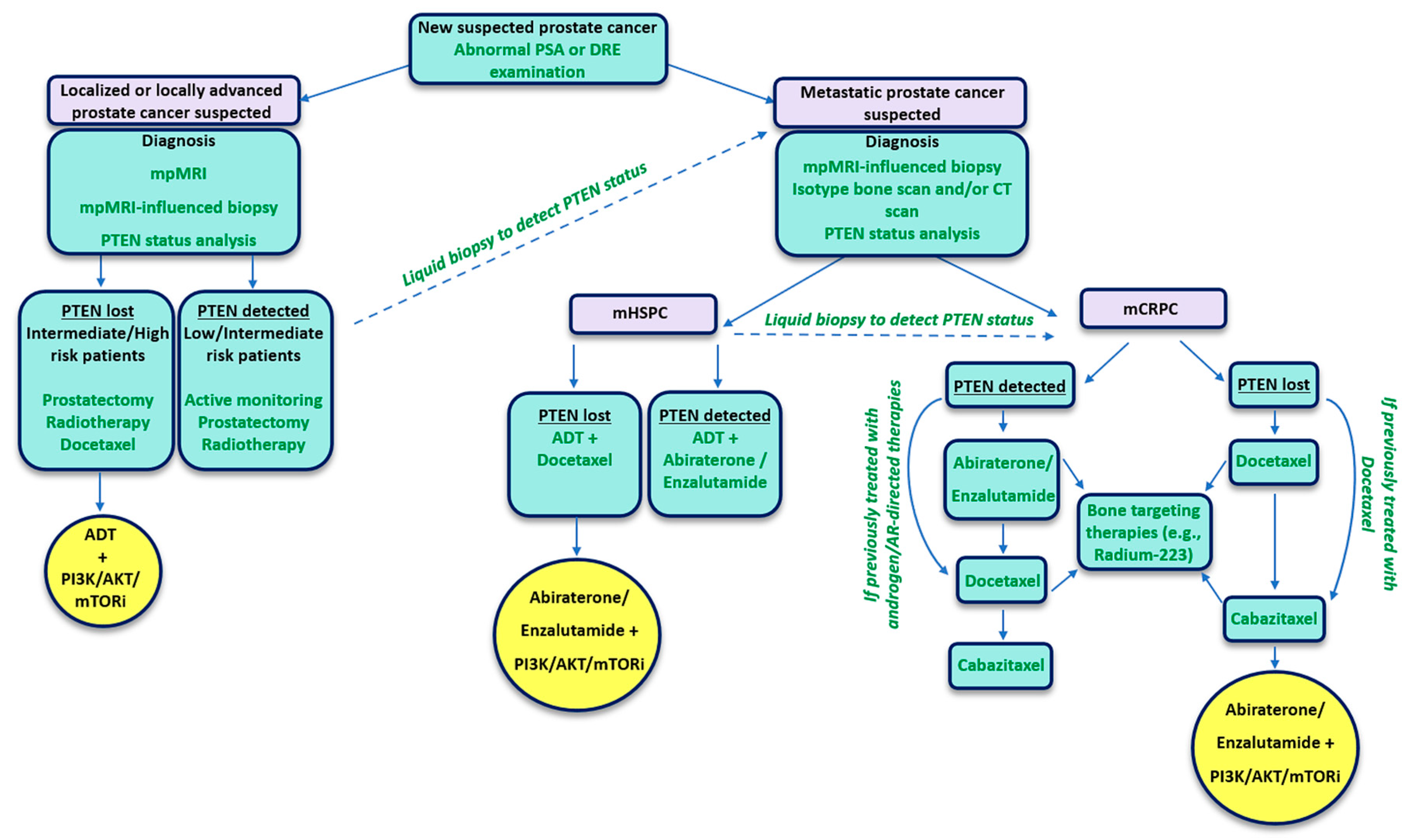
2. PTEN Status as a Predictive Biomarker for Prostate Cancer
3. Targeting PTEN-Deficient Prostate Cancer
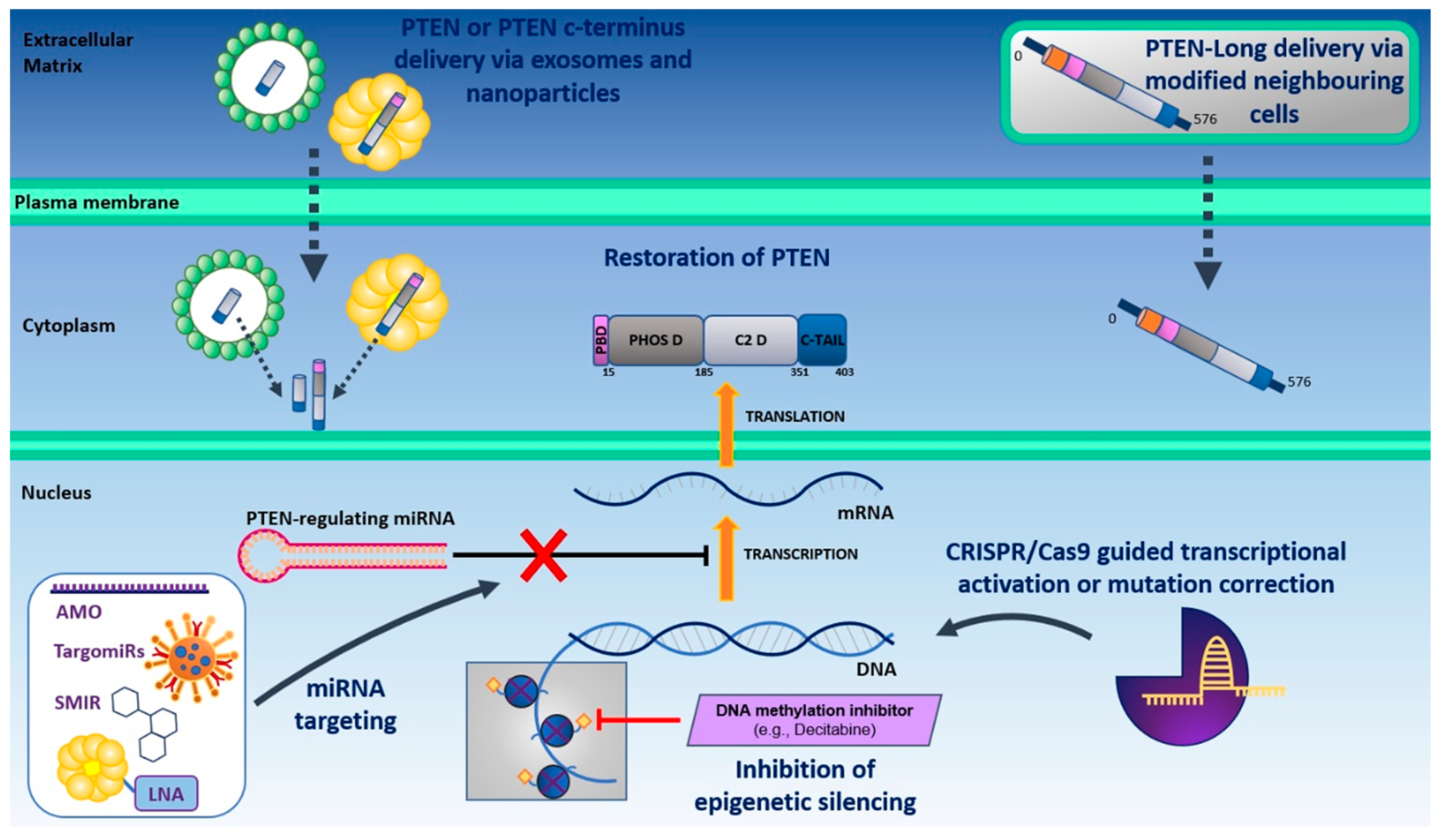
3.1. Direct Restoration of PTEN Function
3.1.1. Direct Delivery of PTEN and PTEN-Long
3.1.2. Restoring PTEN Function by Targeting PTEN-Negative Regulators
3.1.3. miRNA Targeting to Restore PTEN Transcriptional Activity
3.1.4. CRISPR/Cas9-Guided Transcriptional Activation of PTEN
3.2. PI3K Inhibition
3.3. AKT Inhibition
3.4. mTOR Inhibition
3.5. Approaches to Enhance the Efficacy of PI3K–AKT–mTOR-Directed Therapy
3.5.1. SGK Inhibition
3.5.2. PKC Inhibition
3.5.3. GTPase Inhibition
3.5.4. AXL Inhibition
4. PTEN and the DNA Damage Response
5. PTEN and the Tumor Microenvironment
5.1. PTEN and Tumor–Stroma Interactions
5.2. PTEN and the Immune Response
5.3. PTEN and Inflammation
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| ADT | Androgen-deprivation therapy |
| 4EBP1 | 4E (eIF4E)-binding protein 1 |
| AMO | Antisense anti-miR oligonucleotide |
| AR | Androgen receptor |
| BER | Base excision repair |
| BRCA1 | Breast cancer susceptibility protein 1 |
| BRCA2 | Breast cancer susceptibility protein 2 |
| C2D | C2 domain |
| Cas9 | CRISPR-associated protein 9, |
| CHK1 | Cyclin D1 and checkpoint kinase 1 |
| cIAP-1 | Cellular inhibitor of apoptosis protein-1 |
| CRISPR | Clustered regularly interspaced short palindromic repeat |
| CRPC | Castrate-resistant prostate cancer |
| CTC | Circulating tumor cell |
| CXCL8 | C-X-C motif chemokine ligand 8, interleukin 8 |
| DDR | DNA damage response |
| DEPTOR | Disheveled, EGL-10 and pleckstrin (DEP) domain-containing mTOR-interacting protein |
| DHT | Dihydrotestosterone |
| DRE | Digital rectal examination |
| DSBs | Double-strand breaks |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-to-mesenchymal transition |
| ECM | Extracellular matrix |
| FISH | Fluorescence in situ hybridization |
| FOXO | Forkhead box protein O |
| GAS6 | Growth arrest-specific protein 6 |
| GSK3β | Glycogen synthase kinase 3 beta |
| GPCR | G-protein coupled receptor |
| HER2 | Human epidermal growth factor receptor 2 |
| HRR | Homologous recombination repair |
| IHC | Immunohistochemistry |
| IL6 | Interleukin 6 |
| IL8 | Interleukin 8 |
| ITSN | Intersectin |
| LNA | Locked nucleic acid |
| MAN2C1 | α-mannosidase 2C1 |
| MAPK | Mitogen-activated protein kinase |
| MAPKAP1 | Mammalian stress-activated protein kinase-interacting protein 1 |
| mCRPC | Metastatic castrate-resistant prostate cancer |
| MDSC | Myeloid-derived suppressor cells |
| MEF | Mouse embryonic fibroblast |
| miRNA | Micro ribonucleic acid |
| mHSPC | Metastatic hormone-sensitive prostate cancer |
| mLST8 | Mammalian lethal with SEC13 protein 8 |
| MMR | Mismatch repair |
| mpMRI | Multiparametric MRI |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| mTORC2 | Mammalian target of rapamycin complex 2 |
| NER | Nucleotide excision repair |
| NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| NHEJ | Non-homologous end joining |
| NSCLC | Non-small-cell lung carcinoma |
| OncomiRs | Oncogenic miRNAs |
| P | Phosphorylation event |
| PBD | PIP2-binding domain |
| PARP | Poly-ADP ribose polymerase |
| PDK1 | Phosphoinositide-dependent kinase 1 |
| PH domain | Pleckstrin homology domain |
| Phos D | Phosphatase domain |
| PI3K | Phosphoinositide 3-kinase |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PKB | Protein kinase B, AKT |
| PKC | Protein kinase C |
| PKCε | Protein kinase C epsilon |
| PKCζ | Protein kinase C zeta |
| PRAS40 | Proline-rich AKT substrate of 40 kDa |
| PREX1 | PIP3-dependent Rac exchanger 1 |
| PREX2 | PIP3-dependent Rac exchanger 2 |
| PSA | Prostate-specific antigen |
| PTEN | Phosphatase and tensin homologue deleted on chromosome 10 |
| RAPTOR | Regulatory-associated protein of mTOR |
| RBD | Ras-binding domain |
| RHEB | Ras homolog enriched in brain |
| RICTOR | Rapamycin-insensitive companion of mTOR |
| rPFS | Radiographic progression-free survival |
| RFS | Recurrence-free survival |
| RTK | Tyrosine kinase receptor |
| S6K | Ribosomal protein S6 kinase/p70 ribosomal S6 kinase |
| SGK1 | Serum- and glucocorticoid-regulated kinase 1 |
| SGK2 | Serum- and glucocorticoid-regulated kinase 2 |
| SGK3 | Serum- and glucocorticoid-regulated kinase 3 |
| siRNA | Small interfering RNA |
| SMIR | Small-molecule inhibitors of miRNA |
| SRC-3 | Steroid receptor co-activator 3 |
| SSBs | Single-strand breaks |
| TAM | Tumor-associated macrophage |
| TSC1 | Tuberous sclerosis complex 1 |
| TSC2 | Tuberous sclerosis complex 2 |
| TSIs | Tumor–stroma interactions |
| VAV3 | Vav guanine nucleotide exchange factor 3 |
References
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Jamaspishvili, T.; Berman, D.M.; Ross, A.E.; Scher, H.I.; De Marzo, A.M.; Squire, J.A.; Lotan, T.L. Clinical implications of PTEN loss in prostate cancer. Nat. Rev. Urol. 2018, 15, 222–234. [Google Scholar] [CrossRef] [PubMed]
- Leinonen, K.A.; Saramäki, O.R.; Furusato, B.; Kimura, T.; Takahashi, H.; Egawa, S.; Suzuki, H.; Keiger, K.; Hahm, S.H.; Isaacs, W.B.; et al. Loss of PTEN Is Associated with Aggressive Behavior in ERG-Positive Prostate Cancer. Cancer Epidemiol. Biomark. Prev. 2013, 22, 2333–2344. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, M.; Ludkovski, O.; Degrace, D.; Williams, J.L.; Evans, A.; Sircar, K.; Bismar, T.A.; Nuin, P.; Squire, J.A. PTEN genomic deletions that characterize aggressive prostate cancer originate close to segmental duplications. Geneschromosom. Cancer 2011, 51, 149–160. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Heumann, A.; Rico, S.D.; Hicks, J.; Lecksell, K.; Koop, C.; Sauter, G.; Schlomm, T.; Simon, R. PTEN loss detection in prostate cancer: Comparison of PTEN immunohistochemistry and PTEN FISH in a large retrospective prostatectomy cohort. Oncotarget 2017, 8, 65566–65576. [Google Scholar] [CrossRef]
- Lotan, T.L.; Gurel, B.; Sutcliffe, S.; Esopi, D.; Liu, W.; Xu, J.; Hicks, J.L.; Park, B.H.; Humphreys, E.; Partin, A.W.; et al. PTEN Protein Loss by Immunostaining: Analytic Validation and Prognostic Indicator for a High Risk Surgical Cohort of Prostate Cancer Patients. Clin. Cancer Res. 2011, 17, 6563–6573. [Google Scholar] [CrossRef]
- Abeshouse, A.; Ahn, J.; Akbani, R.; Ally, A.; Amin, S.; Andry, C.D.; Annala, M.; Aprikian, A.G.; Armenia, J.; Arora, A.; et al. The Molecular Taxonomy of Primary Prostate Cancer. Cell 2015, 163, 1011–1025. [Google Scholar] [CrossRef]
- Barbieri, C.E.; Baca, S.C.; Lawrence, M.S.; Demichelis, F.; Blattner, M.; Theurillat, J.-P.; White, T.A.; Stojanov, P.; Van Allen, E.; Stransky, N.; et al. Exome sequencing identifies recurrent SPOP, FOXA1 and MED12 mutations in prostate cancer. Nat. Genet. 2012, 44, 685–689. [Google Scholar] [CrossRef]
- Sun, J.; Li, S.; Wang, F.; Fan, C.; Wang, J. Identification of key pathways and genes in PTEN mutation prostate cancer by bioinformatics analysis. BMC Med. Genet. 2019, 20, 1–9. [Google Scholar] [CrossRef]
- Leslie, N.R.; Foti, M. Non-genomic loss of PTEN function in cancer: Not in my genes. Trends Pharmacol. Sci. 2011, 32, 131–140. [Google Scholar] [CrossRef]
- Phin, S.; Moore, M.W.; Cotter, P.D. Genomic Rearrangements of PTEN in Prostate Cancer. Front. Oncol. 2013, 3, 240. [Google Scholar] [CrossRef] [PubMed]
- Whang, Y.E.; Wu, X.; Suzuki, H.; Reiter, R.E.; Tran, C.; Vessella, R.L.; Said, J.W.; Isaacs, W.B.; Sawyers, C.L. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc. Natl. Acad. Sci. USA 1998, 95, 5246–5250. [Google Scholar] [CrossRef] [PubMed]
- Festuccia, C.; Gravina, G.L.; Biordi, L.; Martella, F.; Flati, V.; Ricevuto, E.; Ficorella, C.; Tombolini, V. Epigenetic modulation of PTEN expression during antiandrogenic therapies in human prostate cancer. Int. J. Oncol. 2009, 35, 1133–1139. [Google Scholar] [CrossRef][Green Version]
- Papa, A.; Pandolfi, P.P. The PTEN–PI3K Axis in Cancer. Biomol. 2019, 9, 153. [Google Scholar] [CrossRef]
- Pulido, R. PTEN: A yin-yang master regulator protein in health and disease. Methods 2015, 77–78, 3–10. [Google Scholar] [CrossRef]
- Weng, L.-P.; Brown, J.L.; Eng, C. PTEN coordinates G(1) arrest by down-regulating cyclin D1 via its protein phosphatase activity and up-regulating p27 via its lipid phosphatase activity in a breast cancer model. Hum. Mol. Genet. 2001, 10, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Shen, W.H.; Balajee, A.S.; Wang, J.; Wu, H.; Eng, C.; Pandolfi, P.P.; Yin, Y. Essential Role for Nuclear PTEN in Maintaining Chromosomal Integrity. Cell 2007, 128, 157–170. [Google Scholar] [CrossRef] [PubMed]
- Bassi, C.; Ho, J.; Srikumar, T.; Dowling, R.J.O.; Gorrini, C.; Miller, S.J.; Mak, T.W.; Neel, B.G.; Raught, B.; Stambolic, V. Nuclear PTEN Controls DNA Repair and Sensitivity to Genotoxic Stress. Science 2013, 341, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Leslie, N.R.; Yang, X.; Downes, C.P.; Weijer, K. PtdIns(3,4,5)P3-Dependent and -Independent Roles for PTEN in the Control of Cell Migration. Curr. Biol. 2007, 17, 115–125. [Google Scholar] [CrossRef]
- Wang, S.; Gao, J.; Lei, Q.; Rozengurt, N.; Pritchard, C.; Jiao, J.; Thomas, G.V.; Li, G.; Roy-Burman, P.; Nelson, P.S.; et al. Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 2003, 4, 209–221. [Google Scholar] [CrossRef]
- Pearson, H.B.; Li, J.; Méniel, V.; Fennell, C.; Waring, P.; Montgomery, K.G.; Rebello, R.J.; MacPherson, A.A.; Koushyar, S.; Furic, L.; et al. Identification of Pik3ca Mutation as a Genetic Driver of Prostate Cancer That Cooperates with Pten Loss to Accelerate Progression and Castration-Resistant Growth. Cancer Discov. 2018, 8, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Trotman, L.C.; Niki, M.A.; Dotan, Z.A.; Koutcher, J.; Di Cristofano, A.; Xiao, A.; Khoo, A.S.; Roy-Burman, P.; Greenberg, N.M.; Van Dyke, T.; et al. Pten Dose Dictates Cancer Progression in the Prostate. PLoS Biol. 2003, 1, e59. [Google Scholar] [CrossRef]
- Kwabi-Addo, B.; Giri, D.; Schmidt, K.; Podsypanina, K.; Parsons, R.; Greenberg, N.; Ittmann, M. Haploinsufficiency of the Pten tumor suppressor gene promotes prostate cancer progression. Proc. Natl. Acad. Sci. USA 2001, 98, 11563–11568. [Google Scholar] [CrossRef] [PubMed]
- Ma, X. Targeted Biallelic Inactivation of Pten in the Mouse Prostate Leads to Prostate Cancer Accompanied by Increased Epithelial Cell Proliferation but not by Reduced Apoptosis. Cancer Res. 2005, 65, 5730–5739. [Google Scholar] [CrossRef] [PubMed]
- Backman, S.A.; Ghazarian, D.; So, K.; Sanchez, O.; Wagner, K.-U.; Hennighausen, L.; Suzuki, A.; Tsao, M.-S.; Chapman, W.B.; Stambolic, V.; et al. Early onset of neoplasia in the prostate and skin of mice with tissue-specific deletion of Pten. Proc. Natl. Acad. Sci. USA 2004, 101, 1725–1730. [Google Scholar] [CrossRef]
- Choi, N.; Zhang, B.; Zhang, L.; Ittmann, M.; Xin, L. Adult Murine Prostate Basal and Luminal Cells Are Self-Sustained Lineages that Can Both Serve as Targets for Prostate Cancer Initiation. Cancer Cell 2012, 21, 253–265. [Google Scholar] [CrossRef]
- You, M.J.; Castrillon, D.H.; Bastian, B.C.; O’Hagan, R.C.; Bosenberg, M.W.; Parsons, R.; Chin, L.; Depinho, R.A. Genetic analysis of Pten and Ink4a/Arf interactions in the suppression of tumorigenesis in mice. Proc. Natl. Acad. Sci. USA 2002, 99, 1455–1460. [Google Scholar] [CrossRef]
- Di Cristofano, A.; De Acetis, M.; Koff, A.; Cordon-Cardo, C.; Pandolfi, P.P. Pten and p27KIP1 cooperate in prostate cancer tumor suppression in the mouse. Nat. Genet. 2001, 27, 222–224. [Google Scholar] [CrossRef]
- Blando, J.M.; Carbajal, S.; Abel, E.L.; Beltran, L.; Conti, C.; Fischer, S.; DiGiovanni, J. Cooperation between Stat3 and Akt Signaling Leads to Prostate Tumor Development in Transgenic Mice. Neoplasia 2011, 13, 254-IN12. [Google Scholar] [CrossRef]
- Chen, Z.; Trotman, L.C.; Shaffer, D.; Lin, H.-K.; Dotan, Z.A.; Niki, M.; Koutcher, J.A.; Scher, H.I.; Ludwig, T.; Gerald, W.L.; et al. Crucial role of p53-dependent cellular senescence in suppression of Pten-deficient tumorigenesis. Nat. Cell Biol. 2005, 436, 725–730. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Kobayashi, N.; Ruscetti, M.; Zhi, A.; Tran, L.M.; Huang, J.; Gleave, M.; Wu, H. Pten Loss and RAS/MAPK Activation Cooperate to Promote EMT and Metastasis Initiated from Prostate Cancer Stem/Progenitor Cells. Cancer Res. 2012, 72, 1878–1889. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Kobayashi, T.; Floc’H, N.; Kinkade, C.W.; Aytes, A.; Dankort, D.; Lefebvre, C.; Mitrofanova, A.; Cardiff, R.D.; McMahon, M.; et al. B-Raf Activation Cooperates with PTEN Loss to Drive c-Myc Expression in Advanced Prostate Cancer. Cancer Res. 2012, 72, 4765–4776. [Google Scholar] [CrossRef]
- Mulholland, D.J.; Tran, L.M.; Li, Y.; Cai, H.; Morim, A.; Wang, S.; Plaisier, S.; Garraway, I.P.; Huang, J.; Graeber, T.G.; et al. Cell Autonomous Role of PTEN in Regulating Castration-Resistant Prostate Cancer Growth. Cancer Cell 2011, 19, 792–804. [Google Scholar] [CrossRef] [PubMed]
- Ferraldeschi, R.; Rodrigues, D.N.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rescigno, P.; Ravi, P.; Pezaro, C.; Omlin, A.; Lorente, D.; et al. PTEN Protein Loss and Clinical Outcome from Castration-resistant Prostate Cancer Treated with Abiraterone Acetate. Eur. Urol. 2015, 67, 795–802. [Google Scholar] [CrossRef] [PubMed]
- Mithal, P.; Allott, E.; Gerber, L.; Reid, J.; Welbourn, W.; Tikishvili, E.; Park, J.; Younus, A.; Sangale, Z.; Lanchbury, J.S.; et al. PTEN loss in biopsy tissue predicts poor clinical outcomes in prostate cancer. Int. J. Urol. 2014, 21, 1209–1214. [Google Scholar] [CrossRef]
- Rescigno, P.; Lorente, D.; Dolling, D.; Ferraldeschi, R.; Rodrigues, D.N.; Riisnaes, R.; Miranda, S.; Bianchini, D.; Zafeiriou, Z.; Sideris, S.; et al. Docetaxel Treatment in PTEN- and ERG-aberrant Metastatic Prostate Cancers. Eur. Urol. Oncol. 2018, 1, 71–77. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, A.J.; Halabi, S.; Healy, P.; Alumkal, J.J.; Winters, C.; Kephart, J.; Bitting, R.L.; Hobbs, C.; Soleau, C.F.; Beer, T.M.; et al. Phase II trial of the PI3 kinase inhibitor buparlisib (BKM-120) with or without enzalutamide in men with metastatic castration resistant prostate cancer. Eur. J. Cancer 2017, 81, 228–236. [Google Scholar] [CrossRef]
- Wei, X.X.; Hsieh, A.C.; Kim, W.; Friedlander, T.; Lin, A.M.; Louttit, M.; Ryan, C.J. A Phase I Study of Abiraterone Acetate Combined with BEZ235, a Dual PI3K/mTOR Inhibitor, in Metastatic Castration Resistant Prostate Cancer. Oncol. 2017, 22, 503-e43. [Google Scholar] [CrossRef]
- Graham, L.; Banda, K.; Torres, A.; Carver, B.S.; Chen, Y.; Pisano, K.; Shelkey, G.; Curley, T.; Scher, H.I.; Lotan, T.L.; et al. A phase II study of the dual mTOR inhibitor MLN0128 in patients with metastatic castration resistant prostate cancer. Investig. New Drugs 2018, 36, 458–467. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Netto, G.J.; Rudek, M.A.; Halabi, S.; Wood, D.P.; Creel, P.A.; Mundy, K.; Davis, S.L.; Wang, T.; Albadine, R.; et al. A pharmacodynamic study of rapamycin in men with intermediate- to high-risk localized prostate cancer. Clin. Cancer Res. 2010, 16, 3057–3066. [Google Scholar] [CrossRef]
- Barata, P.; Cooney, M.; Mendiratta, P.; Gupta, R.; Dreicer, R.; Garcia, J.A. Phase I/II study evaluating the safety and clinical efficacy of temsirolimus and bevacizumab in patients with chemotherapy refractory metastatic castration-resistant prostate cancer. Investig. New Drugs 2018, 37, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Courtney, K.D.; Manola, J.B.; ElFiky, A.A.; Ross, R.; Oh, W.K.; Yap, J.T.; Abbeele, A.D.V.D.; Ryan, C.W.; Beer, T.M.; Loda, M.; et al. A Phase I Study of Everolimus and Docetaxel in Patients with Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2015, 13, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Rawla, P. Epidemiology of Prostate Cancer. World J. Oncol. 2019, 10, 63–89. [Google Scholar] [CrossRef] [PubMed]
- Prostate Cancer Treatment (PDQ®)–Health Professional Version. Available online: https://www.cancer.gov/types/prostate/hp/prostate-treatment-pdq (accessed on 5 October 2020).
- NCCN Clinical Practice Guidelines in Oncology Prostate Cancer Version. Available online: https://www.nccn.org/professionals/physician_gls/pdf/prostate_blocks.pdf (accessed on 5 October 2020).
- Van Leenders, G.J.; Van Der Kwast, T.H.; Grignon, D.J.; Evans, A.J.; Kristiansen, G.; Kweldam, C.F.; Litjens, G.; McKenney, J.K.; Melamed, J.; Mottet, N.; et al. The 2019 International Society of Urological Pathology (ISUP) Consensus Conference on Grading of Prostatic Carcinoma. Am. J. Surg. Pathol. 2020, 44, e87–e99. [Google Scholar] [CrossRef] [PubMed]
- Cooperberg, M.R.; Carroll, P.R. Trends in Management for Patients with Localized Prostate Cancer, 1990–2013. JAMA 2015, 314, 80–82. [Google Scholar] [CrossRef]
- Hamdy, F.C.; Donovan, J.L.; Lane, J.A.; Mason, M.; Metcalfe, C.; Holding, P.; Davis, M.; Peters, T.J.; Turner, E.L.; Martin, R.M.; et al. 10-Year Outcomes after Monitoring, Surgery, or Radiotherapy for Localized Prostate Cancer. N. Engl. J. Med. 2016, 375, 1415–1424. [Google Scholar] [CrossRef]
- Loeb, S.; Ross, A.E. Genomic testing for localized prostate cancer. Curr. Opin. Urol. 2017, 27, 495–499. [Google Scholar] [CrossRef]
- Canter, D.J.; Freedland, S.; Rajamani, S.; Latsis, M.; Variano, M.; Halat, S.; Tward, J.; Cohen, T.; Stone, S.; Schlomm, T.; et al. Analysis of the prognostic utility of the cell cycle progression (CCP) score generated from needle biopsy in men treated with definitive therapy. Prostate Cancer Prostatic Dis. 2019, 23, 102–107. [Google Scholar] [CrossRef]
- Eeden, S.K.V.D.; Lu, R.; Zhang, N.; Quesenberry, C.P.; Shan, J.; Han, J.S.; Tsiatis, A.C.; Leimpeter, A.D.; Lawrence, H.J.; Febbo, P.G.; et al. A Biopsy-based 17-gene Genomic Prostate Score as a Predictor of Metastases and Prostate Cancer Death in Surgically Treated Men with Clinically Localized Disease. Eur. Urol. 2018, 73, 129–138. [Google Scholar] [CrossRef]
- Spratt, D.E.; Yousefi, K.; Deheshi, S.; Ross, A.E.; Den, R.B.; Schaeffer, E.M.; Trock, B.J.; Zhang, J.; Glass, A.G.; Dicker, A.P.; et al. Individual Patient-Level Meta-Analysis of the Performance of the Decipher Genomic Classifier in High-Risk Men After Prostatectomy to Predict Development of Metastatic Disease. J. Clin. Oncol. 2017, 35, 1991–1998. [Google Scholar] [CrossRef]
- Tosoian, J.J.; Guedes, L.B.; Morais, C.L.; Mamawala, M.; Ross, A.E.; De Marzo, A.M.; Trock, B.J.; Han, M.; Carter, H.B.; Lotan, T.L. PTEN status assessment in the Johns Hopkins active surveillance cohort. Prostate Cancer Prostatic Dis. 2018, 22, 176–181. [Google Scholar] [CrossRef] [PubMed]
- Guedes, L.B.; Tosoian, J.J.; Hicks, J.; Ross, A.E.; Lotan, T.L. PTEN Loss in Gleason Score 3 + 4 = 7 Prostate Biopsies is Associated with Nonorgan Confined Disease at Radical Prostatectomy. J. Urol. 2017, 197, 1054–1059. [Google Scholar] [CrossRef] [PubMed]
- Picanço-Albuquerque, C.G.; Morais, C.L.; Carvalho, F.L.F.; Peskoe, S.B.; Hicks, J.L.; Ludkovski, O.; Vidotto, T.; Fedor, H.; Humphreys, E.; Han, M.; et al. In prostate cancer needle biopsies, detections of PTEN loss by fluorescence in situ hybridization (FISH) and by immunohistochemistry (IHC) are concordant and show consistent association with upgrading. Virchows Arch. 2016, 468, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Lotan, T.L.; Carvalho, F.L.; Peskoe, S.B.; Hicks, J.L.; Good, J.; Fedor, H.L.; Humphreys, E.; Han, M.; Platz, E.A.; Squire, J.A.; et al. PTEN loss is associated with upgrading of prostate cancer from biopsy to radical prostatectomy. Mod. Pathol. 2014, 28, 128–137. [Google Scholar] [CrossRef]
- Trock, B.J.; Fedor, H.; Gurel, B.; Jenkins, R.B.; Knudsen, B.S.; Fine, S.W.; Said, J.W.; Carter, H.B.; Lotan, T.L.; De Marzo, A.M. PTEN loss and chromosome 8 alterations in Gleason grade 3 prostate cancer cores predicts the presence of un-sampled grade 4 tumor: Implications for active surveillance. Mod. Pathol. 2016, 29, 764–771. [Google Scholar] [CrossRef]
- Wang, Y.; Dai, B. PTEN genomic deletion defines favorable prognostic biomarkers in localized prostate cancer: A systematic review and meta-analysis. Int. J. Clin. Exp. Med. 2015, 8, 5430–5437. [Google Scholar]
- Clinton, T.N.; Bagrodia, A.; Lotan, Y.; Margulis, V.; Raj, G.V.; Woldu, S.L. Tissue-based biomarkers in prostate cancer. Expert Rev. Precis. Med. Drug Dev. 2017, 2, 249–260. [Google Scholar] [CrossRef]
- Lotan, T.L.; Wei, W.; Ludkovski, O.; Morais, C.L.; Guedes, L.B.; Jamaspishvili, T.; Lopez, K.; Hawley, S.T.; Feng, Z.; Fazli, L.; et al. Analytic validation of a clinical-grade PTEN immunohistochemistry assay in prostate cancer by comparison with PTEN FISH. Mod. Pathol. 2016, 29, 904–914. [Google Scholar] [CrossRef]
- Beltran, H.; Yelensky, R.; Frampton, G.M.; Park, K.; Downing, S.R.; Macdonald, T.Y.; Jarosz, M.; Lipson, D.; Tagawa, S.T.; Nanus, D.M.; et al. Targeted Next-generation Sequencing of Advanced Prostate Cancer Identifies Potential Therapeutic Targets and Disease Heterogeneity. Eur. Urol. 2013, 63, 920–926. [Google Scholar] [CrossRef]
- Hamid, A.A.; Gray, K.P.; Huang, Y.; Bowden, M.; Pomerantz, M.M.; Loda, M.; Sweeney, C.J. Loss of PTEN Expression Detected by Fluorescence Immunohistochemistry Predicts Lethal Prostate Cancer in Men Treated with Prostatectomy. Eur. Urol. Oncol. 2019, 2, 475–482. [Google Scholar] [CrossRef]
- Suzuki, H.; Freije, D.; Nusskern, D.R.; Okami, K.; Cairns, P.; Sidransky, D.; Isaacs, W.B.; Bova, G.S. Interfocal heterogeneity of PTEN/MMAC1 gene alterations in multiple metastatic prostate cancer tissues. Cancer Res. 1998, 58, 204–209. [Google Scholar] [PubMed]
- Hariri, N.; Zare, S.; Murphy, J.; Fadare, O. Cost-effectiveness of a Dual (Immunohistochemistry and Fluorescence In Situ Hybridization) HER2/neu Testing Strategy on Invasive Breast Cancers. Appl. Immunohistochem. Mol. Morphol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Harmon, S.A.; Patel, P.G.; Sanford, T.H.; Caven, I.; Iseman, R.; Vidotto, T.; Picanço, C.; Squire, J.A.; Masoudi, S.; Mehralivand, S.; et al. High throughput assessment of biomarkers in tissue microarrays using artificial intelligence: PTEN loss as a proof-of-principle in multi-center prostate cancer cohorts. Mod. Pathol. 2020, 1–12. [Google Scholar] [CrossRef]
- Vidotto, T.; Tiezzi, D.G.; Squire, J.A. Distinct subtypes of genomic PTEN deletion size influence the landscape of aneuploidy and outcome in prostate cancer. Mol. Cytogenet. 2018, 11, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gimm, O.; Perren, A.; Weng, L.-P.; Marsh, D.J.; Yeh, J.J.; Ziebold, U.; Gil, E.; Hinze, R.; Delbridge, L.; Lees, J.A.; et al. Differential Nuclear and Cytoplasmic Expression of PTEN in Normal Thyroid Tissue, and Benign and Malignant Epithelial Thyroid Tumors. Am. J. Pathol. 2000, 156, 1693–1700. [Google Scholar] [CrossRef]
- Jamaspishvili, T.; Patel, P.G.; Niu, Y.; Vidotto, T.; Caven, I.; Livergant, R.; Fu, W.; Kawashima, A.; How, N.; Okello, J.B.; et al. Risk Stratification of Prostate Cancer through Quantitative Assessment of PTEN Loss (qPTEN). J. Natl. Cancer Inst. 2020. [Google Scholar] [CrossRef]
- Bazzichetto, C.; Conciatori, F.; Pallocca, M.; Falcone, I.; Fanciulli, M.; Cognetti, F.; Milella, M.; Ciuffreda, L. PTEN as a Prognostic/Predictive Biomarker in Cancer: An Unfulfilled Promise? Cancers 2019, 11, 435. [Google Scholar] [CrossRef]
- Bononi, A.; Pinton, P. Study of PTEN subcellular localization. Methods 2015, 77–78, 92–103. [Google Scholar] [CrossRef]
- Bianchi-Frias, D.; Basom, R.; Delrow, J.J.; Coleman, I.M.; Dakhova, O.; Qu, X.; Fang, M.; Franco, O.E.; Ericson, N.G.; Bielas, J.H.; et al. Cells Comprising the Prostate Cancer Microenvironment Lack Recurrent Clonal Somatic Genomic Aberrations. Mol. Cancer Res. 2016, 14, 374–384. [Google Scholar] [CrossRef]
- Wozniak, D.J.; Kajdacsy-Balla, A.; Macias, V.; Ball-Kell, S.; Zenner, M.L.; Bie, W.; Tyner, A.L. PTEN is a protein phosphatase that targets active PTK6 and inhibits PTK6 oncogenic signaling in prostate cancer. Nat. Commun. 2017, 8, 1508. [Google Scholar] [CrossRef]
- Ong, C.W.; Maxwell, P.A.; Alvi, M.; McQuaid, S.; Waugh, D.; Mills, I.; Salto-Tellez, M. A gene signature associated with PTEN activation defines good prognosis intermediate risk prostate cancer cases. J. Pathol. Clin. Res. 2018, 4, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Punnoose, A.E.; Ferraldeschi, R.; Szafer-Glusman, E.; Tucker, E.K.; Mohan, S.; Flohr, P.; Riisnaes, R.; Miranda, S.; Figueiredo, I.; Rodrigues, D.N.; et al. PTEN loss in circulating tumour cells correlates with PTEN loss in fresh tumour tissue from castration-resistant prostate cancer patients. Br. J. Cancer 2015, 113, 1225–1233. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Hovelson, D.H.; Kemeny, G.; Halabi, S.; Foo, W.; Anand, M.; Somarelli, J.A.; Tomlins, S.A.; Antonarakis, E.S.; Luo, J.; et al. Discordant and heterogeneous clinically relevant genomic alterations in circulating tumor cells vs plasma DNA from men with metastatic castration resistant prostate cancer. Geneschromosom. Cancer 2019, 59, 225–239. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhou, J.; Xia, S.; Li, T. The impact of PTEN deletion and ERG rearrangement on recurrence after treatment for prostate cancer: A systematic review and meta-analysis. Clin. Transl. Oncol. 2019, 22, 694–702. [Google Scholar] [CrossRef]
- Zafarana, G.; Ishkanian, A.S.; Malloff, C.A.; Locke, J.A.; Sykes, J.; Thoms, J.; Lam, W.L.; Squire, J.A.; Yoshimoto, M.; Ramnarine, V.R.; et al. Copy number alterations of c-MYC and PTEN are prognostic factors for relapse after prostate cancer radiotherapy. Cancer 2012, 118, 4053–4062. [Google Scholar] [CrossRef]
- Barnett, C.M.; Heinrich, M.C.; Lim, J.; Nelson, D.; Beadling, C.; Warrick, A.; Neff, T.; Higano, C.S.; Garzotto, M.; Qian, D.; et al. Genetic profiling to determine risk of relapse-free survival in high-risk localized prostate cancer. Clin. Cancer Res. 2013, 20, 1306–1312. [Google Scholar] [CrossRef]
- Yoshimoto, M.; Cunha, I.W.A.; Coudry, R.; Fonseca, F.P.; Torres, C.H.A.; Soares, F.; Squire, J.A. FISH analysis of 107 prostate cancers shows that PTEN genomic deletion is associated with poor clinical outcome. Br. J. Cancer 2007, 97, 678–685. [Google Scholar] [CrossRef]
- Turner, N.C.; Kingston, B.; Kilburn, L.S.; Kernaghan, S.; Wardley, A.M.; MacPherson, I.R.; Baird, R.D.; Roylance, R.; Stephens, P.; Oikonomidou, O.; et al. Circulating tumour DNA analysis to direct therapy in advanced breast cancer (plasmaMATCH): A multicentre, multicohort, phase 2a, platform trial. Lancet Oncol. 2020, 21, 1296–1308. [Google Scholar] [CrossRef]
- Carm, K.T.; Hoff, A.M.; Bakken, A.C.; Axcrona, U.; Axcrona, K.; Lothe, R.A.; Skotheim, R.I.; Løvf, M. Interfocal heterogeneity challenges the clinical usefulness of molecular classification of primary prostate cancer. Sci. Rep. 2019, 9, 1–6. [Google Scholar] [CrossRef]
- Yun, J.W.; Lee, S.; Ryu, D.; Park, S.; Park, W.-Y.; Joung, J.-G.; Jeong, J. Biomarkers Associated with Tumor Heterogeneity in Prostate Cancer. Transl. Oncol. 2018, 12, 43–48. [Google Scholar] [CrossRef]
- Lotan, T.L.; Wei, W.; Morais, C.L.; Hawley, S.T.; Fazli, L.; Hurtado-Coll, A.; Troyer, D.; McKenney, J.K.; Simko, J.; Carroll, P.R.; et al. PTEN Loss as Determined by Clinical-grade Immunohistochemistry Assay Is Associated with Worse Recurrence-free Survival in Prostate Cancer. Eur. Urol. Focus 2016, 2, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, C.W.; Maxwell, P.J.; Ong, C.W.; Redmond, K.M.; McCann, C.; Neisen, J.; Ward, G.A.; Chessari, G.; Johnson, C.; Crawford, N.T.; et al. PTEN deficiency promotes macrophage infiltration and hypersensitivity of prostate cancer to IAP antagonist/radiation combination therapy. Oncotarget 2016, 7, 7885–7898. [Google Scholar] [CrossRef] [PubMed]
- Juric, D.; Castel, P.; Griffith, M.; Griffith, O.L.; Won, H.H.; Ellis, H.; Ebbesen, S.H.; Ainscough, B.J.; Ramu, A.; Iyer, G.; et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nat. Cell Biol. 2014, 518, 240–244. [Google Scholar] [CrossRef] [PubMed]
- James, N.D.; Sydes, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Spears, M.R.; Ritchie, A.W.S.; Parker, C.C.; Russell, J.M.; Attard, G.; et al. Addition of docetaxel, zoledronic acid, or both to first-line long-term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016, 387, 1163–1177. [Google Scholar] [CrossRef]
- Vale, C.L.; Burdett, S.; Rydzewska, L.H.M.; Albiges, L.; Clarke, N.W.; Fisher, D.; Fizazi, K.; Gravis, G.; James, N.D.; Mason, M.D.; et al. Addition of docetaxel or bisphosphonates to standard of care in men with localised or metastatic, hormone-sensitive prostate cancer: A systematic review and meta-analyses of aggregate data. Lancet Oncol. 2016, 17, 243–256. [Google Scholar] [CrossRef]
- Fizazi, K.; Tran, N.; Fein, L.; Matsubara, N.; Rodriguez-Antolin, A.; Alekseev, B.Y.; Özgüroğlu, M.; Ye, D.; Feyerabend, S.; Protheroe, A.; et al. Abiraterone acetate plus prednisone in patients with newly diagnosed high-risk metastatic castration-sensitive prostate cancer (LATITUDE): Final overall survival analysis of a randomised, double-blind, phase 3 trial. Lancet Oncol. 2019, 20, 686–700. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Szmulewitz, R.Z.; Petrylak, D.P.; Holzbeierlein, J.; Villers, A.; Azad, A.; Alcaraz, A.; Alekseev, B.; Iguchi, T.; Shore, N.D.; et al. ARCHES: A Randomized, Phase III Study of Androgen Deprivation Therapy with Enzalutamide or Placebo in Men With Metastatic Hormone-Sensitive Prostate Cancer. J. Clin. Oncol. 2019, 37, 2974–2986. [Google Scholar] [CrossRef]
- James, N.D.; De Bono, J.S.; Spears, M.R.; Clarke, N.W.; Mason, M.D.; Dearnaley, D.P.; Ritchie, A.W.; Amos, C.L.; Gilson, C.; Jones, R.J.; et al. Abiraterone for Prostate Cancer Not Previously Treated with Hormone Therapy. N. Engl. J. Med. 2017, 377, 338–351. [Google Scholar] [CrossRef]
- Davis, I.D.; Martin, A.J.; Stockler, M.R.; Begbie, S.; Chi, K.N.; Chowdhury, S.; Coskinas, X.; Frydenberg, M.; Hague, W.E.; Horvath, L.G.; et al. Enzalutamide with Standard First-Line Therapy in Metastatic Prostate Cancer. N. Engl. J. Med. 2019, 381, 121–131. [Google Scholar] [CrossRef]
- Bismar, T.A.; Hegazy, S.; Feng, Z.; Yu, D.; Donnelly, B.; Palanisamy, N.; Trock, B.J. Clinical utility of assessing PTEN and ERG protein expression in prostate cancer patients: A proposed method for risk stratification. J. Cancer Res. Clin. Oncol. 2018, 144, 2117–2125. [Google Scholar] [CrossRef]
- Wilkinson, S.; Harmon, S.A.; Terrigino, N.T.; Karzai, F.; Pinto, P.A.; Madan, R.A.; VanderWeele, D.J.; Lake, R.; Atway, R.; Bright, J.R.; et al. A case report of multiple primary prostate tumors with differential drug sensitivity. Nat. Commun. 2020, 11, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Arriaga, J.M.; Abate-Shen, C. Genetically Engineered Mouse Models of Prostate Cancer in the Postgenomic Era. Cold Spring Harb. Perspect. Med. 2018, 9, a030528. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Cardiff, R.D.; Desai, N.; Banach-Petrosky, W.A.; Parsons, R.; Shen, M.M.; Abate-Shen, C. Cooperativity of Nkx3.1 and Pten loss of function in a mouse model of prostate carcinogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 2884–2889. [Google Scholar] [CrossRef] [PubMed]
- Alimonti, A.; Carracedo, A.; Clohessy, J.G.; Trotman, L.C.; Nardella, C.; Egia, A.; Salmena, L.; Sampieri, K.; Haveman, W.J.; Brogi, E.; et al. Subtle variations in Pten dose determine cancer susceptibility. Nat. Genet. 2010, 42, 454–458. [Google Scholar] [CrossRef] [PubMed]
- Shen-Li, H.; Koujak, S.; Szablocs, M.; Parsons, R. Reduction of Pten dose leads to neoplastic development in multiple organs of PtenshRNA mice. Cancer Biol. Ther. 2010, 10, 1194–1200. [Google Scholar] [CrossRef]
- Mitragotri, S.; Burke, P.A.; Langer, R. Overcoming the challenges in administering biopharmaceuticals: Formulation and delivery strategies. Nat. Rev. Drug Discov. 2014, 13, 655–672. [Google Scholar] [CrossRef]
- Altınoğlu, S.A.; Wang, M.; Li, K.Q.; Li, Y.; Xu, Q. Intracellular delivery of the PTEN protein using cationic lipidoids for cancer therapy. Biomater. Sci. 2016, 4, 1773–1780. [Google Scholar] [CrossRef]
- Sun, Y.; Wang, S.; Fan, H. The Effect of Nanoparticle Mediated Phosphatase and Tensin Homologue on Chromosome Ten on Prostate Cancer. J. Biomater. Tissue Eng. 2018, 8, 433–437. [Google Scholar] [CrossRef]
- Islam, M.A.; Xu, Y.; Tao, W.; Ubellacker, J.M.; Lim, M.; Aum, D.; Lee, G.Y.; Zhou, K.; Zope, H.; Yu, M.; et al. Restoration of tumour-growth suppression in vivo via systemic nanoparticle-mediated delivery of PTEN mRNA. Nat. Biomed. Eng. 2018, 2, 850–864. [Google Scholar] [CrossRef]
- Putz, U.; Howitt, J.; Doan, A.; Goh, C.-P.; Low, L.-H.; Silke, J.; Tan, S.-S. The Tumor Suppressor PTEN Is Exported in Exosomes and Has Phosphatase Activity in Recipient Cells. Sci. Signal. 2012, 5, ra70. [Google Scholar] [CrossRef]
- Ahmed, S.F.; Das, N.; Sarkar, M.; Chatterjee, U.; Chatterjee, S.; Ghosh, M.K. Exosome-mediated Delivery of the Intrinsic C-terminus Domain of PTEN Protects It From Proteasomal Degradation and Ablates Tumorigenesis. Mol. Ther. 2015, 23, 255–269. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Fine, B.; Steinbach, N.; Dendy, M.; Rapp, Z.; Shaw, J.; Pappas, K.; Yu, J.S.; Hodakoski, C.; Mense, S.; et al. A Secreted PTEN Phosphatase That Enters Cells to Alter Signaling and Survival. Science 2013, 341, 399–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, P.; Lin, C.; Yu, Q.; Wu, J.; Wang, L.; Cui, Y.; Wang, K.; Gao, Z.; Li, H. Relevance and Therapeutic Possibility of PTEN-Long in Renal Cell Carcinoma. PLoS ONE 2015, 10, e114250. [Google Scholar] [CrossRef]
- Lavictoire, S.J.; Gont, A.; Julian, L.M.; Stanford, W.L.; Vlasschaert, C.A.; Gray, D.; Jomaa, D.; Lorimer, I.A.J. Engineering PTEN-L for Cell-Mediated Delivery. Mol. Ther.-Methods Clin. Dev. 2017, 9, 12–22. [Google Scholar] [CrossRef]
- Malaney, P.; Uversky, V.N.; Davé, V. The PTEN Long N-tail is intrinsically disordered: Increased viability for PTEN therapy. Mol. Biosyst. 2013, 9, 2877–2888. [Google Scholar] [CrossRef]
- Hopkins, B.D.; Hodakoski, C.; Barrows, D.; Mense, S.M.; Parsons, R.E. PTEN function: The long and the short of it. Trends Biochem. Sci. 2014, 39, 183–190. [Google Scholar] [CrossRef]
- He, L.; Fan, C.; Kapoor, A.; Ingram, A.J.; Rybak, A.P.; Austin, R.C.; Dickhout, J.; Cutz, J.-C.; Scholey, J.; Tang, D. α-Mannosidase 2C1 attenuates PTEN function in prostate cancer cells. Nat. Commun. 2011, 2, 307. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Ju, J.Y.; Zhou, Y.Q.; Liu, Y.; Zhu, L.P. Inhibition of α-mannosidaseMan2c1gene expression suppresses growth of esophageal carcinoma cells through mitotic arrest and apoptosis. Cancer Sci. 2008, 99, 2428–2434. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Jin, Y.L.; Shi, G.X.; Liu, Y.; Gao, Y.; Zhao, F.T.; Zhu, L.P. Suppression of 6A8 ?-mannosidase gene expression reduced the potentiality of growth and metastasis of human nasopharyngeal carcinoma. Int. J. Cancer 2003, 108, 189–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Guo, J.-X.; Shao, Z.-Q. miR-21 targets and inhibits tumor suppressor gene PTEN to promote prostate cancer cell proliferation and invasion: An experimental study. Asian Pac. J. Trop. Med. 2017, 10, 87–91. [Google Scholar] [CrossRef]
- Zhou, H.; Zhu, X. MicroRNA-21 and microRNA-30c as diagnostic biomarkers for prostate cancer: A meta-analysis. Cancer Manag. Res. 2019, 11, 2039–2050. [Google Scholar] [CrossRef] [PubMed]
- Poliseno, L.; Salmena, L.; Riccardi, L.; Fornari, A.; Song, M.S.; Hobbs, R.M.; Sportoletti, P.; Varmeh, S.; Egia, A.; Fedele, G.; et al. Identification of the miR-106b 25 MicroRNA Cluster as a Proto-Oncogenic PTEN-Targeting Intron That Cooperates with Its Host Gene MCM7 in Transformation. Sci. Signal. 2010, 3, ra29. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; He, B.; He, J.; Mao, X. Upregulation of miR-153 promotes cell proliferation via downregulation of the PTEN tumor suppressor gene in human prostate cancer. Prostate 2012, 73, 596–604. [Google Scholar] [CrossRef]
- Nip, H.; Dar, A.A.; Saini, S.; Colden, M.; Varahram, S.; Chowdhary, H.; Yamamura, S.; Mitsui, Y.; Tanaka, Y.; Kato, T.; et al. Oncogenic microRNA-4534 regulates PTEN pathway in prostate cancer. Oncotarget 2016, 7, 68371–68384. [Google Scholar] [CrossRef]
- Dhar, S.; Kumar, A.; Rimando, A.M.; Zhang, X.; Levenson, A.S. Resveratrol and pterostilbene epigenetically restore PTEN expression by targeting oncomiRs of the miR-17 family in prostate cancer. Oncotarget 2015, 6, 27214–27226. [Google Scholar] [CrossRef]
- Liu, J.-J.; Zhang, X.; Wu, X.-H. miR-93 Promotes the Growth and Invasion of Prostate Cancer by Upregulating Its Target Genes TGFBR2, ITGB8, and LATS2. Mol. Ther.-Oncolytics 2018, 11, 14–19. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Mu, X.; Yin, Q.; Hu, K. miR-106a contributes to prostate carcinoma progression through PTEN. Oncol. Lett. 2018, 17, 1327–1332. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Liu, X.; Li, Y.; Cao, Y.; Silayiding, A.; Zhang, R.; Wang, J. MicroRNA-498 promotes proliferation, migration, and invasion of prostate cancer cells and decreases radiation sensitivity by targeting PTEN. Kaohsiung J. Med. Sci. 2019, 35, 659–671. [Google Scholar] [CrossRef]
- Griveau, A.; Bejaud, J.; Anthiya, S.; Avril, S.; Autret, D.; Garcion, E. Silencing of miR-21 by locked nucleic acid–lipid nanocapsule complexes sensitize human glioblastoma cells to radiation-induced cell death. Int. J. Pharm. 2013, 454, 765–774. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Cui, P.; Zhou, Y.; Chen, C.; Zhao, J.; Wang, H.; Guo, M.; He, Z.; Xu, L. Antisense Oligonucleotides against miR-21 Inhibit the Growth and Metastasis of Colorectal Carcinoma via the DUSP8 Pathway. Mol. Ther.-Nucleic Acids 2018, 13, 244–255. [Google Scholar] [CrossRef]
- Reid, G.; Kao, S.C.; Pavlakis, N.; Brahmbhatt, H.; MacDiarmid, J.; Clarke, S.; Boyer, M.; Van Zandwijk, N. Clinical development of TargomiRs, a miRNA mimic-based treatment for patients with recurrent thoracic cancer. Epigenomics 2016, 8, 1079–1085. [Google Scholar] [CrossRef] [PubMed]
- Garzon, R.; Marcucci, G.; Croce, C.M. Targeting microRNAs in cancer: Rationale, strategies and challenges. Nat. Rev. Drug Discov. 2010, 9, 775–789. [Google Scholar] [CrossRef] [PubMed]
- Shah, M.Y.; Ferrajoli, A.; Sood, A.K.; Lopez-Berestein, G.; Calin, G.A. microRNA Therapeutics in Cancer—An Emerging Concept. EBioMedicine 2016, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Beg, M.S.; Brenner, A.J.; Sachdev, J.; Borad, M.; Kang, Y.-K.; Stoudemire, J.; Smith, S.; Bader, A.G.; Kim, S.; Hong, D.S. Phase I study of MRX34, a liposomal miR-34a mimic, administered twice weekly in patients with advanced solid tumors. Investig. New Drugs 2016, 35, 180–188. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Bryzgunova, O.E.; Konoshenko, M.; Laktionov, P.P. MicroRNA-guided gene expression in prostate cancer: Literature and database overview. J. Gene Med. 2018, 20, e3016. [Google Scholar] [CrossRef]
- Dillon, L.M.; Miller, T.W. Therapeutic targeting of cancers with loss of PTEN function. Curr. Drug Targets 2014, 15, 65–79. [Google Scholar] [CrossRef]
- Soria, J.-C.; Lee, H.-Y.I.; Lee, J.; Wang, L.; Issa, J.-P.; Kemp, B.L.; Liu, D.; Kurie, J.M.; Mao, L.; Khuri, F.R. Lack of PTEN expression in non-small cell lung cancer could be related to promoter methylation. Clin. Cancer Res. 2002, 8, 1178–1184. [Google Scholar] [PubMed]
- Zhao, Z.; Wu, Q.; Cheng, J.; Qiu, X.; Zhang, J.; Fan, H. Depletion ofDNMT3ASuppressed Cell Proliferation and RestoredPTENin Hepatocellular Carcinoma Cell. J. Biomed. Biotechnol. 2010, 2010. [Google Scholar] [CrossRef]
- Dhar, S.; Kumar, A.; Li, K.; Tzivion, G.; Levenson, A.S. Resveratrol regulates PTEN/Akt pathway through inhibition of MTA1/HDAC unit of the NuRD complex in prostate cancer. Biochim. Biophys. Acta (BBA) Bioenerg. 2015, 1853, 265–275. [Google Scholar] [CrossRef]
- Moses, C.; Nugent, F.; Waryah, C.B.; Garcia-Bloj, B.; Harvey, A.R.; Blancafort, P. Activating PTEN Tumor Suppressor Expression with the CRISPR/dCas9 System. Mol. Ther.-Nucleic Acids 2018, 14, 287–300. [Google Scholar] [CrossRef] [PubMed]
- Fang, N.; Gu, T.; Wang, Y.; Wang, S.; Wang, F.; An, Y.; Wei, W.; Zhang, W.; Guo, X.; Nazarali, A.J.; et al. Expression of PTEN-long mediated by CRISPR/Cas9 can repress U87 cell proliferation. J. Cell. Mol. Med. 2017, 21, 3337–3346. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Mou, H.; Li, S.; Li, Y.; Hough, S.; Tran, K.; Li, J.; Yin, H.; Anderson, D.G.; Sontheimer, E.J.; et al. Adenovirus-Mediated Somatic Genome Editing of Pten by CRISPR/Cas9 in Mouse Liver in Spite of Cas9-Specific Immune Responses. Hum. Gene Ther. 2015, 26, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Annunziato, S.; Kas, S.M.; Nethe, M.; Yücel, H.; Del Bravo, J.; Pritchard, C.; Bin Ali, R.; Van Gerwen, B.; Siteur, B.; Drenth, A.P.; et al. Modeling invasive lobular breast carcinoma by CRISPR/Cas9-mediated somatic genome editing of the mammary gland. Genes Dev. 2016, 30, 1470–1480. [Google Scholar] [CrossRef] [PubMed]
- Garza-Elizondo, M.A.; Rodríguez-Rodríguez, D.R.; Ramírez-Solís, R.; Garza-Rodríguez, M.D.L.; Barrera-Saldaña, H.A. Genome editing: A perspective on the application of CRISPR/Cas9 to study human diseases (Review). Int. J. Mol. Med. 2019, 43, 1559–1574. [Google Scholar] [CrossRef]
- Komor, A.C.; Kim, Y.B.; Packer, M.S.; Zuris, J.A.; Liu, D.R. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nat. Cell Biol. 2016, 533, 420–424. [Google Scholar] [CrossRef]
- Hirakawa, M.P.; Krishnakumar, R.; Timlin, J.A.; Carney, J.P.; Butler, K.S. Gene editing and CRISPR in the clinic: Current and future perspectives. Biosci. Rep. 2020, 40. [Google Scholar] [CrossRef]
- Aghamiri, S.; Talaei, S.; Ghavidel, A.A.; Zandsalimi, F.; Masoumi, S.; Hafshejani, N.H.; Jajarmi, V. Nanoparticles-mediated CRISPR/Cas9 delivery: Recent advances in cancer treatment. J. Drug Deliv. Sci. Technol. 2020, 56, 101533. [Google Scholar] [CrossRef]
- Riedel, M.; Berthelsen, M.F.; Bakiri, L.; Wagner, E.F.; Thomsen, M.K. Virus Delivery of CRISPR Guides to the Murine Prostate for Gene Alteration. J. Vis. Exp. 2018, e57525. [Google Scholar] [CrossRef]
- Zhen, S.; Takahashi, Y.; Narita, S.; Yang, Y.-C.; Li, X. Targeted delivery of CRISPR/Cas9 to prostate cancer by modified gRNA using a flexible aptamer-cationic liposome. Oncotarget 2016, 8, 9375–9387. [Google Scholar] [CrossRef] [PubMed]
- Taylor, B.S.; Schultz, N.; Hieronymus, H.; Gopalan, A.; Xiao, Y.; Carver, B.S.; Arora, V.K.; Kaushik, P.; Cerami, E.; Reva, B.; et al. Integrative Genomic Profiling of Human Prostate Cancer. Cancer Cell 2010, 18, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Shorning, B.Y.; Dass, M.S.; Smalley, M.J.; Pearson, H.B. The PI3K-AKT-mTOR Pathway and Prostate Cancer: At the Crossroads of AR, MAPK, and WNT Signaling. Int. J. Mol. Sci. 2020, 21, 4507. [Google Scholar] [CrossRef] [PubMed]
- Crumbaker, M.; Khoja, L.; Joshua, A.M. AR Signaling and the PI3K Pathway in Prostate Cancer. Cancers 2017, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Amzel, L.M.; Huang, C.-H.; Mandelker, D.; Lengauer, C.; Gabelli, S.B.; Vogelstein, B. Structural comparisons of class I phosphoinositide 3-kinases. Nat. Rev. Cancer 2008, 8, 665–669. [Google Scholar] [CrossRef]
- Jean, S.; Kiger, A.A. Classes of phosphoinositide 3-kinases at a glance. J. Cell Sci. 2014, 127, 923–928. [Google Scholar] [CrossRef]
- Thorpe, L.M.; Yuzugullu, H.; Zhao, J.J. PI3K in cancer: Divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2014, 15, 7–24. [Google Scholar] [CrossRef]
- Chalhoub, N.; Baker, S.J. PTEN and the PI3-Kinase Pathway in Cancer. Annu. Rev. Pathol. Mech. Dis. 2009, 4, 127–150. [Google Scholar] [CrossRef]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef]
- Alessi, D.R.; Andjelkovic, M.; Caudwell, B.; Cron, P.; Morrice, N.; Cohen, P.; Hemmings, B.A. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996, 15, 6541–6551. [Google Scholar] [CrossRef]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; et al. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar] [CrossRef]
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Nie, J.; Ma, X.; Wei, Y.; Peng, Y.; Wei, X. Targeting PI3K in cancer: Mechanisms and advances in clinical trials. Mol. Cancer 2019, 18, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; MacLennan, G.T.; Hartman, U.J.; Fu, P.; Resnick, M.I.; Gupta, S. Activation of PI3K-Akt signaling pathway promotes prostate cancer cell invasion. Int. J. Cancer 2007, 121, 1424–1432. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Ding, M.; Yang, L.; Liu, L.-Z.; Jiang, B.-H. PI3K/PTEN/AKT signaling regulates prostate tumor angiogenesis. Cell. Signal. 2007, 19, 2487–2497. [Google Scholar] [CrossRef]
- Gottschalk, A.R.; Doan, A.; Nakamura, J.L.; Stokoe, D.; Haas-Kogan, D.A. Inhibition of phosphatidylinositol-3-kinase causes increased sensitivity to radiation through a PKB-dependent mechanism. Int. J. Radiat. Oncol. 2005, 63, 1221–1227. [Google Scholar] [CrossRef]
- Gupta, A.K.; Cerniglia, G.J.; Mick, R.; Ahmed, M.S.; Bakanauskas, V.J.; Muschel, R.J.; McKenna, W.G. Radiation sensitization of human cancer cells in vivo by inhibiting the activity of PI3K using LY294002. Int. J. Radiat. Oncol. 2003, 56, 846–853. [Google Scholar] [CrossRef]
- Maira, S.-M.; Pecchi, S.; Huang, A.; Burger, M.; Knapp, M.; Sterker, D.; Schnell, C.; Guthy, D.; Nagel, T.; Wiesmann, M.; et al. Identification and Characterization of NVP-BKM120, an Orally Available Pan-Class I PI3-Kinase Inhibitor. Mol. Cancer Ther. 2011, 11, 317–328. [Google Scholar] [CrossRef]
- González-Billalabeitia, E.; Seitzer, N.; Song, S.J.; Song, M.S.; Patnaik, A.; Liu, X.-S.; Epping, M.T.; Papa, A.; Hobbs, R.M.; Chen, M.; et al. Vulnerabilities of PTEN-TP53-Deficient Prostate Cancers to Compound PARP-PI3K Inhibition. Cancer Discov. 2014, 4, 896–904. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Hall, J.; Durston, M.; Voydanoff, A.; VanSickle, E.; Kelly, S.; Nagulapally, A.B.; Bond, J.; Sholler, G.S. BKM120 induces apoptosis and inhibits tumor growth in medulloblastoma. PLoS ONE 2017, 12, e0179948. [Google Scholar] [CrossRef]
- Speranza, M.C.; Nowicki, M.O.; Behera, P.; Cho, C.-F.; Chiocca, E.A.; Lawler, S. BKM-120 (Buparlisib): A Phosphatidyl-Inositol-3 Kinase Inhibitor with Anti-Invasive Properties in Glioblastoma. Sci. Rep. 2016, 6, 20189. [Google Scholar] [CrossRef]
- Anantharaman, A.; Nguyen, H.G.; Cooperberg, M.R.; Meng, M.V.; Carroll, P.; Friedlander, T.W.; Zhang, L.; Thomas, M.; Febbo, P.G.; Feng, F.Y.-C.; et al. A pharmacodynamic study of pre-prostatectomy buparlisib in men with high-risk, localized prostate cancer. J. Clin. Oncol. 2016, 34, e14110. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Scarsella, L.; Colapietro, A.; Jitariuc, A.; Vitale, F.; Marampon, F.; Ricevuto, E.; Festuccia, C. Dual PI3K/mTOR inhibitor, XL765 (SAR245409), shows superior effects to sole PI3K [XL147 (SAR245408)] or mTOR [rapamycin] inhibition in prostate cancer cell models. Tumor Biol. 2015, 37, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Rowley, B.R.; Bull, C.O.; Schneider, C.; Haegebarth, A.; Schatz, C.A.; Fracasso, P.R.; Wilkie, D.P.; Hentemann, M.; Wilhelm, S.M.; et al. BAY 80-6946 Is a Highly Selective Intravenous PI3K Inhibitor with Potent p110α and p110δ Activities in Tumor Cell Lines and Xenograft Models. Mol. Cancer Ther. 2013, 12, 2319–2330. [Google Scholar] [CrossRef] [PubMed]
- Serra, V.; Markman, B.; Scaltriti, M.; Eichhorn, P.J.; Valero, V.; Guzman, M.; Botero, M.L.; Llonch, E.; Atzori, F.; Di Cosimo, S.; et al. NVP-BEZ235, a Dual PI3K/mTOR Inhibitor, Prevents PI3K Signaling and Inhibits the Growth of Cancer Cells with Activating PI3K Mutations. Cancer Res. 2008, 68, 8022–8030. [Google Scholar] [CrossRef]
- Pongas, G.; Fojo, T. BEZ235: When Promising Science Meets Clinical Reality. Oncologist 2016, 21, 1033–1034. [Google Scholar] [CrossRef]
- Schwartz, S.; Wongvipat, J.; Trigwell, C.B.; Hancox, U.; Carver, B.S.; Rodrik-Outmezguine, V.; Will, M.; Yellen, P.; De Stanchina, E.; Baselga, J.; et al. Feedback Suppression of PI3Kα Signaling in PTEN-Mutated Tumors Is Relieved by Selective Inhibition of PI3Kβ. Cancer Cell 2015, 27, 109–122. [Google Scholar] [CrossRef]
- Jia, S.; Liu, Z.; Zhang, S.; Liu, P.; Zhang, L.; Lee, S.H.; Zhang, J.; Signoretti, S.; Loda, M.; Roberts, T.M.; et al. Essential roles of PI(3)K–p110β in cell growth, metabolism and tumorigenesis. Nat. Cell Biol. 2008, 454, 776–779. [Google Scholar] [CrossRef]
- Mateo, J.; Ganji, G.; Lemech, C.; Burris, H.A.; Han, S.-W.; Swales, K.; Decordova, S.; Deyoung, M.P.; Smith, D.A.; Kalyana-Sundaram, S.; et al. A First-Time-in-Human Study of GSK2636771, a Phosphoinositide 3 Kinase Beta-Selective Inhibitor, in Patients with Advanced Solid Tumors. Clin. Cancer Res. 2017, 23, 5981–5992. [Google Scholar] [CrossRef]
- Chi, K.; Protheroe, A.; Antolin, A.R.; Facchini, G.; Suttmann, H.; Matsubara, N.; Ye, Z.-Q.; Keam, B.; Li, T.; McQuarrie, K.; et al. Benefits of Abiraterone Acetate Plus Prednisone (AA+P) When Added to Androgen Deprivation Therapy (ADT) in LATITUDE on Patient (Pt) Reported Outcomes (PRO). Ann. Oncol. 2017, 28, v269. [Google Scholar] [CrossRef]
- Ni, J.; Liu, Q.; Xie, S.; Carlson, C.B.; Von, T.; Vogel, K.W.; Riddle, S.M.; Benes, C.H.; Eck, M.J.; Roberts, T.M.; et al. Functional characterization of an isoform-selective inhibitor of PI3K-p110β as a potential anticancer agent. Cancer Discov. 2012, 2, 425–433. [Google Scholar] [CrossRef]
- Bonnevaux, H.; Levit, M.N.; Windenberger, F.; Delorme, C.; Virone-Oddos, A.; Lemaitre, O.; Vincent, L.; Halley, F.; Lengauer, C.; Garcia-Echeverria, C. Concomitant Inhibition of PI3K? and BRAF or MEK in PTEN-Deficient/ BRAF-Mutant Melanoma Treatment: Preclinical Assessment of SAR260301 Oral PI3Kβ-Selective Inhibitor. Mol. Cancer Ther. 2016, 15, 1460–1471. [Google Scholar] [CrossRef] [PubMed]
- Bedard, P.L.; Davies, M.A.; Kopetz, S.; Juric, D.; Shapiro, G.I.; Luke, J.J.; Spreafico, A.; Wu, B.; Castell, C.; Gomez, C.; et al. First-in-human trial of the PI3Kβ-selective inhibitor SAR260301 in patients with advanced solid tumors. Cancer 2017, 124, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhao, Y.; Huang, Y.; Yang, Q.; Zeng, X.; Jiang, W.; Liu, J.; Thrasher, J.B.; Forrest, M.L.; Li, B. Nanomicellar TGX221 blocks xenograft tumor growth of prostate cancer in nude mice. Prostate 2015, 75, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Edgar, K.A.; Wallin, J.J.; Berry, M.; Lee, L.B.; Prior, W.W.; Sampath, D.; Friedman, L.S.; Belvin, M. Isoform-Specific Phosphoinositide 3-Kinase Inhibitors Exert Distinct Effects in Solid Tumors. Cancer Res. 2010, 70, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Tai, W.; Shukla, R.S.; Qin, B.; Li, B.; Cheng, K. Development of a Peptide–Drug Conjugate for Prostate Cancer Therapy. Mol. Pharm. 2011, 8, 901–912. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Duan, S.; Zeng, X.; Liu, C.; Davies, N.M.; Li, B.; Forrest, M.L. Prodrug Strategy for PSMA-Targeted Delivery of TGX-221 to Prostate Cancer Cells. Mol. Pharm. 2012, 9, 1705–1716. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Duan, S.; Dong, L.; Wang, Y.; Hu, Q.; Liu, C.; Forrest, M.L.; Holzbeierlein, J.M.; Han, S.; Li, B. Characterization of a novel p110β-specific inhibitor BL140 that overcomes MDV3100-resistance in castration-resistant prostate cancer cells. Prostate 2017, 77, 1187–1198. [Google Scholar] [CrossRef]
- Castel, P.; Scaltriti, M. Mechanisms of Resistance to PI3K and AKT Inhibitors. In Resistance to Targeted Anti-Cancer Therapeutics; Springer: Cham, Switzerland, 2018; Volume 15, pp. 117–146. [Google Scholar]
- Carver, B.S.; Chapinski, C.; Wongvipat, J.; Hieronymus, H.; Chen, Y.; Chandarlapaty, S.; Arora, V.K.; Le, C.; Koutcher, J.; Scher, H.; et al. Reciprocal Feedback Regulation of PI3K and Androgen Receptor Signaling in PTEN-Deficient Prostate Cancer. Cancer Cell 2011, 19, 575–586. [Google Scholar] [CrossRef]
- Sweeney, C.; Percent, I.J.; Babu, S.; Cultrera, J.; Mehlhaff, B.A.; Goodman, O.B.; Morris, D.; Schnadig, I.D.; Albany, C.; Shore, N.D.; et al. Phase 1b/2 study of enzalutamide (ENZ) with LY3023414 (LY) or placebo (PL) in patients (pts) with metastatic castration-resistant prostate cancer (mCRPC) after progression on abiraterone. J. Clin. Oncol. 2019, 37, 5009. [Google Scholar] [CrossRef]
- Massard, C.; Chi, K.N.; Castellano, D.; De Bono, J.; Gravis, G.; Dirix, L.; Machiels, J.-P.; Mita, A.; Mellado, B.; Turri, S.; et al. Phase Ib dose-finding study of abiraterone acetate plus buparlisib (BKM120) or dactolisib (BEZ235) in patients with castration-resistant prostate cancer. Eur. J. Cancer 2017, 76, 36–44. [Google Scholar] [CrossRef]
- Hu, C.; Xia, H.; Bai, S.; Zhao, J.; Edwards, H.; Li, X.; Yang, Y.; Lyu, J.; Wang, G.; Zhan, Y.; et al. CUDC-907, a novel dual PI3K and HDAC inhibitor, in prostate cancer: Antitumour activity and molecular mechanism of action. J. Cell. Mol. Med. 2020, 24, 7239–7253. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.M.; Vessella, R.L.; Morrissey, C.; Brown, L.G.; Coleman, I.M.; Higano, C.S.; Mostaghel, E.A.; Zhang, X.; True, L.D.; Lam, H.-M.; et al. LuCaP Prostate Cancer Patient-Derived Xenografts Reflect the Molecular Heterogeneity of Advanced Disease and Serve as Models for Evaluating Cancer Therapeutics. Prostate 2017, 77, 654–671. [Google Scholar] [CrossRef]
- Jiang, X.; Chen, S.; Asara, J.M.; Balk, S.P. Phosphoinositide 3-Kinase Pathway Activation in Phosphate and Tensin Homolog (PTEN)-deficient Prostate Cancer Cells Is Independent of Receptor Tyrosine Kinases and Mediated by the p110β and p110δ Catalytic Subunits. J. Biol. Chem. 2010, 285, 14980–14989. [Google Scholar] [CrossRef] [PubMed]
- Etzenaki, N.; Papakonstanti, E.A. p110δ PI3 kinase pathway: Emerging roles in cancer. Front. Oncol. 2013, 3, 40. [Google Scholar] [CrossRef]
- Zou, Y.; Qi, Z.; Guo, W.; Zhang, L.; Ruscetti, M.; Shenoy, T.; Liu, N.; Wu, H. Cotargeting the Cell-Intrinsic and Microenvironment Pathways of Prostate Cancer by PI3Kα/β/δ Inhibitor BAY1082439. Mol. Cancer Ther. 2018, 17, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Standaert, M.L.; Bandyopadhyay, G.; Kanoh, Y.; Sajan, M.P.; Farese, R.V. Insulin and PIP3Activate PKC-ζ by Mechanisms That Are Both Dependent and Independent of Phosphorylation of Activation Loop (T410) and Autophosphorylation (T560) Sites. Biochemistry 2001, 40, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Leong, M.L.; Buse, P.; Maiyar, A.C.; Firestone, G.L.; Hemmings, B.A. Serum and glucocorticoid-inducible kinase (SGK) is a target of the PI 3-kinase-stimulated signaling pathway. EMBO J. 1999, 18, 3024–3033. [Google Scholar] [CrossRef]
- Wertheimer, E.; Gutierrez-Uzquiza, A.; Rosemblit, C.; Lopez-Haber, C.; Sosa, M.S.; Kazanietz, M.G. Rac signaling in breast cancer: A tale of GEFs and GAPs. Cell. Signal. 2012, 24, 353–362. [Google Scholar] [CrossRef]
- Wang, Q.; Chen, X.; Hay, N. Akt as a target for cancer therapy: More is not always better (lessons from studies in mice). Br. J. Cancer 2017, 117, 159–163. [Google Scholar] [CrossRef]
- Chin, Y.R.; Yuan, X.; Balk, S.P.; Toker, A. PTEN-Deficient Tumors Depend on AKT2 for Maintenance and Survival. Cancer Discov. 2014, 4, 942–955. [Google Scholar] [CrossRef]
- Chen, M.-L.; Xu, P.-Z.; Peng, X.-D.; Chen, W.S.; Guzman, G.; Yang, X.; Di Cristofano, A.; Pandolfi, P.P.; Hay, N. The deficiency of Akt1 is sufficient to suppress tumor development in Pten+/− mice. Genes Dev. 2006, 20, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Xu, P.-Z.; Chen, M.-L.; Jeon, S.-M.; Peng, X.-D.; Hay, N. The effect Akt2 deletion on tumor development in Pten+/− mice. Oncogene 2011, 31, 518–526. [Google Scholar] [CrossRef]
- Lin, H.-P.; Lin, C.-Y.; Huo, C.; Jan, Y.-J.; Tseng, J.-C.; Jiang, S.S.; Kuo, Y.-Y.; Chen, S.-C.; Wang, C.-T.; Chan, T.-M.; et al. AKT3 promotes prostate cancer proliferation cells through regulation of Akt, B-Raf & TSC1/TSC2. Oncotarget 2015, 6, 27097–27112. [Google Scholar] [CrossRef] [PubMed]
- Bergeron, Y.; Bureau, G.; Laurier-Laurin, M.É.; Asselin, E.; Massicotte, G.; Cyr, M. Genetic Deletion of Akt3 Induces an Endophenotype Reminiscent of Psychiatric Manifestations in Mice. Front. Mol. Neurosci. 2017, 10, 102. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Yu, W.-N.; Chen, X.; Peng, X.-D.; Jeon, S.-M.; Birnbaum, M.J.; Guzman, G.; Hay, N. Spontaneous Hepatocellular Carcinoma after the Combined Deletion of Akt Isoforms. Cancer Cell 2016, 29, 523–535. [Google Scholar] [CrossRef]
- Crabb, S.J.; Birtle, A.J.; Martin, K.; Downs, N.; Ratcliffe, I.; Maishman, T.; Ellis, M.; Griffiths, G.; Thompson, S.; Ksiazek, L.; et al. ProCAID: A phase I clinical trial to combine the AKT inhibitor AZD5363 with docetaxel and prednisolone chemotherapy for metastatic castration resistant prostate cancer. Investig. New Drugs 2017, 35, 599–607. [Google Scholar] [CrossRef] [PubMed]
- Kolinsky, M.; Rescigno, P.; Bianchini, D.; Zafeiriou, Z.; Mehra, N.; Mateo, J.; Michalarea, V.; Riisnaes, R.; Crespo, M.; Figueiredo, I.; et al. A phase I dose-escalation study of enzalutamide in combination with the AKT inhibitor AZD5363 (capivasertib) in patients with metastatic castration-resistant prostate cancer. Ann. Oncol. 2020, 31, 619–625. [Google Scholar] [CrossRef]
- Tamura, K.; Hashimoto, J.; Tanabe, Y.; Kodaira, M.; Yonemori, K.; Seto, T.; Hirai, F.; Arita, S.; Toyokawa, G.; Chen, L.; et al. Safety and tolerability of AZD5363 in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2016, 77, 787–795. [Google Scholar] [CrossRef]
- Banerji, U.; Dean, E.; Pérez-Fidalgo, J.A.; Batist, G.; Bedard, P.L.; You, B.; Westin, S.N.; Kabos, P.; Garrett, M.D.; Tall, M.; et al. A Phase I Open-Label Study to Identify a Dosing Regimen of the Pan-AKT Inhibitor AZD5363 for Evaluation in Solid Tumors and inPIK3CA-Mutated Breast and Gynecologic Cancers. Clin. Cancer Res. 2017, 24, 2050–2059. [Google Scholar] [CrossRef]
- Saura, C.; Roda, D.; Roselló, S.; Oliveira, M.; Macarulla, T.; Pérez-Fidalgo, J.A.; Morales-Barrera, R.; Sanchis-García, J.M.; Musib, L.; Budha, N.; et al. A First-in-Human Phase I Study of the ATP-Competitive AKT Inhibitor Ipatasertib Demonstrates Robust and Safe Targeting of AKT in Patients with Solid Tumors. Cancer Discov. 2016, 7, 102–113. [Google Scholar] [CrossRef]
- Gupta, S.; Argilés, G.; Munster, P.N.; Hollebecque, A.; Dajani, O.; Cheng, J.D.; Wang, R.; Swift, A.; Tosolini, A.; Piha-Paul, S. A Phase I Trial of Combined Ridaforolimus and MK-2206 in Patients with Advanced Malignancies. Clin. Cancer Res. 2015, 21, 5235–5244. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Saura, C.; Jones, S.; Mateo, J.; Hollebecque, A.; Cleary, J.M.; Perez, D.R.; Zhu, J.; Musib, L.C.; Patel, P.H.; Cervantes-Ruiperez, A.; et al. A phase Ib study of the Akt inhibitor GDC-0068 with docetaxel (D) or mFOLFOX-6 (F) in patients (pts) with advanced solid tumors. J. Clin. Oncol. 2012, 30, 3021. [Google Scholar] [CrossRef]
- Zhang, W.; Haines, B.B.; Efferson, C.; Zhu, J.; Ware, C.; Kunii, K.; Tammam, J.; Angagaw, M.; Hinton, M.C.; Keilhack, H.; et al. Evidence of mTOR Activation by an AKT-Independent Mechanism Provides Support for the Combined Treatment of PTEN-Deficient Prostate Tumors with mTOR and AKT Inhibitors. Transl. Oncol. 2012, 5, 422–429. [Google Scholar] [CrossRef][Green Version]
- Velasco, M.A.; Kura, Y.; Yoshikawa, K.; Nishio, K.; Davies, B.R.; Uemura, H. Efficacy of targeted AKT inhibition in genetically engineered mouse models of PTEN-deficient prostate cancer. Oncotarget 2016, 7, 15959–15976. [Google Scholar] [CrossRef]
- Davies, B.R.; Greenwood, H.; Dudley, P.; Crafter, C.; Yu, D.; Zhang, J.; Li, J.; Gao, B.; Ji, Q.; Maynard, J.; et al. Preclinical Pharmacology of AZD5363, an Inhibitor of AKT: Pharmacodynamics, Antitumor Activity, and Correlation of Monotherapy Activity with Genetic Background. Mol. Cancer Ther. 2012, 11, 873–887. [Google Scholar] [CrossRef]
- Sangai, T.; Akcakanat, A.; Chen, H.; Tarco, E.; Wu, Y.; Do, K.-A.; Miller, T.W.; Arteaga, C.L.; Mills, G.B.; Gonzalez-Angulo, A.M.; et al. Biomarkers of Response to Akt Inhibitor MK-2206 in Breast Cancer. Clin. Cancer Res. 2012, 18, 5816–5828. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Sampath, D.; Nannini, M.; Lee, B.B.; Degtyarev, M.; Oeh, J.; Savage, H.; Guan, Z.; Hong, R.; Kassees, R.; et al. Targeting Activated Akt with GDC-0068, a Novel Selective Akt Inhibitor That Is Efficacious in Multiple Tumor Models. Clin. Cancer Res. 2013, 19, 1760–1772. [Google Scholar] [CrossRef]
- Brown, K.K.; Toker, A. The phosphoinositide 3-kinase pathway and therapy resistance in cancer. F1000Prime Rep. 2015, 7, 13. [Google Scholar] [CrossRef]
- Chandarlapaty, S.; Sawai, A.; Scaltriti, M.; Rodrik-Outmezguine, V.; Grbovic-Huezo, O.; Serra, V.; Majumder, P.K.; Baselga, J.; Rosen, N. AKT Inhibition Relieves Feedback Suppression of Receptor Tyrosine Kinase Expression and Activity. Cancer Cell 2011, 19, 58–71. [Google Scholar] [CrossRef]
- Doi, T.; Fujiwara, Y.; Matsubara, N.; Tomomatsu, J.; Iwasa, S.; Tanaka, A.; Endo-Tsukude, C.; Nakagawa, S.; Takahashi, S. Phase I study of ipatasertib as a single agent and in combination with abiraterone plus prednisolone in Japanese patients with advanced solid tumors. Cancer Chemother. Pharmacol. 2019, 84, 393–404. [Google Scholar] [CrossRef]
- De Bono, J.S.; De Giorgi, U.; Rodrigues, D.N.; Massard, C.; Bracarda, S.; Font, A.; Arija, J.A.A.; Shih, K.C.; Radavoi, G.D.; Xu, N.; et al. Randomized Phase II Study Evaluating Akt Blockade with Ipatasertib, in Combination with Abiraterone, in Patients with Metastatic Prostate Cancer with and without PTEN Loss. Clin. Cancer Res. 2019, 25, 928–936. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, C.; Bell-McGuinn, K.M.; Burris, H.A.; Siu, L.L.; Stayner, L.-A.; Wheler, J.J.; Hong, D.S.; Kurkjian, C.; Pant, S.; Santiago-Walker, A.; et al. A phase I, open-label, two-stage study to investigate the safety, tolerability, pharmacokinetics, and pharmacodynamics of the oral AKT inhibitor GSK2141795 in patients with solid tumors. Investig. New Drugs 2018, 36, 1016–1025. [Google Scholar] [CrossRef] [PubMed]
- He, K.; Hu, H.; Ye, S.; Wang, H.; Cui, R.; Yi, L. The effect of metformin therapy on incidence and prognosis in prostate cancer: A systematic review and meta-analysis. Sci. Rep. 2019, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, B.D.; Pauli, C.; Du, X.; Wang, D.G.; Li, X.; Wu, D.; Amadiume, S.C.; Goncalves, M.D.; Hodakoski, C.; Lundquist, M.R.; et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nat. Cell Biol. 2018, 560, 499–503. [Google Scholar] [CrossRef]
- Efeyan, A.; Sabatini, D.M. mTOR and cancer: Many loops in one pathway. Curr. Opin. Cell Biol. 2010, 22, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Hua, H.; Kong, Q.; Zhang, H.; Wang, J.; Luo, T.; Jiang, Y. Targeting mTOR for cancer therapy. J. Hematol. Oncol. 2019, 12, 71. [Google Scholar] [CrossRef]
- Chen, X.; Xiong, X.; Cui, D.; Yang, F.; Wei, D.; Li, H.; Shu, J.; Bi, Y.; Dai, X.; Gong, L.; et al. DEPTOR is an in vivo tumor suppressor that inhibits prostate tumorigenesis via the inactivation of mTORC1/2 signals. Oncogene 2019, 39, 1557–1571. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged Rapamycin Treatment Inhibits mTORC2 Assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Huang, J.; Manning, B.D. A complex interplay between Akt, TSC2 and the two mTOR complexes. Biochem. Soc. Trans. 2009, 37, 217–222. [Google Scholar] [CrossRef]
- Blando, J.; Portis, M.; Benavides, F.; Alexander, A.; Mills, G.; Dave, B.; Conti, C.J.; Kim, J.; Walker, C.L. PTEN Deficiency Is Fully Penetrant for Prostate Adenocarcinoma in C57BL/6 Mice via mTOR-Dependent Growth. Am. J. Pathol. 2009, 174, 1869–1879. [Google Scholar] [CrossRef]
- Wu, L.; Birle, D.C.; Tannock, I.F. Effects of the Mammalian Target of Rapamycin Inhibitor CCI-779 Used Alone or with Chemotherapy on Human Prostate Cancer Cells and Xenografts. Cancer Res. 2005, 65, 2825–2831. [Google Scholar] [CrossRef]
- Grünwald, V.; Degraffenried, L.; Russel, D.E.; Friedrichs, W.; Ray, R.B.; Hidalgo, M. Inhibitors of mTOR reverse doxorubicin resistance conferred by PTEN status in prostate cancer cells. Cancer Res. 2002, 62, 6141–6145. [Google Scholar] [PubMed]
- Neshat, M.S.; Mellinghoff, I.K.; Tran, C.; Stiles, B.; Thomas, G.; Petersen, R.; Frost, P.; Gibbons, J.J.; Wu, H.; Sawyers, C.L. Enhanced sensitivity of PTEN-deficient tumors to inhibition of FRAP/mTOR. Proc. Natl. Acad. Sci. USA 2001, 98, 10314–10319. [Google Scholar] [CrossRef] [PubMed]
- Podsypanina, K.; Lee, R.T.; Politis, C.; Hennessy, I.; Crane, A.; Puc, J.; Neshat, M.; Wang, H.; Yang, L.; Gibbons, J.; et al. An inhibitor of mTOR reduces neoplasia and normalizes p70/S6 kinase activity in Pten+/− mice. Proc. Natl. Acad. Sci. USA 2001, 98, 10320–10325. [Google Scholar] [CrossRef] [PubMed]
- Milam, M.R.; Celestino, J.; Wu, W.; Broaddus, R.R.; Schmeler, K.M.; Slomovitz, B.M.; Soliman, P.T.; Gershenson, D.M.; Wang, H.; Ellenson, L.H.; et al. Reduced progression of endometrial hyperplasia with oral mTOR inhibition in the Pten heterozygote murine model. Am. J. Obstet. Gynecol. 2007, 196, 247.e1–247.e5. [Google Scholar] [CrossRef]
- Kenerson, H.L.; Yeh, M.M.; Kazami, M.; Jiang, X.; Riehle, K.J.; McIntyre, R.L.; Park, J.O.; Kwon, S.; Campbell, J.S.; Yeung, R.S. Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology 2013, 144, 1055–1065. [Google Scholar] [CrossRef]
- Ma, L.; Teruya-Feldstein, J.; Behrendt, N.; Chen, Z.; Noda, T.; Hino, O.; Cordon-Cardo, C.; Pandolfi, P.P. Genetic analysis of Pten and Tsc2 functional interactions in the mouse reveals asymmetrical haploinsufficiency in tumor suppression. Genes Dev. 2005, 19, 1779–1786. [Google Scholar] [CrossRef]
- Manning, B.D.; Logsdon, M.N.; Lipovsky, A.I.; Abbott, D.; Kwiatkowski, D.J.; Cantley, L.C. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005, 19, 1773–1778. [Google Scholar] [CrossRef]
- Nellist, M.; Van Slegtenhorst, M.A.; Goedbloed, M.; Ouweland, A.M.W.V.D.; Halley, D.J.J.; Van Der Sluijs, P. Characterization of the Cytosolic Tuberin-Hamartin Complex. J. Biol. Chem. 1999, 274, 35647–35652. [Google Scholar] [CrossRef]
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.-H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR Complex 2 Is Required for the Development of Prostate Cancer Induced by Pten Loss in Mice. Cancer Cell 2009, 15, 148–159. [Google Scholar] [CrossRef]
- Rodrik-Outmezguine, V.S.; Okaniwa, M.; Yao, Z.; Novotny, C.J.; McWhirter, C.; Banaji, A.; Won, H.; Wong, W.; Berger, H.W.M.; De Stanchina, W.W.E.; et al. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nat. Cell Biol. 2016, 534, 272–276. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.-Y. mTOR kinase inhibitors as potential cancer therapeutic drugs. Cancer Lett. 2013, 340, 1–8. [Google Scholar] [CrossRef]
- Dai, Y.; Zhao, L.; Siemann, D.W. Abstract B59: Dual mTOR kinase inhibitor reverses rapamycin resistance in prostate cancer cells. Drug Resist. Modif. 2015, 14, 59. [Google Scholar] [CrossRef]
- La Manna, F.; De Menna, M.; Patel, N.; Karkampouna, S.; De Filippo, M.; Klima, I.; Kloen, P.; Beimers, L.; Thalmann, G.N.; Pelger, R.C.M.; et al. Dual-mTOR Inhibitor Rapalink-1 Reduces Prostate Cancer Patient-Derived Xenograft Growth and Alters Tumor Heterogeneity. Front. Oncol. 2020, 10, 1012. [Google Scholar] [CrossRef]
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nat. Cell Biol. 2012, 485, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Sandi, C.; Ramos-Montoya, A.; Felisbino, S.L.; Jurmeister, S.; Madhu, B.; Wadhwa, K.; Griffiths, J.R.; Richards, F.M.; Jodrell, D.I.E.; Neal, D.; et al. Abstract A123: Preclinical evaluation of dual mTOR inhibitor, AZD2014, in prostate cancer. Mol. Cancer Ther. 2015, 14. [Google Scholar] [CrossRef]
- Pacey, S.; Shah, N.; Davies, B.; Bratt, O.; Warren, A.; Baird, R.D.; Gnanapragasam, V.; Ingle, S.; Stearn, S.; Machin, A.; et al. A pharmacodynamic biomarker study of vistusertib (AZD2014), an mTORC1/2 inhibitor, given prior to radical prostatectomy (CANCAP02). J. Clin. Oncol. 2018, 36, 5081. [Google Scholar] [CrossRef]
- Amato, R.J.; Jac, J.; Mohammad, T.; Saxena, S. Pilot Study of Rapamycin in Patients with Hormone-Refractory Prostate Cancer. Clin. Genitourin. Cancer 2008, 6, 97–102. [Google Scholar] [CrossRef]
- Narayan, V.; Vapiwala, N.; Mick, R.; Subramanian, P.; Christodouleas, J.P.; Bekelman, J.E.; Deville, C.; Rajendran, R.; Haas, N.B. Phase 1 Trial of Everolimus and Radiation Therapy for Salvage Treatment of Biochemical Recurrence in Prostate Cancer Patients Following Prostatectomy. Int. J. Radiat. Oncol. 2017, 97, 355–361. [Google Scholar] [CrossRef]
- Kruczek, K.; Ratterman, M.; Tolzien, K.; Sulo, S.; Lestingi, T.M.; Nabhan, C. A phase II study evaluating the toxicity and efficacy of single-agent temsirolimus in chemotherapy-naïve castration-resistant prostate cancer. Br. J. Cancer 2013, 109, 1711–1716. [Google Scholar] [CrossRef]
- Armstrong, A.J.; Shen, T.; Halabi, S.; Kemeny, G.; Bitting, R.L.; Kartcheske, P.; Embree, E.; Morris, K.; Winters, C.; Jaffe, T.; et al. A Phase II Trial of Temsirolimus in Men with Castration-Resistant Metastatic Prostate Cancer. Clin. Genitourin. Cancer 2013, 11, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Vaishampayan, U.; Shevrin, D.; Stein, M.; Heilbrun, L.; Land, S.; Stark, K.; Li, J.; Dickow, B.; Heath, E.; Smith, D.; et al. Phase II Trial of Carboplatin, Everolimus, and Prednisone in Metastatic Castration-resistant Prostate Cancer Pretreated with Docetaxel Chemotherapy: A Prostate Cancer Clinical Trial Consortium Study. Urology 2015, 86, 1206–1211. [Google Scholar] [CrossRef] [PubMed]
- Rathkopf, D.; Larson, S.M.; Anand, A.; Morris, M.J.; Slovin, S.F.; Shaffer, D.R.; Heller, G.; Carver, B.; Rosen, N.; Scher, H.I. Everolimus combined with gefitinib in patients with metastatic castration-resistant prostate cancer: Phase 1/2 results and signaling pathway implications. Cancer 2015, 121, 3853–3861. [Google Scholar] [CrossRef] [PubMed]
- Koshkin, V.S.; Mir, M.C.; Barata, P.; Gul, A.; Gupta, R.; Stephenson, A.J.; Kaouk, J.; Berglund, R.; Magi-Galluzzi, C.; Klein, E.A.; et al. Randomized phase II trial of neoadjuvant everolimus in patients with high-risk localized prostate cancer. Investig. New Drugs 2019, 37, 559–566. [Google Scholar] [CrossRef]
- George, D.J.; Halabi, S.; Healy, P.; Jonasch, D.; Anand, M.; Rasmussen, J.; Wood, S.Y.; Spritzer, C.; Madden, J.F.; Armstrong, A.J. Phase 2 clinical trial of TORC1 inhibition with everolimus in men with metastatic castration-resistant prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2020, 38, 79.e15–79.e22. [Google Scholar] [CrossRef]
- McHugh, D.J.; Chudow, J.; DeNunzio, M.; Slovin, S.F.; Danila, D.C.; Morris, M.J.; Scher, H.I.; Rathkopf, D.E. A Phase I Trial of IGF-1R Inhibitor Cixutumumab and mTOR Inhibitor Temsirolimus in Metastatic Castration-resistant Prostate Cancer. Clin. Genitourin. Cancer 2020, 18, 171–178.e2. [Google Scholar] [CrossRef]
- Gonzalez-Angulo, A.M.; Meric-Bernstam, F.; Chawla, S.; Falchook, G.; Hong, D.; Akcakanat, A.; Chen, H.; Naing, A.; Fu, S.; Wheler, J.; et al. Weekly nab-Rapamycin in Patients with Advanced Nonhematologic Malignancies: Final Results of a Phase I Trial. Clin. Cancer Res. 2013, 19, 5474–5484. [Google Scholar] [CrossRef]
- Bohan, P.M.K.; Cindass, J.L.; Chick, R.C.; Vreeland, T.J.; Hale, D.F.; Hickerson, A.; Clifton, G.T.; Peoples, G.E.; Liss, M. Results of a phase Ib trial of encapsulated rapamycin in prostate cancer patients under active surveillance to prevent progression. J. Clin. Oncol. 2020, 38, 34. [Google Scholar] [CrossRef]
- Templeton, A.; Dutoit, V.; Cathomas, R.; Rothermundt, C.; Bärtschi, D.; Dröge, C.; Gautschi, O.; Borner, M.; Fechter, E.; Stenner-Liewen, F.; et al. Phase 2 Trial of Single-agent Everolimus in Chemotherapy-naive Patients with Castration-resistant Prostate Cancer (SAKK 08/08). Eur. Urol. 2013, 64, 150–158. [Google Scholar] [CrossRef]
- Kmak, J.A.; Agarwal, N.; He, Y.; Heilmann, A.M.; Miller, V.A.; Ross, J.S.; Pal, S.K.; Ali, S.M.; Kilari, D. Exceptional Response to Everolimus in a Patient with Metastatic Castrate-Resistant Prostate Cancer Harboring a PTEN Inactivating Mutation. Case Rep. Oncol. 2020, 13, 456–461. [Google Scholar] [CrossRef]
- Chow, H.; Ghosh, P.M.; White, R.D.; Evans, C.P.; Dall’Era, M.A.; Yap, S.A.; Li, Y.; Beckett, L.A.; Lara, P.N.; Pan, C.-X. A phase 2 clinical trial of everolimus plus bicalutamide for castration-resistant prostate cancer. Cancer 2016, 122, 1897–1904. [Google Scholar] [CrossRef]
- Nakabayashi, M.; Werner, L.; Courtney, K.D.; Buckle, G.; Oh, W.K.; Bubley, G.J.; Hayes, J.H.; Weckstein, D.; ElFiky, A.; Sims, D.M.; et al. Phase II trial of RAD001 and bicalutamide for castration-resistant prostate cancer. BJU Int. 2012, 110, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Sherk, A.B.; Frigo, D.E.; Schnackenberg, C.G.; Bray, J.D.; Laping, N.J.; Trizna, W.; Hammond, M.; Patterson, J.R.; Thompson, S.K.; Kazmin, D.; et al. Development of a Small-Molecule Serum- and Glucocorticoid-Regulated Kinase-1 Antagonist and Its Evaluation as a Prostate Cancer Therapeutic. Cancer Res. 2008, 68, 7475–7483. [Google Scholar] [CrossRef]
- Bago, R.; Sommer, E.; Castel, P.; Crafter, C.; Bailey, F.P.; Shpiro, N.; Baselga, J.; Cross, D.A.; Eyers, P.; Alessi, D.R. The hVps34- SGK 3 pathway alleviates sustained PI3K/Akt inhibition by stimulating mTORC 1 and tumour growth. EMBO J. 2016, 35, 1902–1922. [Google Scholar] [CrossRef] [PubMed]
- Di Cristofano, A. SGK1: The dark side of PI3K signaling. Curr. Top. Dev. Biol. 2017, 123, 49–71. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Cohen, P. Activation of serum- and glucocorticoid-regulated protein kinase by agonists that activate phosphatidylinositide 3-kinase is mediated by 3-phosphoinositide-dependent protein kinase-1 (PDK1) and PDK2. Biochem. J. 1999, 339, 319–328. [Google Scholar] [CrossRef]
- García-Martínez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef]
- Castel, P.; Ellis, H.; Bago, R.; Toska, E.; Razavi, P.; Carmona, F.J.; Kannan, S.; Verma, C.S.; Dickler, M.; Chandarlapaty, S.; et al. PDK1-SGK1 Signaling Sustains AKT-Independent mTORC1 Activation and Confers Resistance to PI3Kα Inhibition. Cancer Cell 2016, 30, 229–242. [Google Scholar] [CrossRef] [PubMed]
- Brunet, A.; Park, J.; Tran, H.; Hu, L.S.; Hemmings, B.A.; Greenberg, M.E. Protein Kinase SGK Mediates Survival Signals by Phosphorylating the Forkhead Transcription Factor FKHRL1 (FOXO3a). Mol. Cell. Biol. 2001, 21, 952–965. [Google Scholar] [CrossRef]
- Zhang, B.-H.; Tang, E.D.; Zhu, T.; Greenberg, M.E.; Vojtek, A.B.; Guan, K.-L. Serum- and Glucocorticoid-inducible Kinase SGK Phosphorylates and Negatively Regulates B-Raf. J. Biol. Chem. 2001, 276, 31620–31626. [Google Scholar] [CrossRef]
- Sommer, E.M.; Dry, H.; Cross, D.; Guichard, S.; Davies, B.R.; Alessi, D.R. Elevated SGK1 predicts resistance of breast cancer cells to Akt inhibitors. Biochem. J. 2013, 452, 499–508. [Google Scholar] [CrossRef]
- Liu, W.; Wang, X.; Wang, Y.; Dai, Y.; Xie, Y.; Ping, Y.; Yin, B.; Yu, P.; Liu, Z.; Duan, X.; et al. SGK1 inhibition-induced autophagy impairs prostate cancer metastasis by reversing EMT. J. Exp. Clin. Cancer Res. 2018, 37, 1–12. [Google Scholar] [CrossRef]
- Orlacchio, A.; Ranieri, M.; Brave, M.; Arciuch, V.A.; Forde, T.; De Martino, D.; Anderson, K.E.; Hawkins, P.T.; Di Cristofano, A. SGK1 Is a Critical Component of an AKT-Independent Pathway Essential for PI3K-Mediated Tumor Development and Maintenance. Cancer Res. 2017, 77, 6914–6926. [Google Scholar] [CrossRef]
- Tovell, H.; Testa, A.; Zhou, H.; Shpiro, N.; Crafter, C.; Ciulli, A.; Alessi, D.R. Design and Characterization of SGK3-PROTAC1, an Isoform Specific SGK3 Kinase PROTAC Degrader. ACS Chem. Biol. 2019, 14, 2024–2034. [Google Scholar] [CrossRef]
- Ikenoue, T.; Inoki, K.; Yang, Q.; Zhou, X.; Guan, K.-L. Essential function of TORC2 in PKC and Akt turn motif phosphorylation, maturation and signalling. EMBO J. 2008, 27, 1919–1931. [Google Scholar] [CrossRef] [PubMed]
- Schnaper, H. Signal transduction through protein kinase C. Pediatr. Nephrol. 2000, 14, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Corbit, K.C.; Soh, J.-W.; Yoshida, K.; Eves, E.M.; Weinstein, I.B.; Rosner, M.R. Different Protein Kinase C Isoforms Determine Growth Factor Specificity in Neuronal Cells. Mol. Cell. Biol. 2000, 20, 5392–5403. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Chauhan, A.; Brockerhoff, H.; Chauhan, V. Activation of Protein Kinase C by Phosphatidylinositol 3,4,5-Trisphosphate. Biochem. Biophys. Res. Commun. 1993, 195, 104–112. [Google Scholar] [CrossRef]
- Garg, R.B.; Benedetti, L.G.; Abera, M.B.; Wang, H.; Abba, M.C.; Kazanietz, M.G. Protein kinase C and cancer: What we know and what we do not. Oncogene 2013, 33, 5225–5237. [Google Scholar] [CrossRef]
- Ghosh, P.M.; Bedolla, R.; Mikhailova, M.; Kreisberg, J.I. RhoA-dependent murine prostate cancer cell proliferation and apoptosis: Role of protein kinase Czeta. Cancer Res. 2002, 62, 2630–2636. [Google Scholar]
- Yao, S.; Bee, A.; Brewer, D.S.; Dodson, A.; Beesley, C.; Ke, Y.; Ambroisine, L.; Fisher, G.; Møller, H.; Dickinson, T.; et al. PRKC-ζ Expression Promotes the Aggressive Phenotype of Human Prostate Cancer Cells and Is a Novel Target for Therapeutic Intervention. Genes Cancer 2010, 1, 444–464. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Valencia, T.; Abu-Baker, S.; Linares, J.; Lee, S.J.; Yajima, T.; Chen, J.; Eroshkin, A.; Castilla, E.A.; Brill, L.M.; et al. c-Myc phosphorylation by PKCζ represses prostate tumorigenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 6418–6423. [Google Scholar] [CrossRef] [PubMed]
- Fan, H.-H.; Li, L.; Zhang, Y.-M.; Yang, J.; Li, M.-C.; Zeng, F.; Deng, F. PKCζ in prostate cancer cells represses the recruitment and M2 polarization of macrophages in the prostate cancer microenvironment. Tumor Biol. 2017, 39. [Google Scholar] [CrossRef]
- Zang, G.; Mu, Y.; Gao, L.; Bergh, A.; Landström, M. PKCζ facilitates lymphatic metastatic spread of prostate cancer cells in a mice xenograft model. Oncogene 2019, 38, 4215–4231. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Blando, J.M.; Perez, C.J.; Abba, M.C.; Benavides, F.; Kazanietz, M.G. Protein Kinase C Epsilon Cooperates with PTEN Loss for Prostate Tumorigenesis through the CXCL13-CXCR5 Pathway. Cell Rep. 2017, 19, 375–388. [Google Scholar] [CrossRef]
- Garg, R.; Blando, J.M.; Perez, C.J.; Lal, P.; Feldman, M.D.; Smyth, E.M.; Ricciotti, E.; Grosser, T.; Benavides, F.; Kazanietz, M.G. COX-2 mediates pro-tumorigenic effects of PKCε in prostate cancer. Oncogene 2018, 37, 4735–4749. [Google Scholar] [CrossRef]
- Gutierrez-Uzquiza, A.; Lopez-Haber, C.; Jernigan, D.L.; Fatatis, A.; Kazanietz, M.G. PKC Is an Essential Mediator of Prostate Cancer Bone Metastasis. Mol. Cancer Res. 2015, 13, 1336–1346. [Google Scholar] [CrossRef]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond AKT. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef]
- Ridley, A.J. Rho family proteins: Coordinating cell responses. Trends Cell Biol. 2001, 11, 471–477. [Google Scholar] [CrossRef]
- Aoki, K.; Nakamura, T.; Fujikawa, K.; Matsuda, M. Local Phosphatidylinositol 3,4,5-Trisphosphate Accumulation Recruits Vav2 and Vav3 to Activate Rac1/Cdc42 and Initiate Neurite Outgrowth in Nerve Growth Factor-stimulated PC12 Cells. Mol. Biol. Cell 2005, 16, 2207–2217. [Google Scholar] [CrossRef]
- Fritsch, R.; De Krijger, I.; Fritsch, K.; George, R.; Reason, B.; Kumar, M.S.; Diefenbacher, M.; Stamp, G.; Downward, J. RAS and RHO Families of GTPases Directly Regulate Distinct Phosphoinositide 3-Kinase Isoforms. Cell 2013, 153, 1050–1063. [Google Scholar] [CrossRef] [PubMed]
- Yuzugullu, H.; Baitsch, L.; Von, T.; Steiner, A.; Tong, H.; Ni, J.; Clayton, L.K.; Bronson, R.; Roberts, T.M.; Gritsman, K.; et al. A PI3K p110β–Rac signalling loop mediates Pten-loss-induced perturbation of haematopoiesis and leukaemogenesis. Nat. Commun. 2015, 6, 8501. [Google Scholar] [CrossRef] [PubMed]
- Liliental, J.; Moon, S.Y.; Lesche, R.; Mamillapalli, R.; Li, D.; Zheng, Y.; Sun, H.; Wu, H. Genetic deletion of the Pten tumor suppressor gene promotes cell motility by activation of Rac1 and Cdc42 GTPases. Curr. Biol. 2000, 10, 401–404. [Google Scholar] [CrossRef]
- Mense, S.M.; Barrows, D.; Hodakoski, C.; Steinbach, N.; Schoenfeld, D.; Su, W.; Hopkins, B.D.; Su, T.; Fine, B.; Hibshoosh, H.; et al. PTEN inhibits PREX2-catalyzed activation of RAC1 to restrain tumor cell invasion. Sci. Signal. 2015, 8, ra32. [Google Scholar] [CrossRef] [PubMed]
- Zins, K.; Lucas, T.; Reichl, P.; Abraham, D.; Aharinejad, S. A Rac1/Cdc42 GTPase-Specific Small Molecule Inhibitor Suppresses Growth of Primary Human Prostate Cancer Xenografts and Prolongs Survival in Mice. PLoS ONE 2013, 8, e74924. [Google Scholar] [CrossRef]
- Gao, Y.; Dickerson, J.B.; Guo, F.; Zheng, J.; Zheng, Y. Rational design and characterization of a Rac GTPase-specific small molecule inhibitor. Proc. Natl. Acad. Sci. USA 2004, 101, 7618–7623. [Google Scholar] [CrossRef]
- Qin, J.; Xie, Y.; Wang, B.; Hoshino, M.; Wolff, D.W.; Zhao, J.; Scofield, M.A.; Dowd, F.J.; Lin, M.-F.; Tu, Y. Upregulation of PIP3-dependent Rac exchanger 1 (P-Rex1) promotes prostate cancer metastasis. Oncogene 2009, 28, 1853–1863. [Google Scholar] [CrossRef]
- Hirai, K.; Nomura, T.; Yamasaki, M.; Inoue, T.; Narimatsu, T.; Nakada, P.C.; Tsukamoto, P.Y.; Matsuura, K.; Sato, F.; Moriyama, M.; et al. The Vav3 oncogene enhances the malignant potential of prostate cancer cells under chronic hypoxia. Urol. Oncol. Semin. Orig. Investig. 2014, 32, 101–109. [Google Scholar] [CrossRef]
- Aguilar, B.J.; Zhao, Y.; Zhou, H.; Huo, S.; Chen, Y.-H.; Lu, Q. Inhibition of Cdc42–intersectin interaction by small molecule ZCL367 impedes cancer cell cycle progression, proliferation, migration, and tumor growth. Cancer Biol. Ther. 2019, 20, 740–749. [Google Scholar] [CrossRef]
- Friesland, A.; Zhao, Y.; Chen, Y.-H.; Wang, L.; Zhou, H.; Lu, Q. Small molecule targeting Cdc42–intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. USA 2013, 110, 1261–1266. [Google Scholar] [CrossRef]
- Yamaguchi, M.; Takagi, K.; Sato, A.; Miki, Y.; Miyashita, M.; Sasano, H.; Suzuki, T. Rac1 activation in human breast carcinoma as a prognostic factor associated with therapeutic resistance. Breast Cancer 2020, 27, 919–928. [Google Scholar] [CrossRef]
- Bid, H.K.; Roberts, R.D.; Manchanda, P.K.; Houghton, P.J. RAC1: An Emerging Therapeutic Option for Targeting Cancer Angiogenesis and Metastasis. Mol. Cancer Ther. 2013, 12, 1925–1934. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Chakrabarti, J.; Li, Y.; Kalim, K.W.; Zhang, M.; Zhang, L.; Zheng, Y.; Guo, F. Rational Targeting of Cdc42 Overcomes Drug Resistance of Multiple Myeloma. Front. Oncol. 2019, 9, 958. [Google Scholar] [CrossRef]
- Maldonado, M.D.M.; Medina, J.I.; Velazquez, L.; Dharmawardhane, S. Targeting Rac and Cdc42 GEFs in Metastatic Cancer. Front. Cell Dev. Biol. 2020, 8, 201. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Yin, L.; Qiao, G.; Li, Y.; Li, B.; Bai, Y.; Feng, F. Inhibition of Rac1 reverses enzalutamide resistance in castration-resistant prostate cancer. Oncol. Lett. 2020, 20, 2997–3005. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Kenney, S.R.; Cook, L.; Adams, S.F.; Rutledge, T.; Romero, E.; Oprea, T.I.; Sklar, L.A.; Bedrick, E.; Wiggins, C.L.; et al. A Novel Pharmacologic Activity of Ketorolac for Therapeutic Benefit in Ovarian Cancer Patients. Clin. Cancer Res. 2015, 21, 5064–5072. [Google Scholar] [CrossRef]
- Paccez, J.D.; Vasques, G.J.; Correa, R.G.; Vasconcellos, J.F.; Duncan, K.; Gu, X.; Bhasin, M.K.; Libermann, T.A.; Zerbini, L.F. The receptor tyrosine kinase Axl is an essential regulator of prostate cancer proliferation and tumor growth and represents a new therapeutic target. Oncogene 2012, 32, 689–698. [Google Scholar] [CrossRef]
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 1–22. [Google Scholar] [CrossRef]
- Lew, E.D.; Oh, J.; Burrola, P.G.; Lax, I.; Zagórska, A.; Través, P.G.; Schlessinger, J.; Lemke, G. Differential TAM receptor–ligand–phospholipid interactions delimit differential TAM bioactivities. eLife 2014, 3, e03385. [Google Scholar] [CrossRef]
- Sainaghi, P.P.; Castello, L.; Bergamasco, L.; Galletti, M.; Bellosta, P.; Avanzi, G.C. Gas6 induces proliferation in prostate carcinoma cell lines expressing the Axl receptor. J. Cell. Physiol. 2005, 204, 36–44. [Google Scholar] [CrossRef]
- Lee, Y.; Lee, M.; Kim, S. Gas6 induces cancer cell migration and epithelial–mesenchymal transition through upregulation of MAPK and Slug. Biochem. Biophys. Res. Commun. 2013, 434, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Pedersen, E.A.; Patel, L.R.; Ziegler, A.M.; Havens, A.M.; Jung, Y.; Wang, J.; Zalucha, S.; Loberg, R.D.; Pienta, K.J.; et al. GAS6/AXL Axis Regulates Prostate Cancer Invasion, Proliferation, and Survival in the Bone Marrow Niche. Neoplasia 2010, 12, 116-IN4. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, H.D.; Valkenburg, K.C.; Amend, S.R.; Hicks, J.L.E.; Parsana, P.; Torga, G.; De Marzo, A.M.; Pienta, K.J. AXL Is a Putative Tumor Suppressor and Dormancy Regulator in Prostate Cancer. Mol. Cancer Res. 2018, 17, 356–369. [Google Scholar] [CrossRef]
- Turkington, R.; Cairns, L.; Stevenson, L.; Douglas, R.; McCabe, N.; Kennedy, R.; Harrison, T. AXL Mediates Resistance to AKT inhibition in Oesophageal Adenocarcinoma. In Proceedings of the NCRI Cancer Conference 2016, Liverpool, UK, 6 November 2016. [Google Scholar]
- Elkabets, M.; Pazarentzos, E.; Juric, D.; Jinsheng, L.; Pelossof, R.A.; Brook, S.; Benzaken, A.O.; Rodon, J.; Morse, N.; Yan, J.J.; et al. AXL Mediates Resistance to PI3Kα Inhibition by Activating the EGFR/PKC/mTOR Axis in Head and Neck and Esophageal Squamous Cell Carcinomas. Cancer Cell 2015, 27, 533–546. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wang, Z.-J.; De, W.; Zheng, M.; Xu, W.-Z.; Wu, H.-F.; Armstrong, A.; Zhu, J.-G. Targeting AXL overcomes resistance to docetaxel therapy in advanced prostate cancer. Oncotarget 2017, 8, 41064–41077. [Google Scholar] [CrossRef]
- Mita, M.M.; Gordon, M.; Rosen, L.; Kapoor, N.; Choy, G.; Redkar, S.; Taverna, P.; Oganesian, A.; Sahai, A.; Azab, M.; et al. Phase 1B study of amuvatinib in combination with five standard cancer therapies in adults with advanced solid tumors. Cancer Chemother. Pharmacol. 2014, 74, 195–204. [Google Scholar] [CrossRef]
- Qi, W.; Cooke, L.S.; Stejskal, A.; Riley, C.; Della Croce, K.; Saldanha, J.W.; Bearss, D.; Mahadevan, D. MP470, a novel receptor tyrosine kinase inhibitor, in combination with Erlotinib inhibits the HER family/PI3K/Akt pathway and tumor growth in prostate cancer. BMC Cancer 2009, 9, 142. [Google Scholar] [CrossRef]
- Wu, G.; Ma, Z.; Cheng, Y.; Hu, W.; Deng, C.; Jiang, S.; Li, T.; Chen, F.; Yang, Y. Targeting Gas6/TAM in cancer cells and tumor microenvironment. Mol. Cancer 2018, 17, 20. [Google Scholar] [CrossRef]
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136. [Google Scholar] [CrossRef]
- Loges, S.; Gjertsen, B.T.; Heuser, M.; Ben-Batalla, I.; Micklem, D.R.; Jorg, C.; Kebenko, M.; Fiedler, W.; Cortes, J.E. A first-in-patient phase I study of BGB324, a selective Axl kinase inhibitor in patients with refractory/relapsed AML and high-risk MDS. J. Clin. Oncol. 2016, 34, 2561. [Google Scholar] [CrossRef]
- Liu, H.; Feng, X.; Ennis, K.N.; Behrmann, C.A.; Sarma, P.; Jiang, T.T.; Kofuji, S.; Niu, L.; Stratton, Y.; Thomas, H.E.; et al. Pharmacologic Targeting of S6K1 in PTEN-Deficient Neoplasia. Cell Rep. 2017, 18, 2088–2095. [Google Scholar] [CrossRef] [PubMed]
- Zuo, Q.; Liu, J.; Huang, L.; Qin, Y.; Hawley, T.; Seo, C.; Merlino, G.; Yu, Y. AXL/AKT axis mediated-resistance to BRAF inhibitor depends on PTEN status in melanoma. Oncogene 2018, 37, 3275–3289. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA Damage Response: Making It Safe to Play with Knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef]
- O’Connor, M.J. Targeting the DNA Damage Response in Cancer. Mol. Cell 2015, 60, 547–560. [Google Scholar] [CrossRef]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Hsieh, P.; Yamane, K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef] [PubMed]
- Krejci, L.; Altmannova, V.; Spirek, M.; Zhao, X. Homologous recombination and its regulation. Nucleic Acids Res. 2012, 40, 5795–5818. [Google Scholar] [CrossRef]
- Li, X.; Heyer, W.-D. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008, 18, 99–113. [Google Scholar] [CrossRef]
- Davis, A.J.; Chen, D.J. DNA double strand break repair via non-homologous end-joining. Transl. Cancer Res. 2013, 2, 130–143. [Google Scholar]
- Ceccaldi, R.; Rondinelli, B.; D’Andrea, A.D. Repair Pathway Choices and Consequences at the Double-Strand Break. Trends Cell Biol. 2016, 26, 52–64. [Google Scholar] [CrossRef]
- Ming, M.; He, Y.-Y. PTEN in DNA damage repair. Cancer Lett. 2012, 319, 125–129. [Google Scholar] [CrossRef] [PubMed]
- Mansour, W.Y.; Tennstedt, P.; Volquardsen, J.; Oing, C.; Kluth, M.; Hube-Magg, C.; Borgmann, K.; Simon, R.; Petersen, C.; Dikomey, E.; et al. Loss of PTEN-assisted G2/M checkpoint impedes homologous recombination repair and enhances radio-curability and PARP inhibitor treatment response in prostate cancer. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Fraser, M.; Zhao, H.; Luoto, K.R.; Lundin, C.; Coackley, C.; Chan, N.; Joshua, A.M.; Bismar, T.A.; Evans, A.; Helleday, T.; et al. PTEN Deletion in Prostate Cancer Cells Does Not Associate with Loss of RAD51 Function: Implications for Radiotherapy and Chemotherapy. Clin. Cancer Res. 2011, 18, 1015–1027. [Google Scholar] [CrossRef] [PubMed]
- Puc, J.; Keniry, M.; Li, H.S.; Pandita, T.K.; Choudhury, A.D.; Memeo, L.; Mansukhani, M.; Murty, V.V.; Gaciong, Z.; Meek, S.E.; et al. Lack of PTEN sequesters CHK1 and initiates genetic instability. Cancer Cell 2005, 7, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Ming, M.; Feng, L.; Shea, C.R.; Soltani, K.; Zhao, B.; Han, W.; Smart, R.C.; Trempus, C.S.; He, Y.-Y. PTEN positively regulates UVB-induced DNA damage repair. Cancer Res. 2011, 71, 5287–5295. [Google Scholar] [CrossRef]
- Mendes-Pereira, A.M.; Martin, S.A.; Brough, R.; McCarthy, A.; Taylor, J.R.; Kim, J.; Waldman, T.; Lord, C.J.; Ashworth, A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol. Med. 2009, 1, 315–322. [Google Scholar] [CrossRef]
- Sandhu, S.K.; Schelman, W.R.; Wilding, G.; Moreno, V.; Baird, R.D.; Miranda, S.; Hylands, L.; Riisnaes, R.; Forster, M.; Omlin, A.; et al. The poly(ADP-ribose) polymerase inhibitor niraparib (MK4827) in BRCA mutation carriers and patients with sporadic cancer: A phase 1 dose-escalation trial. Lancet Oncol. 2013, 14, 882–892. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Rodrigues, D.N.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Szanto, A.; Hellebrand, E.E.; Bognar, Z.; Tucsek, Z.; Szabo, A.; Gallyas, F.; Sumegi, B.; Várbiró, G. PARP-1 inhibition-induced activation of PI-3-kinase-Akt pathway promotes resistance to taxol. Biochem. Pharmacol. 2009, 77, 1348–1357. [Google Scholar] [CrossRef]
- Palfi, A.; Toth, A.; Kulcsar, G.; Hantó, K.; Deres, P.; Bartha, E.; Halmosi, R.; Szabados, E.; Czopf, L.; Kálai, T.; et al. The Role of Akt and Mitogen-Activated Protein Kinase Systems in the Protective Effect of Poly(ADP-Ribose) Polymerase Inhibition in Langendorff Perfused and in Isoproterenol-Damaged Rat Hearts. J. Pharmacol. Exp. Ther. 2005, 315, 273–282. [Google Scholar] [CrossRef]
- Tapodi, A.; Bognar, Z.; Szabo, C.; Gallyas, F.; Sumegi, B.; Hocsak, E. PARP inhibition induces Akt-mediated cytoprotective effects through the formation of a mitochondria-targeted phospho-ATM-NEMO-Akt-mTOR signalosome. Biochem. Pharmacol. 2019, 162, 98–108. [Google Scholar] [CrossRef] [PubMed]
- Tapodi, A.; Debreceni, B.; Hanto, K.; Bognar, Z.; Wittmann, I.; Gallyas, F.; Varbiro, G.; Sumegi, B. Pivotal Role of Akt Activation in Mitochondrial Protection and Cell Survival by Poly(ADP-ribose)polymerase-1 Inhibition in Oxidative Stress. J. Biol. Chem. 2005, 280, 35767–35775. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Li, C.; Zhang, Y.; Wang, M.; Jiang, N.; Xiang, L.; Li, T.; Roberts, T.M.; Zhao, J.J.; Cheng, H.; et al. Combined inhibition of PI3K and PARP is effective in the treatment of ovarian cancer cells with wild-type PIK3CA genes. Gynecol. Oncol. 2016, 142, 548–556. [Google Scholar] [CrossRef]
- Yap, T.A.; Kristeleit, R.; Michalarea, V.; Pettitt, S.J.; Lim, J.S.J.; Carreira, S.; Roda, D.; Miller, R.; Riisnaes, R.; Miranda, S.; et al. Phase I trial of the poly(ADP-ribose) polymerase (PARP) inhibitor olaparib and AKT inhibitor capivasertib in patients with BRCA1/2 and non-BRCA1/2 mutant cancers. Cancer Discov. 2020, 10, 1528–1543. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, Y.H.; García-García, C.; Serra, V.; He, L.; Torres-Lockhart, K.; Prat, A.; Anton, P.; Cozar, P.; Guzmán, M.; Grueso, J.; et al. PI3K Inhibition Impairs BRCA1/2 Expression and Sensitizes BRCA-Proficient Triple-Negative Breast Cancer to PARP Inhibition. Cancer Discov. 2012, 2, 1036–1047. [Google Scholar] [CrossRef] [PubMed]
- Mo, W.; Liu, Q.; Lin, C.C.-J.; Dai, H.; Peng, Y.; Liang, Y.; Peng, G.; Meric-Bernstam, F.; Mills, G.B.; Li, K.; et al. mTOR Inhibitors Suppress Homologous Recombination Repair and Synergize with PARP Inhibitors via Regulating SUV39H1 in BRCA-Proficient Triple-Negative Breast Cancer. Clin. Cancer Res. 2015, 22, 1699–1712. [Google Scholar] [CrossRef]
- Konstantinopoulos, P.A.; Barry, W.T.; Birrer, M.; Westin, S.N.A.; Cadoo, K.I.; Shapiro, G.; Mayer, E.L.E.; O’Cearbhaill, R.; Coleman, R.L.; Kochupurakkal, B.; et al. Olaparib and α-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: A dose-escalation and dose-expansion phase 1b trial. Lancet Oncol. 2019, 20, 570–580. [Google Scholar] [CrossRef]
- Wang, M.; Zhao, J.; Zhang, L.; Wei, F.; Lian, Y.; Wu, Y.; Gong, Z.; Zhang, S.; Zhou, J.; Cao, K.; et al. Role of tumor microenvironment in tumorigenesis. J. Cancer 2017, 8, 761–773. [Google Scholar] [CrossRef]
- Levesque, C.; Nelson, P.S. Cellular Constituents of the Prostate Stroma: Key Contributors to Prostate Cancer Progression and Therapy Resistance. Cold Spring Harb. Perspect. Med. 2017, 8, a030510. [Google Scholar] [CrossRef]
- Valkenburg, K.C.; De Groot, A.E.; Pienta, K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018, 15, 366–381. [Google Scholar] [CrossRef]
- Francoa, P.I.R.; Rodrigues, A.P.; De Menezes, L.B.; Miguel, M.P. Tumor microenvironment components: Allies of cancer progression. Pathol.-Res. Pract. 2020, 216, 152729. [Google Scholar] [CrossRef]
- Shree, T.; Olson, O.C.; Elie, B.T.; Kester, J.C.; Garfall, A.L.; Simpson, K.; Bell-McGuinn, K.M.; Zabor, E.C.; Brogi, E.; Joyce, J.A. Macrophages and cathepsin proteases blunt chemotherapeutic response in breast cancer. Genes Dev. 2011, 25, 2465–2479. [Google Scholar] [CrossRef]
- Jamal, M.; Rath, B.H.; Tsang, P.S.; Camphausen, K.; Tofilon, P.J. The Brain Microenvironment Preferentially Enhances the Radioresistance of CD133+ Glioblastoma Stem-like Cells. Neoplasia 2012, 14, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Predina, J.; Eruslanov, E.; Judy, B.; Kapoor, V.; Cheng, G.; Wang, L.-C.; Sun, J.; Moon, E.K.; Fridlender, Z.G.; Albelda, S.; et al. Changes in the local tumor microenvironment in recurrent cancers may explain the failure of vaccines after surgery. Proc. Natl. Acad. Sci. USA 2012, 110, E415–E424. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Yuan, J.; Righi, E.; Kamoun, W.S.; Ancukiewicz, M.; Nezivar, J.; Santosuosso, M.; Martin, J.D.; Martin, M.R.; Vianello, F.; et al. Vascular normalizing doses of antiangiogenic treatment reprogram the immunosuppressive tumor microenvironment and enhance immunotherapy. Proc. Natl. Acad. Sci. USA 2012, 109, 17561–17566. [Google Scholar] [CrossRef]
- Klemm, F.; Joyce, J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015, 25, 198–213. [Google Scholar] [CrossRef]
- Daverey, A.; Drain, A.P.; Kidambi, S. Physical Intimacy of Breast Cancer Cells with Mesenchymal Stem Cells Elicits Trastuzumab Resistance through Src Activation. Sci. Rep. 2015, 5, 13744. [Google Scholar] [CrossRef]
- Ayala, G.E.A.; Tuxhorn, J.; Wheeler, T.M.; Frolov, A.; Scardino, P.T.; Ohori, M.; Wheeler, M.; Spitler, J.; Rowley, D.R. Reactive stroma as a predictor of biochemical-free recurrence in prostate cancer. Clin. Cancer Res. 2003, 9, 4792–4801. [Google Scholar]
- Mueller, K.L.; Madden, J.M.; Zoratti, G.L.; Kuperwasser, C.; List, K.; Boerner, J.L. Fibroblast-secreted hepatocyte growth factor mediates epidermal growth factor receptor tyrosine kinase inhibitor resistance in triple-negative breast cancers through paracrine activation of Met. Breast Cancer Res. 2012, 14, R104. [Google Scholar] [CrossRef] [PubMed]
- Kharaziha, P.; Rodriguez, P.L.; Li, Q.; Rundqvist, H.; Björklund, A.-C.; Augsten, M.; Ullen, A.; Egevad, L.; Wiklund, P.G.; Nilsson, S.; et al. Targeting of distinct signaling cascades and cancer-associated fibroblasts define the efficacy of Sorafenib against prostate cancer cells. Cell Death Dis. 2012, 3, e262. [Google Scholar] [CrossRef]
- Mouw, J.K.; Yui, Y.; Damiano, L.O.; Bainer, R.; Lakins, J.N.; Acerbi, I.; Ou, G.; Wijekoon, A.C.; Levental, K.R.; Gilbert, P.M.; et al. Tissue mechanics modulate microRNA-dependent PTEN expression to regulate malignant progression. Nat. Med. 2014, 20, 360–367. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Bai, Y.; He, Y.; Zhao, Y.; Chen, J.; Ma, L.; Pan, Y.; Hinten, M.; Zhang, J.; Karnes, R.J.; et al. PTEN Loss Promotes Intratumoral Androgen Synthesis and Tumor Microenvironment Remodeling via Aberrant Activation of RUNX2 in Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2017, 24, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Trimboli, A.J.; Cantemir-Stone, C.Z.; Li, F.; Wallace, J.A.; Merchant, A.; Creasap, N.; Thompson, J.C.; Caserta, E.; Wang, H.; Chong, J.-L.; et al. Pten in stromal fibroblasts suppresses mammary epithelial tumours. Nat. Cell Biol. 2009, 461, 1084–1091. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.E.; Hammer, A.M.; Cho, Y.; Sizemore, G.M.; Cukierman, E.; Yee, L.D.; Ghadiali, S.N.; Ostrowski, M.C.; Leight, J.L. Stromal PTEN Regulates Extracellular Matrix Organization in the Mammary Gland. Neoplasia 2018, 21, 132–145. [Google Scholar] [CrossRef]
- Valencia, T.; Kim, J.Y.; Abu-Baker, S.; Moscat-Pardos, J.; Ahn, C.S.; Reina-Campos, M.; Duran, A.; Castilla, E.A.; Metallo, C.M.; Diaz-Meco, M.T.; et al. Metabolic Reprogramming of Stromal Fibroblasts through p62-mTORC1 Signaling Promotes Inflammation and Tumorigenesis. Cancer Cell 2014, 26, 121–135. [Google Scholar] [CrossRef]
- Conciatori, F.; Bazzichetto, C.; Falcone, I.; Ciuffreda, L.; Ferretti, G.; Vari, S.; Ferraresi, V.; Cognetti, F.; Milella, M. PTEN Function at the Interface between Cancer and Tumor Microenvironment: Implications for Response to Immunotherapy. Int. J. Mol. Sci. 2020, 21, 5337. [Google Scholar] [CrossRef]
- Piro, G.; Carbone, C.; Carbognin, L.; Pilotto, S.; Ciccarese, C.; Iacovelli, R.; Milella, M.; Bria, E.; Tortora, G. Revising PTEN in the Era of Immunotherapy: New Perspectives for an Old Story. Cancers 2019, 11, 1525. [Google Scholar] [CrossRef]
- Dong, Y.; Richards, J.-A.; Gupta, R.; Aung, P.P.; Emley, A.; Kluger, Y.; Dogra, S.K.; Mahalingam, M.; Wajapeyee, N. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene 2013, 33, 4632–4642. [Google Scholar] [CrossRef]
- Vidotto, T.; Saggioro, F.P.; Jamaspishvili, T.; Chesca, D.L.; De Albuquerque, C.G.P.; Reis, R.B.; Graham, C.H.; Berman, D.M.; Siemens, D.R.; Squire, J.A.; et al. PTEN-deficient prostate cancer is associated with an immunosuppressive tumor microenvironment mediated by increased expression of IDO1 and infiltrating FoxP3+ T regulatory cells. Prostate 2019, 79, 969–979. [Google Scholar] [CrossRef]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing Chemotherapy Efficacy in Pten -Deficient Prostate Tumors by Activating the Senescence-Associated Antitumor Immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef]
- Silvestri, I.; Tortorella, E.; Giantulli, S.; Scarpa, S.; Sciarra, A. Immunotherapy in Prostate Cancer: Recent Advances and Future Directions. EMJ Urol. 2019, 7, 51–61. [Google Scholar]
- Holl, E.K.; McNamara, M.A.; Healy, P.; Anand, M.; Concepcion, R.S.; Breland, C.D.; Dumbudze, I.; Tutrone, R.; Shore, N.; Armstrong, A.J.; et al. Prolonged PSA stabilization and overall survival following sipuleucel-T monotherapy in metastatic castration-resistant prostate cancer patients. Prostate Cancer Prostatic Dis. 2019, 22, 588–592. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Piulats, J.M.; Gross-Goupil, M.; Goh, J.; Ojamaa, K.; Hoimes, C.J.; Vaishampayan, U.; Berger, R.; Sezer, A.; Alanko, T.; et al. Pembrolizumab for Treatment-Refractory Metastatic Castration-Resistant Prostate Cancer: Multicohort, Open-Label Phase II KEYNOTE-199 Study. J. Clin. Oncol. 2020, 38, 395–405. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Miao, D.; Demetri, G.D.; Adeegbe, D.; Rodig, S.J.; Shukla, S.; Lipschitz, M.; Amin-Mansour, A.; Raut, C.P.; Carter, S.L.; et al. Loss of PTEN Is Associated with Resistance to Anti-PD-1 Checkpoint Blockade Therapy in Metastatic Uterine Leiomyosarcoma. Immunity 2017, 46, 197–204. [Google Scholar] [CrossRef]
- Peng, W.; Chen, J.Q.; Liu, C.; Malu, S.; Creasy, C.; Tetzlaff, M.T.; Xu, C.; McKenzie, J.A.; Zhang, C.; Liang, X.; et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2015, 6, 202–216. [Google Scholar] [CrossRef]
- Parikh, A.R.; Ali, S.M.; Schrock, A.B.A.; Albacker, L.A.; Miller, V.; Stephens, P.J.; Crilley, P.; Markman, M. Response to rapamycin analogs but not PD-1 inhibitors in PTEN-mutated metastatic non-small-cell lung cancer with high tumor mutational burden. Lung Cancer Targets Ther. 2018, 9, 45–47. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, A.X.; Gartrell, R.D.; Silverman, A.M.; Aparicio, L.; Chu, T.; Bordbar, D.; Shan, D.; Samanamud, J.; Mahajan, A.; et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat. Med. 2019, 25, 462–469. [Google Scholar] [CrossRef]
- Peng, W.; Williams, L.J.; Xu, C.; Melendez, B.; McKenzie, J.A.; Chen, Y.; Jackson, H.L.; Voo, K.S.; Mbofung, R.M.; Leahey, S.E.; et al. Anti-OX40 Antibody Directly Enhances The Function of Tumor-Reactive CD8+ T Cells and Synergizes with PI3Kβ Inhibition in PTEN Loss Melanoma. Clin. Cancer Res. 2019, 25, 6406–6416. [Google Scholar] [CrossRef]
- Tawbi, H.A.-H.; Peng, W.; Phillips, S.; Milton, D.R.; Amaria, R.N.; Diab, A.; Glitza, I.C.; Patel, S.P.; Wong, M.K.; Yee, C.; et al. Safety results from phase I/II study of the PI3Kβ inhibitor GSK2636771 (G) in combination with pembrolizumab (P) in patients (pts) with PD-1 refractory metastatic melanoma (MM) and PTEN loss. J. Clin. Oncol. 2020, 38, e22000. [Google Scholar] [CrossRef]
- Sfanos, K.S.; De Marzo, A.M. Prostate cancer and inflammation: The evidence. Histopathology 2011, 60, 199–215. [Google Scholar] [CrossRef]
- Gurel, B.; Lucia, M.S.; Thompson, I.M.; Goodman, P.J.; Tangen, C.M.; Kristal, A.R.; Parnes, H.L.; Hoque, A.; Lippman, S.M.; Sutcliffe, S.; et al. Chronic Inflammation in Benign Prostate Tissue Is Associated with High-Grade Prostate Cancer in the Placebo Arm of the Prostate Cancer Prevention Trial. Cancer Epidemiol. Biomark. Prev. 2014, 23, 847–856. [Google Scholar] [CrossRef]
- Kwon, O.-J.; Zhang, L.; Ittmann, M.M.; Xin, L. Prostatic inflammation enhances basal-to-luminal differentiation and accelerates initiation of prostate cancer with a basal cell origin. Proc. Natl. Acad. Sci. USA 2013, 111, E592–E600. [Google Scholar] [CrossRef]
- De Bono, J.; Guo, C.; Gurel, B.; De Marzo, A.M.; Sfanos, K.S.; Mani, R.S.; Gil, J.; Drake, C.G.; Alimonti, A. Prostate carcinogenesis: Inflammatory storms. Nat. Rev. Cancer 2020, 20, 1–15. [Google Scholar] [CrossRef]
- Calcinotto, A.; Spataro, C.; Zagato, E.; Di Mitri, D.; Gil, V.; Crespo, M.; De Bernardis, G.; Losa, M.; Mirenda, M.; Pasquini, E.; et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nat. Cell Biol. 2018, 559, 363–369. [Google Scholar] [CrossRef]
- Escamilla, J.; Schokrpur, S.; Liu, C.; Priceman, S.J.; Moughon, D.; Jiang, Z.; Pouliot, F.; Magyar, C.; Sung, J.L.; Xu, J.; et al. CSF1 Receptor Targeting in Prostate Cancer Reverses Macrophage-Mediated Resistance to Androgen Blockade Therapy. Cancer Res. 2015, 75, 950–962. [Google Scholar] [CrossRef]
- Hossain, D.M.S.; Pal, S.; Moreira, D.; Duttagupta, P.; Zhang, Q.; Won, H.; Jones, J.; D’Apuzzo, M.; Forman, S.; Kortylewski, M. TLR9-Targeted STAT3 Silencing Abrogates Immunosuppressive Activity of Myeloid-Derived Suppressor Cells from Prostate Cancer Patients. Clin. Cancer Res. 2015, 21, 3771–3782. [Google Scholar] [CrossRef]
- Lehmann, B.D.; Paine, M.S.; Brooks, A.M.; McCubrey, J.A.; Renegar, R.H.; Wang, R.; Terrian, D.M. Senescence-Associated Exosome Release from Human Prostate Cancer Cells. Cancer Res. 2008, 68, 7864–7871. [Google Scholar] [CrossRef]
- Zhang, B.; Fu, D.; Xu, Q.; Cong, X.; Wu, C.; Zhong, X.; Ma, Y.; Lv, Z.; Chen, F.; Han, L.; et al. The senescence-associated secretory phenotype is potentiated by feedforward regulatory mechanisms involving Zscan4 and TAK1. Nat. Commun. 2018, 9, 1–19. [Google Scholar] [CrossRef]
- Garcia, A.J.; Ruscetti, M.; Arenzana, T.L.; Tran, L.M.; Bianci-Frias, D.; Sybert, E.; Priceman, S.J.; Wu, L.; Nelson, P.S.; Smale, S.T.; et al. Pten Null Prostate Epithelium Promotes Localized Myeloid-Derived Suppressor Cell Expansion and Immune Suppression during Tumor Initiation and Progression. Mol. Cell. Biol. 2014, 34, 2017–2028. [Google Scholar] [CrossRef]
- Maxwell, P.J.; Coulter, J.A.; Walker, S.M.; McKechnie, M.; Neisen, J.; McCabe, N.; Kennedy, R.D.; Salto-Tellez, M.; Albanese, C.; Waugh, D.J. Potentiation of inflammatory CXCL8 signalling sustains cell survival in PTEN-deficient prostate carcinoma. Eur. Urol. 2012, 64, 177–188. [Google Scholar] [CrossRef]
- Maxwell, P.J.; Neisen, J.; Messenger, J.; Waugh, D.J. Tumor-derived CXCL8 signaling augments stroma-derived CCL2-promoted proliferation and CXCL12-mediated invasion of PTEN-deficient prostate cancer cells. Oncotarget 2014, 5, 4895–4908. [Google Scholar] [CrossRef]
- Sun, Y.; Campisi, J.; Higano, C.S.; Beer, T.M.; Porter, P.L.; Coleman, I.; True, L.D.; Nelson, P.S. Treatment-induced damage to the tumor microenvironment promotes prostate cancer therapy resistance through WNT16B. Nat. Med. 2012, 18, 1359–1368. [Google Scholar] [CrossRef]
- Di Mitri, D.; Toso, A.; Alimonti, A. Tumor-infiltrating myeloid cells drive senescence evasion and chemoresistance in tumors. OncoImmunology 2015, 4, 988473. [Google Scholar] [CrossRef]
- Parisotto, M.; Grelet, E.; El Bizri, R.; Metzger, D. Senescence controls prostatic neoplasia driven by Pten loss. Mol. Cell. Oncol. 2018, 6, 1511205. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; DeMaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020, 6, 838–857. [Google Scholar] [CrossRef]
- Bavik, C.; Coleman, I.; Dean, J.P.; Knudsen, B.; Plymate, S.; Nelson, P.S. The Gene Expression Program of Prostate Fibroblast Senescence Modulates Neoplastic Epithelial Cell Proliferation through Paracrine Mechanisms. Cancer Res. 2006, 66, 794–802. [Google Scholar] [CrossRef]
- Taddei, M.L.; Cavallini, L.; Comito, G.; Giannoni, E.; Folini, M.; Marini, A.; Gandellini, P.; Morandi, A.; Pintus, G.; Raspollini, M.R.; et al. Senescent stroma promotes prostate cancer progression: The role of miR-210. Mol. Oncol. 2014, 8, 1729–1746. [Google Scholar] [CrossRef]
- Laberge, R.-M.; Sun, Y.; Orjalo, A.V.; Patil, C.K.; Freund, A.; Zhou, L.; Curran, S.C.; Davalos, A.R.; Wilson-Edell, K.A.; Liu, S.; et al. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol. 2015, 17, 1049–1061. [Google Scholar] [CrossRef]
- Kang, C. Senolytics and Senostatics: A Two-Pronged Approach to Target Cellular Senescence for Delaying Aging and Age-Related Diseases. Mol. Cells 2019, 42, 821–827. [Google Scholar]
- Liu, S.; Zhang, Q.; Chen, C.; Ge, D.; Qu, Y.; Chen, R.; Fan, Y.-M.; Li, N.; Tang, W.W.; Zhang, W.; et al. Hyperinsulinemia enhances interleukin-17-induced inflammation to promote prostate cancer development in obese mice through inhibiting glycogen synthase kinase 3-mediated phosphorylation and degradation of interleukin-17 receptor. Oncotarget 2016, 7, 13651–13666. [Google Scholar] [CrossRef]
- Hayashi, T.; Fujita, K.; Nojima, S.; Hayashi, Y.; Nakano, K.; Ishizuya, Y.; Wang, C.; Yamamoto, Y.; Kinouchi, T.; Matsuzaki, K.; et al. High-Fat Diet-Induced Inflammation Accelerates Prostate Cancer Growth via IL6 Signaling. Clin. Cancer Res. 2018, 24, 4309–4318. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, M.; Fujita, K.; Nonomura, N. Influence of Diet and Nutrition on Prostate Cancer. Int. J. Mol. Sci. 2020, 21, 1447. [Google Scholar] [CrossRef] [PubMed]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Turnham, D.J.; Bullock, N.; Dass, M.S.; Staffurth, J.N.; Pearson, H.B. The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer. Cells 2020, 9, 2342. https://doi.org/10.3390/cells9112342
Turnham DJ, Bullock N, Dass MS, Staffurth JN, Pearson HB. The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer. Cells. 2020; 9(11):2342. https://doi.org/10.3390/cells9112342
Chicago/Turabian StyleTurnham, Daniel J., Nicholas Bullock, Manisha S. Dass, John N. Staffurth, and Helen B. Pearson. 2020. "The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer" Cells 9, no. 11: 2342. https://doi.org/10.3390/cells9112342
APA StyleTurnham, D. J., Bullock, N., Dass, M. S., Staffurth, J. N., & Pearson, H. B. (2020). The PTEN Conundrum: How to Target PTEN-Deficient Prostate Cancer. Cells, 9(11), 2342. https://doi.org/10.3390/cells9112342