The Role of Aryl-Hydrocarbon Receptor (AhR) in Osteoclast Differentiation and Function

Abstract
1. Introduction
2. Role of AhR in Osteoclast Differentiation and Function
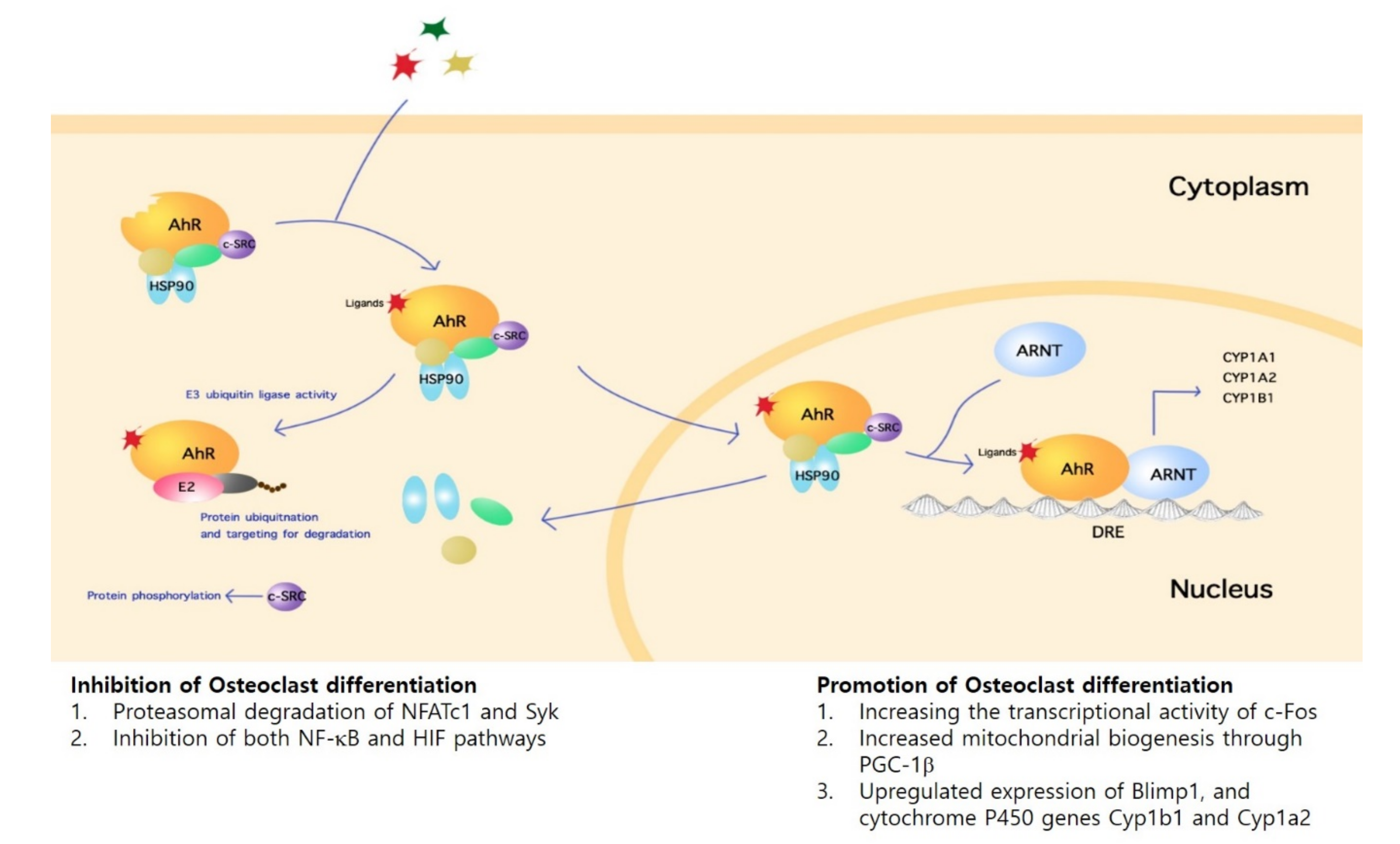
3. Regulatory Mechanisms of AhR in Osteoclasts
3.1. Inhibitory Mechanisms
3.1.1. Direct Mechanisms
3.1.2. Indirect Mechanisms
3.2. Promotion Mechanisms
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kewley, R.J.; Whitelaw, M.L.; Chapman-Smith, A. The mammalian basic helix-loop-helix/PAS family of transcriptional regulators. Int. J. Biochem. Cell Biol. 2004, 36, 189–204. [Google Scholar] [CrossRef]
- Stockinger, B.; Meglio, P.D.; Gialitakis, M.; Duarte, J.H. The Aryl Hydrocarbon Receptor: Multitasking in the Immune System. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Trikha, P.; Lee, D.A. The role of AhR in transcriptional regulation of immune cell development and function. Biochim. Biophys. Acta (BBA)-Rev. Cancer 2020, 1873, 188335. [Google Scholar] [CrossRef]
- Goettel, J.A.; Gandhi, R.; Kenison, J.E.; Yeste, A.; Murugaiyan, G.; Sambanthamoorthy, S.; Griffith, A.E.; Patel, B.; Shouval1, D.S.; Weiner, H.L.; et al. AHR Activation Is Protective against Colitis Driven by T Cells in Humanized Mice. Cell Rep. 2016, 17, 1318–1329. [Google Scholar] [CrossRef]
- Gandhi, R.; Kumar, D.; Burns, E.J.; Nadeau, M.; Dake, B.; Laroni, A.; Kozoriz, D.; Weiner, H.L.; Quintana, F.J. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell-like and Foxp3(+) regulatory T cells. Nat. Immunol. 2010, 11, 846–853. [Google Scholar] [CrossRef]
- Gagliani, N.; Amezcua Vesely, M.C.; Iseppon, A.; Brockmann, L.; Xu, H.; Palm, N.W.; De Zoete, M.R.; Licona-Limón, P.; Paiva, R.S.; Ching, T.; et al. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature 2015, 523, 221–225. [Google Scholar] [CrossRef]
- Bankoti, J.; Burnett, A.; Navarro, S.; Miller, A.K.; Rase, B.; Shepherd, D.M. Effects of TCDD on the fate of naive dendritic cells. Toxicol. Sci. 2010, 115, 422–434. [Google Scholar] [CrossRef]
- Bankoti, J.; Rase, B.; Simones, T.; Shepherd, D.M. Functional and phenotypic effects of AhR activation in inflammatory dendritic cells. Toxicol. Appl. Pharmacol. 2010, 246, 18–28. [Google Scholar] [CrossRef]
- Quintana, F.J.; Murugaiyan, G.; Farez, M.F.; Mitsdoerffer, M.; Tukpah, A.M.; Burns, E.J.; Weiner, H.L. An endogenous aryl hydrocarbon receptor ligand acts on dendritic cells and T cells to suppress experimental autoimmune encephalomyelitis. Proc. Natl. Acad. Sci. USA 2010, 107, 20768–20773. [Google Scholar] [CrossRef]
- Boitano, A.E.; Wang, J.; Romeo, R.; Bouchez, L.C.; Parker, A.E.; Sutton, S.E.; Walker, J.R.; Flaveny, C.A.; Perdew, G.H.; Denison, M.S.; et al. Aryl hydrocarbon receptor antagonists promote the expansion of human hematopoietic stem cells. Science 2010, 329, 1345–1348. [Google Scholar] [CrossRef] [PubMed]
- Ilvesaro, J.; Pohjanvirta, R.; Tuomisto, J.; Viluksela, M.; Tuukkanen, J. Bone resorption by aryl hydrocarbon receptor-expressing osteoclasts is not disturbed by TCDD in short-term cultures. Life Sci. 2005, 77, 1351–1366. [Google Scholar] [CrossRef] [PubMed]
- Fujii-Kuriyama, Y.; Mimura, J. Molecular mechanisms of AhR functions in the regulation of cytochrome P450 genes. Biochem. Biophys. Res. Commun. 2005, 338, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Dong, B.; Cheng, W.; Li, W.; Zheng, J.; Wu, D.; Matsumura, F.; Vogel, C.F.A. FRET analysis of protein tyrosine kinase c-Src activation mediated via aryl hydrocarbon receptor. Biochim. Biophys. Acta 2011, 1810, 427–431. [Google Scholar] [CrossRef] [PubMed]
- Ohtake, F.; Baba, A.; Takada, I.; Okada, M.; Iwasaki, K.; Miki, H.; Takahashi, S.; Kouzmenko, A.; Nohara, K.; Chiba, T.; et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature 2007, 446, 562–566. [Google Scholar] [CrossRef]
- Ovrevik, J.; Lag, M.; Lecureur, V.; Gilot, D.; Lagadic-Gossmann, D.; Refsnes, M.; Schwarze, P.E.; Skuland, T.; Becher, R.; Holme, J.A. AhR and Arnt differentially regulate NF-kappaB signaling and chemokine responses in human bronchial epithelial cells. Cell Commun. Signal. 2014, 12, 48. [Google Scholar] [CrossRef]
- Schneider, A.J.; Branam, A.M.; Peterson, R.E. Intersection of AHR and Wnt signaling in development, health, and disease. Int. J. Mol. Sci. 2014, 15, 17852–17885. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Kurita, H.; Carreira, V.; Ko, C.I.; Fan, Y.; Zhang, X.; Biesiada, J.; Medvedovic, M.; Puga, A. Ah Receptor Activation by Dioxin Disrupts Activin, BMP, and WNT Signals During the Early Differentiation of Mouse Embryonic Stem Cells and Inhibits Cardiomyocyte Functions. Toxicol. Sci. 2016, 149, 346–357. [Google Scholar] [CrossRef]
- Wincent, E.; Stegeman, J.J.; Jonsson, M.E. Combination effects of AHR agonists and Wnt/beta-catenin modulators in zebrafish embryos: Implications for physiological and toxicological AHR functions. Toxicol. Appl. Pharmacol. 2015, 284, 163–179. [Google Scholar] [CrossRef]
- Occhi, G.; Barollo, S.; Regazzo, D.; Bertazza, L.; Galuppini, F.; Guzzardo, V.; Jaffrain-Rea, M.L.; Vianello, F.; Ciato, D.; Ceccato, F.; et al. A constitutive active MAPK/ERK pathway due to BRAFV600E positively regulates AHR pathway in PTC. Oncotarget 2015, 6, 32104–32114. [Google Scholar] [CrossRef][Green Version]
- Nguyen, N.T.; Hanieh, H.; Nakahama, T.; Kishimoto, T. The roles of aryl hydrocarbon receptor in immune responses. Int. Immunol. 2013, 25, 335–343. [Google Scholar] [CrossRef] [PubMed]
- Herlin, M.; Finnila, M.A.; Zioupos, P.; Aula, A.; Risteli, J.; Miettinen, H.M.; Jämsä, T.; Tuukkanen, J.; Korkalainen, M.; Håkansson, H.; et al. New insights to the role of aryl hydrocarbon receptor in bone phenotype and in dioxin-induced modulation of bone microarchitecture and material properties. Toxicol. Appl. Pharmacol. 2013, 273, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.Y.; Kondo, T.; Matsumoto, T.; Fujii-Kuriyama, Y.; Imai, Y. Aryl hydrocarbon receptor catabolic activity in bone metabolism is osteoclast dependent in vivo. Biochem. Biophys. Res. Commun. 2014, 450, 416–422. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Hata, S.; Ono, K.; Suzuki, T.; Ito, K.; Kumamoto, H.; Sasano, H. Roles of Aryl Hydrocarbon Receptor in Aromatase-Dependent Cell Proliferation in Human Osteoblasts. Int. J. Mol. Sci. 2017, 18, 2159. [Google Scholar] [CrossRef]
- Liu, W.C.; Shyu, J.F.; Lim, P.S.; Fang, T.C.; Lu, C.L.; Zheng, C.M.; Hou, Y.C.; Wu, C.C.; Lin, Y.F.; Lu, K.C. Concentration and Duration of Indoxyl Sulfate Exposure Affects Osteoclastogenesis by Regulating NFATc1 via Aryl Hydrocarbon Receptor. Int. J. Mol. Sci. 2020, 21, 3486. [Google Scholar] [CrossRef]
- Iqbal, J.; Sun, L.; Cao, J.; Yuen, T.; Lu, P.; Bab, I.; Leu, N.A.; Srinivasan, S.; Wagage, S.; Hunter, C.A.; et al. Smoke carcinogens cause bone loss through the aryl hydrocarbon receptor and induction of Cyp1 enzymes. Proc. Natl. Acad. Sci. USA 2013, 110, 11115–11120. [Google Scholar] [CrossRef]
- Voronov, I.; Heersche, J.N.; Casper, R.F.; Tenenbaum, H.C.; Manolson, M.F. Inhibition of osteoclast differentiation by polycyclic aryl hydrocarbons is dependent on cell density and RANKL concentration. Biochem. Pharmacol. 2005, 70, 300–307. [Google Scholar] [CrossRef]
- Korkalainen, M.; Kallio, E.; Olkku, A.; Nelo, K.; Ilvesaro, J.; Tuukkanen, J.; Mahonen, A.; Viluksela, M. Dioxins interfere with differentiation of osteoblasts and osteoclasts. Bone 2009, 44, 1134–1142. [Google Scholar] [CrossRef]
- Wejheden, C.; Brunnberg, S.; Larsson, S.; Lind, P.M.; Andersson, G.; Hanberg, A. Transgenic mice with a constitutively active aryl hydrocarbon receptor display a gender-specific bone phenotype. Toxicol. Sci. 2010, 114, 48–58. [Google Scholar] [CrossRef]
- Boverhof, D.R.; Kwekel, J.C.; Humes, D.G.; Burgoon, L.D.; Zacharewski, T.R. Dioxin induces an estrogen-like, estrogen receptor-dependent gene expression response in the murine uterus. Mol. Pharmacol. 2006, 69, 1599–1606. [Google Scholar] [CrossRef] [PubMed]
- Safe, S.; Connor, K.; Gaido, K. Methods for xenoestrogen testing. Toxicol. Lett. 1998, 102, 665–670. [Google Scholar] [CrossRef]
- Lorenzo, J. Sexual Dimorphism in Osteoclasts. Cells 2020, 9, E2086. [Google Scholar] [CrossRef] [PubMed]
- DuSell, C.D.; Nelson, E.R.; Wittmann, B.M.; Fretz, J.A.; Kazmin, D.; Thomas, R.S.; Pike, J.W.; McDonnell, D.P. Regulation of aryl hydrocarbon receptor function by selective estrogen receptor modulators. Mol. Endocrinol. 2010, 24, 33–46. [Google Scholar] [CrossRef] [PubMed]
- Jamsa, T.; Viluksela, M.; Tuomisto, J.T.; Tuomisto, J.; Tuukkanen, J. Effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin on bone in two rat strains with different aryl hydrocarbon receptor structures. J. Bone Miner. Res. 2001, 16, 1812–1820. [Google Scholar] [CrossRef]
- Miettinen, H.M.; Pulkkinen, P.; Jamsa, T.; Koistinen, J.; Simanainen, U.; Tuomisto, J.; Tuukkanen, J.; Viluksela, M. Effects of in utero and lactational TCDD exposure on bone development in differentially sensitive rat lines. Toxicol. Sci. 2005, 85, 1003–1012. [Google Scholar] [CrossRef]
- Asagiri, M.; Takayanagi, H. The molecular understanding of osteoclast differentiation. Bone 2007, 40, 251–264. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Gierthy, J.F.; Silkworth, J.B.; Tassinari, M.; Stein, G.S.; Lian, J.B. 2,3,7,8-Tetrachlorodibenzo-p-dioxin inhibits differentiation of normal diploid rat osteoblasts in vitro. J. Cell Biochem. 1994, 54, 231–238. [Google Scholar] [CrossRef]
- Naruse, M.; Ishihara, Y.; Miyagawa-Tomita, S.; Koyama, A.; Hagiwara, H. 3-Methylcholanthrene, which binds to the arylhydrocarbon receptor, inhibits proliferation and differentiation of osteoblasts in vitro and ossification in vivo. Endocrinology 2002, 143, 3575–3581. [Google Scholar] [CrossRef] [PubMed]
- Koskela, A.; Viluksela, M.; Keinanen, M.; Tuukkanen, J.; Korkalainen, M. Synergistic effects of tributyltin and 2,3,7,8-tetrachlorodibenzo-p- dioxin on differentiating osteoblasts and osteoclasts. Toxicol. Appl. Pharmacol. 2012, 263, 210–217. [Google Scholar] [CrossRef]
- Izawa, T.; Arakaki, R.; Mori, H.; Tsunematsu, T.; Kudo, Y.; Tanaka, E.; Ishimaru, N. The Nuclear Receptor AhR Controls Bone Homeostasis by Regulating Osteoclast Differentiation via the RANK/c-Fos Signaling Axis. J. Immunol. 2016, 197, 4639–4650. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.Y.; Pang, W.J.; Yang, G.S. Aryl hydrocarbon receptors in osteoclast lineage cells are a negative regulator of bone mass. PLoS ONE 2015, 10, e0117112. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.F.; Lv, Q.; Xia, Y.; Yue, M.F.; Shi, C.; Xia, Y.F.; Chou, G.; Wang, Z.; Dai, Y. Norisoboldine, an Anti-Arthritis Alkaloid Isolated from Radix Linderae, Attenuates Osteoclast Differentiation and Inflammatory Bone Erosion in an Aryl Hydrocarbon Receptor-Dependent Manner. Int. J. Biol Sci. 2015, 11, 1113–1126. [Google Scholar] [CrossRef] [PubMed]
- Voronov, I.; Li, K.; Tenenbaum, H.C.; Manolson, M.F. Benzo[a]pyrene inhibits osteoclastogenesis by affecting RANKL-induced activation of NF-kappaB. Biochem. Pharmacol. 2008, 75, 2034–2044. [Google Scholar] [CrossRef] [PubMed]
- Naruse, M.; Otsuka, E.; Ishihara, Y.; Miyagawa-Tomita, S.; Hagiwara, H. Inhibition of osteoclast formation by 3-methylcholanthrene, a ligand for arylhydrocarbon receptor: Suppression of osteoclast differentiation factor in osteogenic cells. Biochem. Pharmacol. 2004, 67, 119–127. [Google Scholar] [CrossRef]
- Jia, Y.; Tao, Y.; Lv, C.; Xia, Y.; Wei, Z.; Dai, Y. Tetrandrine enhances the ubiquitination and degradation of Syk through an AhR-c-src-c-Cbl pathway and consequently inhibits osteoclastogenesis and bone destruction in arthritis. Cell Death Dis. 2019, 10, 38. [Google Scholar] [CrossRef]
- Fu, J.; Nogueira, S.V.; Drongelen, V.V.; Coit, P.; Ling, S.; Rosloniec, E.F.; Sawalha, A.H.; Holoshitz, J. Shared epitope-aryl hydrocarbon receptor crosstalk underlies the mechanism of gene-environment interaction in autoimmune arthritis. Proc. Natl. Acad. Sci. USA 2018, 115, 4755–4760. [Google Scholar] [CrossRef]
- Csanaky, I.L.; Lickteig, A.J.; Klaassen, C.D. Aryl hydrocarbon receptor (AhR) mediated short-term effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) on bile acid homeostasis in mice. Toxicol. Appl. Pharmacol. 2018, 343, 48–61. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, K.; Nakashima, T.; Hayashi, M.; Fukunaga, T.; Kato, S.; Kodama, T.; Takahashi, S.; Calame, K.; Takayanagi, H. Blimp1-mediated repression of negative regulators is required for osteoclast differentiation. Proc. Natl. Acad. Sci. USA 2010, 107, 3117–3122. [Google Scholar] [CrossRef]


{kind=link}
| Study | Experimental Model | Agonist | Antagonist | Effect on Osteoclastogenesis | Mechanism of Action |
|---|---|---|---|---|---|
| In vitro studies | |||||
| Koskela 2012 [40] | BM cells | TCDD | N/A | Inhibition | N/A |
| Izawa 2016 [41] | BM cells | BaP | N/A | Promotion | Via c-Fos–NFATc1 and mitochondrial biogenesis through PGC-1β |
| Yu 2015 [42] | BM cells | 3-MC | N/A | Promotion | Blimp1, Cyp1b1, and Cyp1a2 expression was downregulated in the absence of AhR |
| Voronov 2005 [27] | RAW264.7 cells | BaP | Resveratrol | Inhibition | N/A |
| Liu 2020 [25] | RAW 264.7 cells | Indoxyl-sulfate | CH223191 or siRNA | Agonist dose and duration dependent: -Promotion (short-term, low-dose) -Inhibition (long-term, high-dose) | Different IS levels switch the role of AhR from that of a ligand-activated transcription factor to that of an E3 ubiquitin ligase |
| DuSell 2010 [33] | RAW264.7 cells | 4-OHT (4-hydroxy-TAM) | α-naphthoflavone siRNA | Inhibition | N/A |
| Wei 2015 [43] | RAW 264.7 cells | Norisoboldine (NOR) | Resveratrol α-naphthoflavone | Inhibition | Inhibition of both NF-κB and HIF pathways |
| Voronov 2008 [44] | RAW264.7 cells | BaP | N/A | Inhibition | Consequence of crosstalk between AhR and RANKL signaling pathways competing for the common transcription factor NF-kB |
| Naruse 2004 [45] | Mouse spleen cells and clonal osteogenic stromal ST2 cells | 3-MC | N/A | Inhibition | Via the inhibition of RANKL expression in osteoblastic cells |
| Korkalainen 2009 [28] | Haematopoietic stem cells | TCDD | N/A | Inhibition | N/A |
| Ilvesaro 2009 [12] | Rat osteoclasts from long bones | TCDD | N/A | No effect | N/A |
| Iqbal 2013 [26] | In vitro: BM cells, RAW-C3 cells In vivo: mice with BaP, TCDD oral gavage | BaP, TCDD | N/A | Promotion | N/A |
| Jia 2019 [46] | In vitro: BM cells RAW264.7 cells In vivo: CIA rats | Tetrandrine, DIM | CH223191 or siRNA | Inhibition | Enhanced ubiquitination and degradation of Syk through the AhR/c-src/c-Cbl signaling pathway |
| Fu 2018 [47] | In vitro: BM cells In vivo: SE transgenic mice | 6-formylindolo[3,2-b]carbazole (FICZ), TCDD | N/A | Promotion | Interaction between SE and AhR agonists during osteoclastogenesis is mediated by the NF-κB signaling pathway |
| In vivo studies | |||||
| Csanaky 2018 [48] | Juvenile mice with oral gavage of TCDD | TCDD | N/A | Inhibition | N/A |
| Yu 2014 [23] | AhR(ΔOc/ΔOc) mice | 3-MC | N/A | Promotion | N/A |
| Wejheden 2010 [29] | CA-AhR mice | N/A | N/A | Promotion | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, R.; Madhavaram, S.; Ji, J.D. The Role of Aryl-Hydrocarbon Receptor (AhR) in Osteoclast Differentiation and Function. Cells 2020, 9, 2294. https://doi.org/10.3390/cells9102294
Park R, Madhavaram S, Ji JD. The Role of Aryl-Hydrocarbon Receptor (AhR) in Osteoclast Differentiation and Function. Cells. 2020; 9(10):2294. https://doi.org/10.3390/cells9102294
Chicago/Turabian StylePark, Robin, Shreya Madhavaram, and Jong Dae Ji. 2020. "The Role of Aryl-Hydrocarbon Receptor (AhR) in Osteoclast Differentiation and Function" Cells 9, no. 10: 2294. https://doi.org/10.3390/cells9102294
APA StylePark, R., Madhavaram, S., & Ji, J. D. (2020). The Role of Aryl-Hydrocarbon Receptor (AhR) in Osteoclast Differentiation and Function. Cells, 9(10), 2294. https://doi.org/10.3390/cells9102294