1. Introduction
Although the splicing process was discovered more than 30 years ago [
1], the mechanisms of spliceosome assembly and splicing regulation are not well understood. To date, over 100 proteins that regulate spliceosome assembly and function have been identified [
2]. Understanding the role of these proteins in splicing is important because several disorders such as retinitis pigmentosa (RP) [
3], amyotrophic lateral sclerosis, spinal muscular atrophy [
4], and chronic lymphocytic leukemia [
5] are caused by mutations in splicing factors.
We have characterized one of these genes,
sart1, which was isolated from an N-ethyl-N-nitrosourea (ENU) mutagenesis forward genetic screen in the enhancer trap zebrafish line
Et(
cp:EGFP)
sj2 to identify mutants in choroid plexus (CP) development [
6]. In this genetic screen, zebrafish larvae were observed at 4 days postfertilization (dpf) to look for phenotypes in the CP. The green fluorescent protein (GFP), was expressed in CP epithelia so mutants in CP development could be easily observed under a fluorescent microscope. Mutants identified from the screen were classified based on intensity of GFP expression in the CP, localization of epithelial cells, and overall CP size. Previously referred to as
cp27.5, this line with a mutation in
sart1 had variable GFP expression, small epithelial aggregates, and an expanded CP. The mutants also had expanded ventricles, small eyes, and increased CNS barrier permeability. Mutants were embryonic lethal by late day 4. Using bulked segregant analysis, we were able to map the mutation to chromosome 21. In our current study, we identified the mutation in
sart1 using exome sequencing, which has been utilized as an effective and time-efficient strategy to identify mutations from lines discovered in zebrafish forward genetic screens [
7].
Sart1, also known as U4/U6.U5 tri-snRNP-associated protein 1, squamous cell carcinoma antigen recognized by T-cells, HAF in mice, and Snu66 in yeast, is a complex protein involved in U4/U6.U5 tri-sRNP recruitment to the spliceosome [
8]. There are two protein isoforms of Sart1, one is an 800 amino acid sequence located in the nucleus of proliferating cells and contains a leucine zipper motif suggesting it may bind to DNA [
9]. Another 259 amino acid protein is synthesized by a frame shift in an internal ribosomal entry site that has been shown to be expressed in the cytosol in squamous cell carcinomas [
9].
While splicing genes are thought to be ubiquitously expressed, recent evidence suggests that certain components of the spliceosome may be temporally or spatially regulated. Studies have identified spliceosome components such as U2 that have high RNA levels, specifically in the cerebellum, and mutations in U2 result in neurodegeneration [
10]. In zebrafish, we observed that
sart1 is expressed specifically in the brain and upregulated in
cp27.5 mutants. This provides further evidence that splicing components may be regulated or expressed in specific tissues resulting in tissue-specific phenotypes.
In addition, we have demonstrated that this mutation results in up or downregulation of numerous gene classes. Using RNA-Seq analysis, we identified that sart1 is upregulated in mutants. It also results in upregulation of genes essential to the apoptotic pathway such as tumor protein 53 (tp53) and the proto-oncogene mdm2. Conversely, this mutation causes genes regulating eye development and function to be downregulated. This analysis confirmed our observations of retinal lamination loss and increased cell death in the eyes of cp27.5 mutants. We also observed an upregulation of other spliceosome components such as sm-like 7 (lsm7) and pre-mRNA processing factor 31 (prpf31) which may indicate a compensation mechanism due to defects in sart1 function. While previous studies have identified the major role of Sart1 as recruiting the U4/U5/U6 tri-sRNP to the spliceosome, we provide evidence that it may directly affect specific classes of genes such as those involved in apoptosis along with eye formation and maintenance. Understanding Sart1 regulation of gene expression will reveal how spliceosome components not only play a role in splicing, but also other cellular processes, such as apoptosis, and pathological conditions, such as degeneration and cancer.
2. Materials and Methods
2.1. Isolating cp27.5 Mutants
cp27.5 mutants were previously identified from an ENU mutagenesis screen identifying mutants in CP development. CP morphology and function of
cp27.5 mutants were characterized by Henson et al. [
6].
2.2. Fish Lines and Maintenance
Zebrafish were maintained in accordance with established protocols and all experiments were approved by the St. Jude Children’s Research Hospital Institutional Animal Care and Use Committee. Zebrafish were maintained at 28.5 °C on a 14 h light/dark cycle. Embryos used for imaging were collected in egg water (0.03% Instant Ocean) and treated at 24 h postfertilization (hpf) with 0.003% 1-phenyl-2-thiourea (PTU) (Sigma) to prevent pigment formation. Fish strains used for this study include Et(cp:EGFP)sj2, cp27.5, and TL.
2.3. WES and Analysis
cp27.5 wild-type and mutant larvae (20 each) were collected in MeOH at 3 dpf. The 20 larvae were generated from 3 different heterozygous parents. DNA was extracted using the MagAttract High Molecular Weight (HMW) DNA kit (Qiagen Sciences LLC, Germanton, MD, USA) following the manufacturer’s instructions. Values for DNA concentration and purity were obtained by a ND-1000 NanoDrop Spectrophotometer and Qubit assay (Invitrogen, Carlsbad, CA, USA). Concentration was adjusted to the required 3 µg/120 µL for sequence capture. Sequence capture and sequencing was done by the Genome Sequencing Facility Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital. Paired end 100 cycle sequencing was run on a HiSeq 2500 using TruSeq SBS v3 chemistry according to the manufacturer’s instructions (Illumina, San Diego, CA, USA).
From the raw sequencing data, the quality control, read mapping, and variant calling were performed using CLC Genomic Workbench v6.5 (CLC Bio, Aarhus, Denmark). In brief, the reads were trimmed against the sequencing adapters, and only reads with a sequencing quality greater than 20 and a read length greater than 50 bp were retained. The filtered reads were then aligned to the zebrafish reference genome sequence (Zv9 assembly, 2010), and the lists of single-nucleotide variants (SNV) and indels were generated. To identify the causative mutation responsible for the phenotype, we compared the variants of mutant with those of wild type, and a region enriched with mutant-specific homozygosities was observed at a peak of homozygosity scores defined by the percentage of mutant-specific homozygous variants within a 1-Mb window across the genome. A candidate mutation was further pinpointed by screening the region for nonsense and essential splice mutations. A diagram of whole-exome sequencing (WES) workflow and analysis is represented in
Figure 1.
2.4. Reverse Transcriptase (RT) PCR
Larvae were collected on ice in 50 µL Trizol (Life Technologies, Carlsbad, CA, USA) and stored at −80 °C until use. Samples were homogenized and a phenol-chloroform extraction was used to isolate RNA. After the RNA precipitation using isopropanol, the SuperScript® III Reverse Transcriptase kit (Life Technologies, Carlsbad, CA, USA) was used according to the manufacturer’s instructions to prepare cDNA. cDNA was diluted 1:5 in water for PCR. RT-PCR was done using the AccuPrime Taq DNA Polymerase System (Invitrogen, Carlsbad, CA, USA). Samples were analyzed on a 1% agarose gel. Primer sequences included: sart1F1: 5′-CGTTTTTAAGCCAAAGTCTGTGCTG-3′; sart1R1: 5′-CACCTCCTTCTTTCTCGTCATCCTT-3′.
2.5. sart1 mRNA Rescue
cDNA from 3 dpf wild-type larvae was amplified using the AccuPrime Taq DNA Polymerase System to produce a ~2.5 kb fragment of sart1 open reading frame (ORF). The sart1 fragment was cloned into the PCRII TOPO vector using the PCRII TOPO TA cloning kit dual promoter (Life Technologies, Carlsbad, CA, USA) and transformed into TOP 10F’ chemically competent Escherichia coli (Invitrogen, Carlsbad, CA, USA). Plasmid DNA was purified using the DNA Miniprep kit (Qiagen Sciences LLC, Germantown, MD, USA). The plasmid was then cut using EcoRV and Spe1 restriction enzymes (New England BioLabs, Ipswich, MA, USA) to release the sart1 fragment. The PCS2+ vector was cut using SnaB1 and Xba1 and a ligation reaction was performed to insert sart1 into the PCS2+ vector. After molecular cloning of PCS2+:sart1, DNA was purified using the DNA Miniprep Kit (Qiagen Sciences LLC, Germantown, MD, USA). Purified DNA was linearized using Not1 (New England BioLabs) and RNase A was added to the reaction.
Following the protocols as outlined in the mMessage machine kit (Ambion, Life Technologies, Austin, TX, USA), transcription-quality plasmid DNA was prepared and RNA was synthesized from the linearized DNA plasmid. For the rescue, approximately 150 pg of sart1 mRNA was injected into single-celled embryos produced from cp27.5 heterozygous parents. Embryos were observed daily to look for rescue of the mutant phenotype. A scoring system based on the phenotype at 4 dpf was used to determine the rescue efficiency. Larvae were scored as wild-type, mutant, or abnormal. Embryos were considered abnormal if there was an observable defect that did not resemble a wild-type or mutant phenotype. After scoring the larvae based on phenotype, samples were also collected and genotyped using PCR and Sanger DNA sequencing to determine how many mutant larvae were rescued. The percentage rescued was based on the number of mutants (based on genotype) that had wild-type phenotypes. Abnormal phenotypes for mutants were considered a partial rescue since most “abnormalities” were minor compared to phenotypes of uninjected mutants, such as reduced head size or slight heart edema.
2.6. Whole Mount In Situ Hybridization
The RNA probe was synthesized from the PCRII TOPO vector containing the 2.5 kb
sart1 fragment as mentioned above using the DIG RNA Labeling Kit (SP6/T7) (Roche, Basel, Switzerland). A probe for
deltaC was also synthesized to use as a control. The probes were purified using the illustra Probe Quant G-50 micro columns (GE Healthcare, Chicago, IL, USA) and resuspended in 80 µL hybridization buffer. Hybridization buffer was prepared as described in Thisse and Thisse [
11]. Probes were analyzed on a 1% agarose gel to determine the quality of the RNA.
cp27.5 wild-type and mutant larvae were collected at 3 dpf, anesthetized in 0.02% tricaine, and fixed in 4% paraformaldehyde (PFA) (Electron Microscopy Sciences, Hatfield, PA, USA). Larvae were stored at 4 °C for 16 h overnight. The whole mount in situ hybridization protocol was performed as previously described in Thisse and Thisse [
11].
2.7. Immunohistochemistry (IHC)
Larvae were anesthetized in 0.02% tricaine and fixed in 4% PFA at 4 °C overnight and washed the next day in 1× phosphate buffered serum (PBS) (Calbiochem, San Diego, CA, USA). Samples were sunk in 30% sucrose/PBS at 4 °C overnight and embedded in Tissue-Tek Optimal Cutting Temperature (O.C.T.) Compound (Sakura-Finetek, Torrance, CA, USA), frozen on dry ice, and stored at −80 °C. Tissue sections were washed in PBS and PBST [PBS/0.03% Triton X-100 (Sigma-Aldrich, St. Louis, MO, USA)] and incubated in blocking buffer (PBST with 5% goat serum (Gibco, Dublin, Ireland) and 1% BSA (Sigma-Aldrich, St. Louis, MO, USA)). Primary antibodies were incubated at 4 °C overnight followed by secondary antibody incubation for 2 h at room temperature. Primary antibodies included rabbit antiactivated Caspase 3 (1:200; Cell Signaling Technology, Danvers, MA, USA), rabbit anti-Glut1 (1:200, Novus Biologicals, Littleton, CO, USA), mouse anti-SV2 (1:300 Developmental Studies Hybridoma Bank, Iowa City, IA, USA), mouse anti-Zpr1 (1:100; ZIRC, Eugene, OR, USA), mouse anti-HuC/HuD neuronal protein (1:500; Life Technologies, Carlsbad, CA, USA), and rabbit anti-S100β (1:1000; Dako, Carpinteria, CA, USA). Secondary antibodies included Alexa Fluor goat antirabbit 488 (1:200; Invitrogen, Carlsbad, CA, USA) and Alexa Fluor goat antimouse 555 (1:200; Invitrogen, Carlsbad, CA, USA). Sections were counterstained with 1 µg/mL DAPI (Roche, Basel, Switzerland). Images were taken on a Nikon E800 microscope and analyzed using NIS-Elements AR Version 3.2.14 software.
2.8. Whole Mount IHC
Embryos were incubated in egg water with 0.003% PTU to prevent pigment formation. Larvae were anesthetized in 0.02% tricaine and fixed in 4% PFA overnight. Samples were washed in 1× PBS followed by 1× PBST and treated with 20 µg/mL Proteinase K (New England Biolabs, Ipswich, MA, USA). The reaction was stopped by adding 10% lamb serum (Gibco, Dublin, Ireland) followed by additional washes in PBST. Samples were blocked with 10% lamb serum for 1–4 h and incubated in primary antibodies followed by secondary antibodies. Antibodies used included rabbit anti-GFP (1:100; Invitrogen, Carlsbad, CA, USA), rabbit anti-activated Caspase-3 (1:100; Cell Signaling Technology, Danvers, MA, USA), mouse anti-Zpr1 (1:50; ZIRC, Eugene, OR, USA), and mouse anti-Cldn5 (1:100; Invitrogen, Carlsbad, CA, USA), Alexa Fluor goat anti-rabbit 488 (1:200; Invitrogen, Carlsbad, CA, USA) and Alexa Fluor goat anti-mouse 555 (1:200; Invitrogen, Carlsbad, CA, USA). Samples were imaged on Nikon C1Si laser scanning confocal microscope. Z-stacks were compiled to create maximum intensity projection images using Nikon NIS-Elements Version 3.1 imaging software.
2.9. Western Blot
An equal number of wild-type and mutant larvae were each collected in homogenization buffer (2×) (Mini Complete tablet, Roche, Basel, Switzerland) containing 0.002% DNase (Ambion Life Technologies, Austin, TX, USA). After homogenization, SDS/gel loading buffer containing DTT was added to the homogenate. Samples were then placed in boiling water for 5 min. Samples and the Precision Plus Kaleidoscope Protein Standard ladder (BioRad, Hercules, CA, USA) were run on a 4–15% polyacrylamide gel (BioRad, Hercules, CA, USA). Proteins were transferred to a nitrocellulose membrane using the Trans-Blot Turbo Transfer System (BioRad, Hercules, CA, USA). Following protein transfer, the nitrocellulose membrane was blocked in 5% milk. Membranes were incubated in primary antibody at 4 °C overnight. The membrane was then washed in 1× Tris-buffered saline/0.1% Tween (TBST) at RT and incubated in secondary antibody for 1 h at RT. After incubation, the membranes were washed in 1× TBST and imaged using LI-COR Odyssey 1393 and Odyssey Infrared Imaging System Version 3.0. Primary antibodies included rabbit anti-p53 (1:500; GeneTex, Irvine, CA, USA) and mouse anti-actin (1:1000; Sigma-Aldrich, St. Louis, MO, USA). Secondary antibodies included goat anti-rabbit IRDye ® 680 (1:20,000; LI-COR Biosciences, Lincoln, NE, USA) and goat anti-mouse IRDye ® 800 CW (1:15,000; LI-COR Biosciences, Lincoln, NE, USA).
2.10. Doxycycline Treatment
cp27.5 wild-type and mutant larvae were treated at 24 hpf with 50 µg/mL doxycycline (ClonTech Mountain View, CA, USA). Larvae were observed at 3 dpf to look for GFP expression in the CNS and imaged using a Nikon SMZ1500 epifluorescence stereoscope and Nikon NIS-Elements Version 3.1 software.
2.11. Fluorescent Tracer Injections
Wild-type and mutant cp27.5 larvae were anesthetized in 0.02% tricaine and coinjected intravenously into the common cardinal vein with approximately 1.8 nL of 10 mg/mL 3-kDa Cascade blue dextran, 10-kDa rhodamine dextran, and a 40-kDa anionic fluorescein dextran (Invitrogen, Carlsbad, CA, USA). Injections were performed using a micromanipulator and a pneumatic picopump (World Precision Instruments, Sarasota County, FL, USA). Immediately after injection, larvae were embedded in 1.2% low-melting-point agarose (Invitrogen, Carlsbad, CA, USA) made in egg water. Larvae were imaged within 30 min postinjection on a Nikon C1Si confocal microscope and analyzed using Nikon EZC1 Version 3.91 software.
2.12. RNA-Seq Analysis
RNA was extracted as described before previously (see
Section 2.4) from wild-type and mutant
cp27.5 larvae at 72 hpf. Samples were submitted to the Genome Sequencing Facility Hartwell Center for Bioinformatics and Biotechnology at St. Jude Children’s Research Hospital for Quality Control (QC) and quantification analysis and sequencing. Approximately 500 ng total RNA was used to generate libraries with the TruSeq RNA v2 kit according to the manufacturer’s instructions (Illumina, San Diego, CA). PolyA-selected RNAs from wild-type and mutant zebrafish were sequenced using Illumina HiSeq 2500. The raw 100-bp pair end reads were mapped to zebrafish genome (Zv9) using Spliced Transcripts Alignment to a Reference (STAR) [
12]. Duplicated reads were marked using Picard (
http://picard.sourceforge.net/command-line-overview.shtml). The detailed mapping statistics are listed in
Table 1.
Considering genome duplication, FPKM (fragments per kilobase of exon per million fragments mapped) for each RefSeq gene was calculated as follows: first fragments mapped to each exon region of a gene, regardless of their genomic location, were marked and summed up as the gene counts. The final FPKM value for the gene was calculated as:
where F
gene is the total fragments mapped to a gene, F
allexon is total fragments mapped to exonic region in Zv9, and L
gene is size of unique exonic bases for the gene. Differential expression analysis was done using purely fold change between the mutant and wild-type cells. Gene Ontology (GO) analysis was done using the Database for Annotation, Visualization and Integrated Discovery (DAVID) [
13], with differentially expressed genes that have moderate expression level (FPKM > 0.1).
2.13. Quantitative Real Time PCR (qRT-PCR)
RNA was extracted from 9
cp27.5 wild-type and 9
cp27.5 mutant larvae and cDNA was synthesized using the same method as described in previously (see
Section 2.4). RNA concentrations were normalized between wild-type and mutant. Samples were treated with DNAse I (Ambion Life Technologies, Austin, TX, USA) followed by treatment with 25 mM EDTA (Ambion). 20 ng of cDNA was used per reaction for quantitative real-time PCR (qRT-PCR). qRT-PCR was performed using SYBR
® Green PCR Master Mix (Life Technologies, Carlsbad, CA, USA) on a 7500 Fast Real Time PCR machine (Applied Biosystems, Foster City, CA, USA) and analyzed using 7500 Software v2.0.6. Relative gene expression was calculated using the Delta Delta CT (ddCt) method relative to the reference gene
β-actin. Error bars were based on the mean of 3 rounds of qRT-PCR using standard error. The primer templates used for each gene are listed in
Table 2.
2.14. Identification of Novel Junctions in cp27.5 Mutants
From the RNA-Seq data, tp53 and mdm2 were analyzed to identify novel junctions or alternative splice sites in cp27.5 mutants. Junctions were identified using a CompBio pipeline. From there, the low count junctions were filtered and normalized using Limma-Voom. The criteria for selecting differential exon junctions included the average intensity (log2 RPM) > 2 and a log2ratio > 2. The reads mapped to exon junctions were extracted and normalized to counts per million (RPM). Different usage of each junction between the wild type and mutant sample was evaluated and candidate junctions were selected with fold change greater than 4 and average RPM of the 2 samples more than 4.
2.15. Data Availability
The strains available upon request included
Et(
cp:EGFP)
sj2 which express GFP in the CP. The
cp27.5 strain was no longer available.
Supplementary Files are available at FigShare.
Supplementary File S1 contains the list of genes with at least 1 read observed in either one of the samples (wild type or mutant), genes expressed where the FPKM > 0.1 in both samples, gene ontology for biological processes and molecular function, the Kyoto Encyclopedia of Genes and Genomes (KEGG), and a list of upregulated spliceosome-related genes.. The data is deposited at Figshare:
https://doi.org/10.6084/m9.figshare.12980063.v1. Code and software used to generate the whole exome sequencing and RNA-Seq data can be found at:
4. Discussion
The current study describes a zebrafish mutant with a point mutation in
sart1 that results in abnormal CP development [
6] in addition to defects throughout the brain and eye. In addition to splicing,
Sart1 has been implicated in a number of different processes such as cell cycle arrest and apoptosis and has been suggested as a target for gene therapy due to its role as an antigen recognized by cytotoxic T-lymphocytes (CTLs) in certain types of cancer [
17]. More recently,
Sart1 has been shown to contribute to drug resistance in cancer cells [
18,
29]. While the goal of this study was to characterize
sart1 and identify other genes affected by this mutation, future experiments plan to discover the role of Sart1 in overall cellular processes such as cell death; in addition to better understanding its traditional role in splicing.
In our study, we identified the point mutation in
sart1 using WES. We were able to validate along with previously published studies [
7,
30] that WES is a suitable method for identifying genes in zebrafish. WES allowed us to effectively identify what gene contained the mutation and that the mutation was a transition. In future genetic mapping experiments, we plan to use WES as our main method for identifying the map position and type of mutation. Studies have cited that more than 9000 mutants have been discovered as a result of forward genetic screens in zebrafish, but many of the mutants have not been further studied and 62% of them are not cloned. This is due to limitations involved in positional cloning including the time, money, and effort to identify the gene [
31]. Therefore, we propose that WES provides a more efficient method than positional cloning to identify mutants generated from forward genetic screens.
Additional validation to show that the phenotypes were due to a mutation in
sart1 was done by performing mRNA rescue experiments.
sart1 mRNA prevented forebrain necrosis at 48 hpf, which is the earliest detectable phenotype in
cp27.5 mutants. We were able to rescue phenotypes that began at 3–4 dpf such as heart edema, small head and eyes, reduced heartbeat, and circulation. From this analysis, a number of wild-type larvae were scored as abnormal. This could be due to a dominant negative effect of the
sart1 mutation. However, in
Figure 2B representing the whole exome sequencing data, we have shown that the zebrafish larvae screened as “wild-type” are heterozygous because some of the fish in the sample had the G → A transition. We believe that if a dominant-negative mutation was present, the heterozygous fish would have similar phenotypes if not the same phenotypes, as the homozygous mutants because the phenotypes can be observed quite easily and at early developmental time points. We suggest that abnormal phenotypes for wild-type larvae may be due to off target effects from the injection process. When sequencing wild-type and mutant DNA from
sart1 mRNA injections, we identified only 20% as mutant rather than 25% according to Mendelian genetics. We attributed the decreased percentage due to damaged embryos from microinjection that may not have survived past early development. For uninjected samples, we observed between 23 and 25% mutant.
Prior to 48 hpf, mutants were undistinguishable from wild-type. The maternally derived transcript as identified by RT-PCR was present shortly after fertilization which may explain why we were unable to isolate mutant embryos prior to 48 hpf because of maternal contribution. Previous studies performed by Thisse and Thisse have also identified
sart1 expression shortly after fertilization during early cleavage thereby also confirming that the transcripts are maternal [
14]. Interestingly, by whole mount in situ hybridization and RNA-Seq analysis, we determined that the point mutation in
sart1 results in upregulation of the transcript. Upregulation of mutated splicing genes have been demonstrated in other studies such as
prpf4, which interacts with the U4/U6 di-snRNP and stabilizes the complex [
15]. A mutation from a proline to leucine in
prpf4 led to its increased expression and also upregulation of other splicing factors including
sart1. Similar to our study, they detected a loss of photoreceptors in the zebrafish eye and a downregulation of vision related genes such as
opn1lw1, another opsin gene [
15].
Additionally, the loss of expression in other vision-related proteins, such as Zpr1, SV2, HuC, and S100β, is not surprising due to previous studies showing that mutations in splicing factors can result in photoreceptor degeneration in diseases such as RP [
15] where the most noticeable phenotype is a loss of photoreceptor cells [
32]. Interestingly, for proteins expressed throughout the CNS, such as Zpr1 and SV2, there were differences in expression between the brain and eye suggesting that
sart1 may be regulating expression of certain genes in a tissue-specific manner. A recent study cited six genes related to splicing that have mutations in patients with RP including
PRPF31,
PRPF8,
PRPF3,
PAP-1,
SNRNP200, and
PRPF6. These mutations are responsible for autosomal dominant RP in more than 12% of cases [
33]. Of these genes, three including
PRPF8,
PRPF31, and
PRPF3 are involved in forming and recruiting the U4/U6.U5 tri-snRNP to the spliceosome [
32]. The exact cause of how mutations in splicing genes result in RP is still unknown, however, several models or hypothesis have been suggested by Mordes et al. [
32]. Jia et al. also proposed a hypothesis where mis-splicing due to mutations in U2, a major spliceosome component, results in a buildup of mis-spliced mRNA that translates into dysfunctional proteins. Another hypothesis by Jia et al. proposes that introns retained within the mRNA sequence cause the spliceosome to remain at the intron rather than be released, resulting in alternative splicing and a toxic feedback loop where mRNA continues to be mis-spliced and cell death occurs [
10].
In addition to splicing factors, mutations in other genes such as
crx encoding a transcription factor, and
pde genes involved in phototransduction, both of which were downregulated in
cp27.5 mutants, also contribute to RP [
32]. We have to consider that perhaps the downregulation of vision-related genes is simply due to a developmental delay in these mutants. However, we do not believe that developmental delay can be contributing to the downregulation of genes in
cp27.5 mutants because the expression of these genes is not downregulated in every tissue. For example, mutants lacked Zpr1 expression in the retina, although expression was present in the pineal gland (
Figure 6A,B) and SV2 exhibited similar expression in the brain between wild-type and mutants, but was decreased in the eye in
cp27.5 mutants. We would anticipate a decreased expression of these markers in all tissues had the downregulation been due to developmental delay.
In
cp27.5 mutants, we detected increased cell death within the eye as observed by activated Caspase 3 antibody staining. From the RNA-Seq data, we also detected upregulation of
tp53 and
mdm2. This is interesting because Mdm2 acts as an inhibitor of p53, so we did not expect that both would be upregulated. While upregulation of
tp53 explains the increased cell death found in the eye and brain, increased expression of
mdm2 may indicate that it is trying to compensate for increased
tp53 expression. However, in previous studies, a feedback loop has been shown to occur where p53 can activate Mdm2 [
34]. During cellular stress, the cell death pathway is initiated causing p53 levels to increase and Mdm2 levels to decrease. As a result of decreased Mdm2, p53 initiates
mdm2 transcription. As Mdm2 levels then increase, Mdm2 in turn acts to inhibit p53 [
34]. Future studies will investigate whether
mdm2 is functional in inhibiting the p53 protein and whether p53 activates Mdm2 and forms a negative feedback loop. We also believe that the mutated
sart1 is affecting splicing of these apoptotic genes. We have identified novel splice site junctions in
p53 and
mdm2 in
cp27.5 mutants (
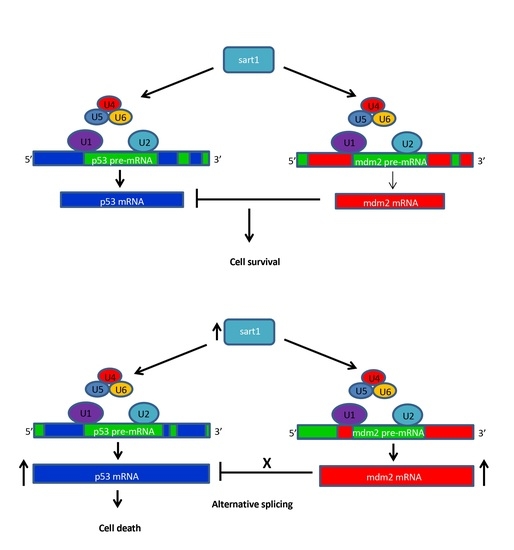
Figure 9) suggesting that these novel splice junctions result in new protein isoforms that cannot be properly regulated. To confirm this hypothesis, a western blot analysis detected an alternative p53 protein synthesized in
cp27.5 mutants as identified by a prominent larger band for p53 than the predicted 43 kDa in mutant samples. While this band was also detected in wild-type samples, expression was much less compared to mutants. We believe this is due to heterozygous siblings present in the wild type sample. We suggest from our data that alternative splicing due to mutated Sart1 causes additional p53 proteins to be synthesized in
cp27.5 mutants that cannot be inhibited by Mdm2 thereby resulting in increased cell death (
Figure 10). We also hypothesize that novel splice site junctions in
mdm2 may result in a defective protein that is unable to inhibit p53 function. As to why cell death is localized to the eye and brain has yet to be determined, but it is possible that Sart1 is affecting splicing in a tissue specific manner.
cp27.5 was originally identified from a genetic screen isolating mutants with CP developmental defects. Interestingly, one of the genes we identified from the RNA-Seq data was
clu. This gene is expressed specifically within the zebrafish CP in early development [
19]. It acts as a chaperone and is a prosurvival gene in its secreted form by binding to Bax and preventing Bax from traveling to the mitochondria [
35]. In its nuclear form, which originates from alternative splicing, it acts as a prodeath gene by promoting cell death in a Caspase-3
independent manner [
35,
36]. This agrees with our findings showing
clu and Caspase-3 upregulation in
cp27.5 mutants. Future studies will investigate whether the nuclear form of
clu is responsible for cell death in
cp27.5 mutants and whether cell death is specific to the CP. Upregulation of
clu from the RNA-Seq analysis may be due to alternative splicing of
clu resulting in its overexpression and therefore increased cell death. Alternatively, as mentioned previously, the secreted form of
clu can act as a prosurvival protein [
35]. If the secreted protein is upregulated in mutants, it may act in competition with proapoptotic genes such as
tp53 and compensating for cell death in the CP. However, even though
clu was found to be upregulated at the transcript level, future studies will need to determine whether it is translated into a functional protein.
Another protein demonstrated in previous studies to be expressed in the CP is Cldn5 [
6]. From the RNA-Seq data, we determined that
cldn5a is also slightly upregulated in
cp27.5 mutants. While we were able to detect Cldn5 expression in mutants by whole mount IHC, future experiments will need to determine if it is functional. Cldn5 appeared to be mislocalized in mutants due to an absence of co-expression with GFP. Tracer studies revealed abnormal vasculature in the CNS indicating that the
sart1 mutation may be strictly CNS specific and does not affect peripheral tight junctions or vasculature.
While the genetic pathways downstream of Sart1 in cp27.5 mutants have yet to be determined, the current study has revealed dysregulation of genes involved in apoptosis, splicing, vision, tight junctions, and matrix metalloproteinases. RNA-Seq analysis has provided substantial insights into genes and potential pathways regulated by Sart1. This study and others have demonstrated that mutations in splicing machinery or proteins associated with the spliceosome have widespread affects in other signaling processes in addition to splicing defects. Additionally, differences between phenotypes observed in the CNS versus the periphery may suggest that Sart1 regulates the expression of certain genes in a tissue-specific manner.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
