Targeting Notch Trafficking and Processing in Cancers


Abstract
:1. Introduction
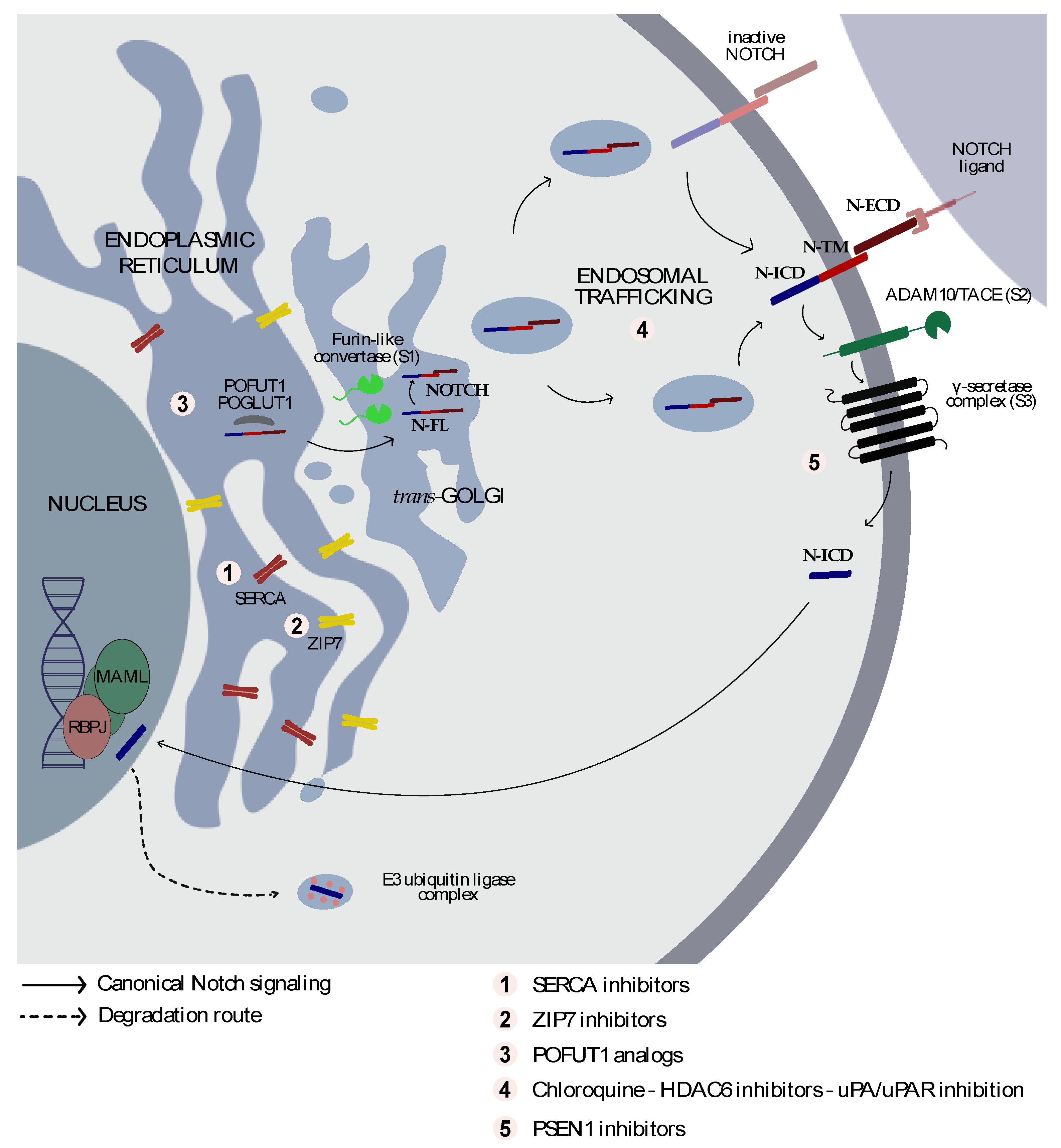
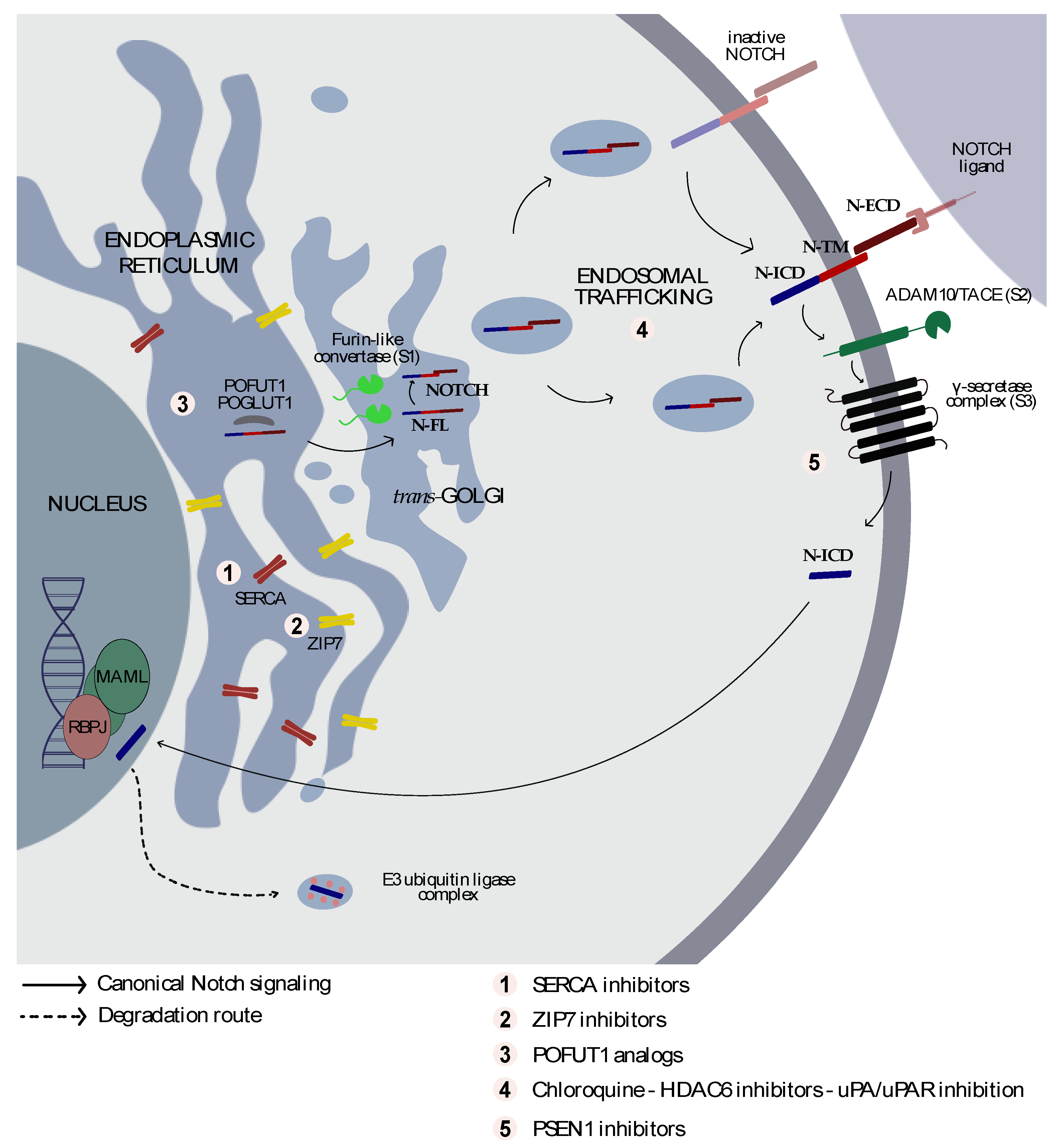
2. The NOTCH Structure
3. From Golgi to the Cell Membrane
4. From the Cell Membrane to the Nucleus
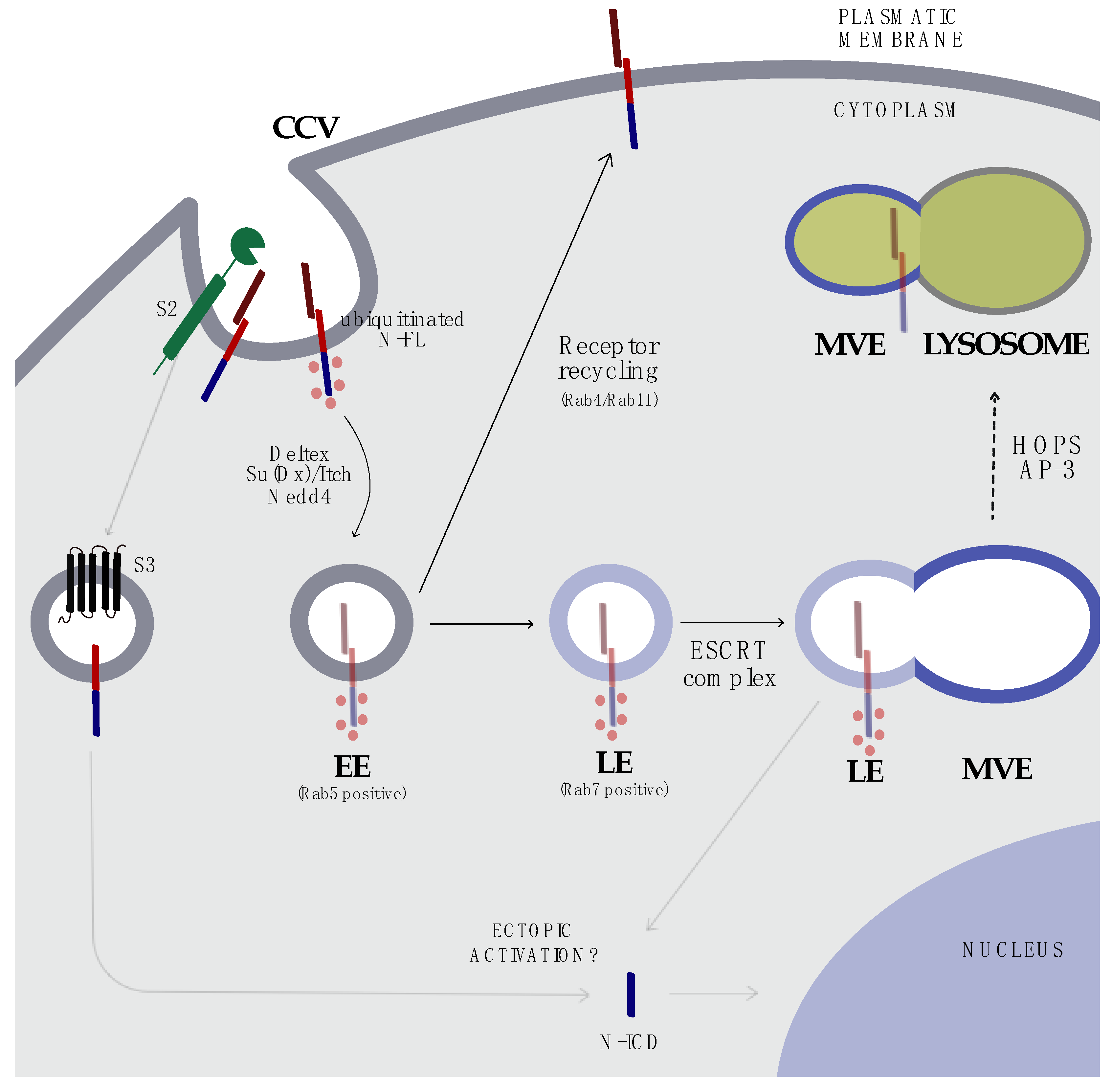
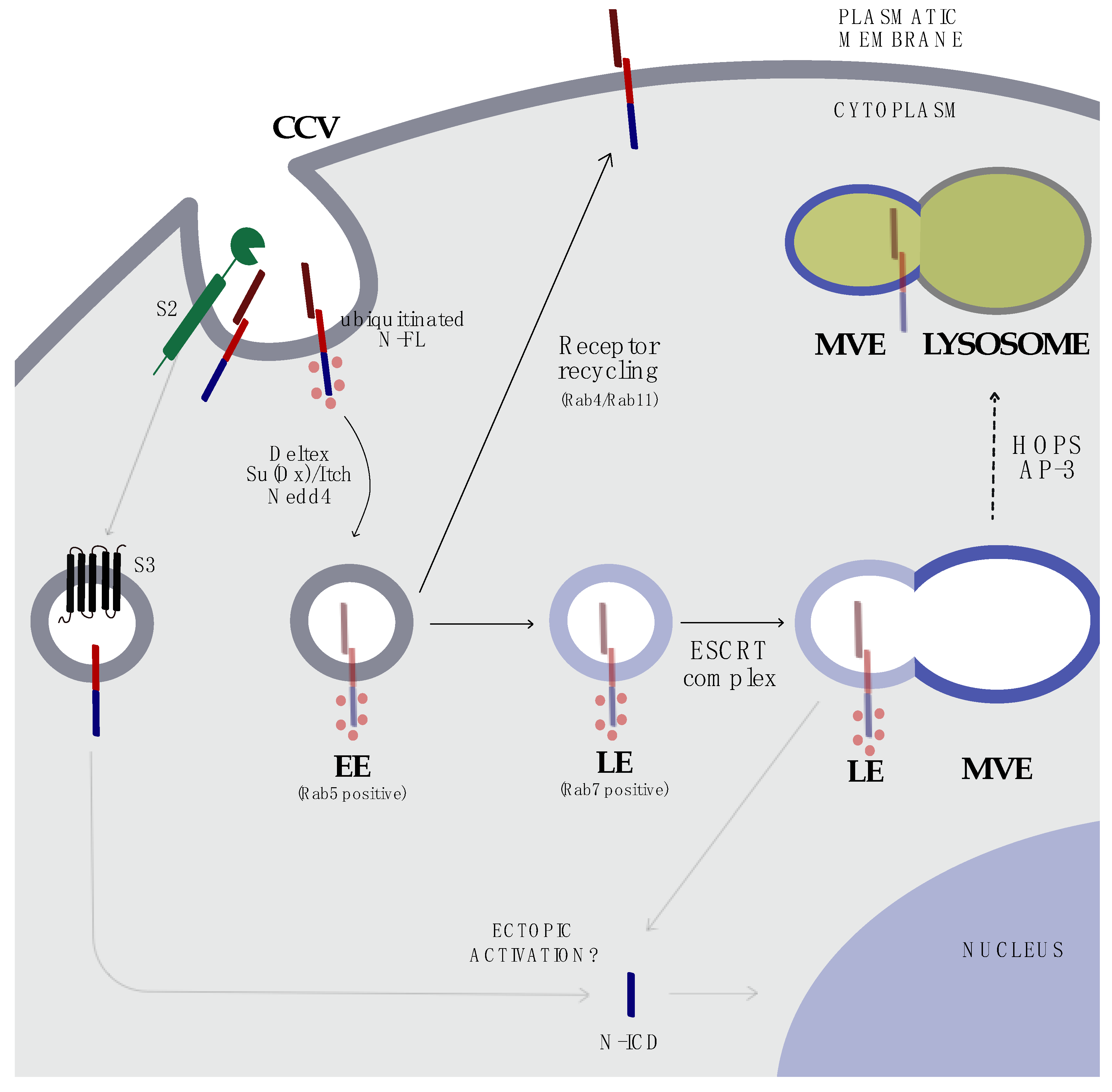
5. Endocytosis and Trafficking
6. Notch Signaling in Cancer
6.1. NOTCH as an Oncogene
6.2. Notch as a Tumor Suppressor
7. Targeting Notch Trafficking
8. Targeting the Endosomal Compartment
9. Improving the GS Complex Inhibition Strategy
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dexter, J.S. The analysis of a case of continuous variation in Drosophila by a study of its linkage relations. Am. Nat. 1914, 48, 712–758. [Google Scholar] [CrossRef]
- Morgan, T.H. The Origin of Nine Wing Mutations in Drosophila. Science 1911, 33, 496–499. [Google Scholar] [CrossRef] [PubMed]
- Morgan, T.H. Sex Limited Inheritance in Drosophila. Science 1910, 32, 120–122. [Google Scholar] [CrossRef] [Green Version]
- Morgan, T.H. The Theory of the Gene. Am. Nat. 1917, 51, 513–544. [Google Scholar] [CrossRef]
- Kidd, S.; Kelley, M.R.; Young, M.W. Sequence of the notch locus of Drosophila melanogaster: Relationship of the encoded protein to mammalian clotting and growth factors. Mol. Cell Biol. 1986, 6, 3094–3108. [Google Scholar] [CrossRef] [Green Version]
- Wharton, K.A.; Johansen, K.M.; Xu, T.; Artavanis-Tsakonas, S. Nucleotide sequence from the neurogenic locus notch implies a gene product that shares homology with proteins containing EGF-like repeats. Cell 1985, 43, 567–581. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling in context. Nat. Rev. Mol. Cell. Biol. 2016, 17, 722–735. [Google Scholar] [CrossRef]
- Gridley, T. Notch signaling and inherited disease syndromes. Hum. Mol. Genet. 2003, 12, R9–R13. [Google Scholar] [CrossRef]
- Takebe, N.; Miele, L.; Harris, P.J.; Jeong, W.; Bando, H.; Kahn, M.; Yang, S.X.; Ivy, S.P. Targeting Notch, Hedgehog, and Wnt pathways in cancer stem cells: Clinical update. Nat. Rev. Clin. Oncol. 2015, 12, 445–464. [Google Scholar] [CrossRef]
- Lai, E.C. Notch signaling: Control of cell communication and cell fate. Development 2004, 131, 965–973. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Irizarry, C.; Carpenter, A.C.; Weng, A.P.; Pear, W.S.; Aster, J.C.; Blacklow, S.C. Notch subunit heterodimerization and prevention of ligand-independent proteolytic activation depend, respectively, on a novel domain and the LNR repeats. Mol. Cell Biol. 2004, 24, 9265–9273. [Google Scholar] [CrossRef] [Green Version]
- Kopan, R.; Ilagan, M.X. The canonical Notch signaling pathway: Unfolding the activation mechanism. Cell 2009, 137, 216–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, W.R.; Roy, M.; Vardar-Ulu, D.; Garfinkel, M.; Mansour, M.R.; Aster, J.C.; Blacklow, S.C. Structure of the Notch1-negative regulatory region: Implications for normal activation and pathogenic signaling in T-ALL. Blood 2009, 113, 4381–4390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okajima, T.; Irvine, K.D. Regulation of notch signaling by o-linked fucose. Cell 2002, 111, 893–904. [Google Scholar] [CrossRef] [Green Version]
- Acar, M.; Jafar-Nejad, H.; Takeuchi, H.; Rajan, A.; Ibrani, D.; Rana, N.A.; Pan, H.; Haltiwanger, R.S.; Bellen, H.J. Rumi is a CAP10 domain glycosyltransferase that modifies Notch and is required for Notch signaling. Cell 2008, 132, 247–258. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Kantharia, J.; Sethi, M.K.; Bakker, H.; Haltiwanger, R.S. Site-specific O-glucosylation of the epidermal growth factor-like (EGF) repeats of notch: Efficiency of glycosylation is affected by proper folding and amino acid sequence of individual EGF repeats. J. Biol. Chem. 2012, 287, 33934–33944. [Google Scholar] [CrossRef] [Green Version]
- Rebay, I.; Fleming, R.J.; Fehon, R.G.; Cherbas, L.; Cherbas, P.; Artavanis-Tsakonas, S. Specific EGF repeats of Notch mediate interactions with Delta and Serrate: Implications for Notch as a multifunctional receptor. Cell 1991, 67, 687–699. [Google Scholar] [CrossRef]
- Wang, Y.; Shao, L.; Shi, S.; Harris, R.J.; Spellman, M.W.; Stanley, P.; Haltiwanger, R.S. Modification of epidermal growth factor-like repeats with O-fucose. Molecular cloning and expression of a novel GDP-fucose protein O-fucosyltransferase. J.Biol. Chem. 2001, 276, 40338–40345. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Haltiwanger, R.S. O-fucosylation of notch occurs in the endoplasmic reticulum. J. Biol. Chem. 2005, 280, 11289–11294. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, H.; Yu, H.; Hao, H.; Takeuchi, M.; Ito, A.; Li, H.; Haltiwanger, R.S. O-Glycosylation modulates the stability of epidermal growth factor-like repeats and thereby regulates Notch trafficking. J. Biol. Chem. 2017, 292, 15964–15973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sethi, M.K.; Buettner, F.F.; Ashikov, A.; Krylov, V.B.; Takeuchi, H.; Nifantiev, N.E.; Haltiwanger, R.S.; Gerardy-Schahn, R.; Bakker, H. Molecular cloning of a xylosyltransferase that transfers the second xylose to O-glucosylated epidermal growth factor repeats of notch. J. Biol. Chem. 2012, 287, 2739–2748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urata, Y.; Saiki, W.; Tsukamoto, Y.; Sago, H.; Hibi, H.; Okajima, T.; Takeuchi, H. Xylosyl Extension of O-Glucose Glycans on the Extracellular Domain of NOTCH1 and NOTCH2 Regulates Notch Cell Surface Trafficking. Cells 2020, 9, 1220. [Google Scholar] [CrossRef] [PubMed]
- Cui, Q.; Xing, J.; Gu, Y.; Nan, X.; Ma, W.; Chen, Y.; Zhao, H. GXYLT2 accelerates cell growth and migration by regulating the Notch pathway in human cancer cells. Exp. Cell Res. 2019, 376, 1–10. [Google Scholar] [CrossRef]
- Hicks, C.; Johnston, S.H.; diSibio, G.; Collazo, A.; Vogt, T.F.; Weinmaster, G. Fringe differentially modulates Jagged1 and Delta1 signalling through Notch1 and Notch2. Nat. Cell Biol. 2000, 2, 515–520. [Google Scholar] [CrossRef]
- Panin, V.M.; Papayannopoulos, V.; Wilson, R.; Irvine, K.D. Fringe modulates Notch-ligand interactions. Nature 1997, 387, 908–912. [Google Scholar] [CrossRef]
- Haines, N.; Irvine, K.D. Glycosylation regulates Notch signalling. Nat. Rev. Mol. Cell Biol. 2003, 4, 786–797. [Google Scholar] [CrossRef]
- Takeuchi, H.; Schneider, M.; Williamson, D.B.; Ito, A.; Takeuchi, M.; Handford, P.A.; Haltiwanger, R.S. Two novel protein O-glucosyltransferases that modify sites distinct from POGLUT1 and affect Notch trafficking and signaling. Proc. Natl. Acad. Sci. USA 2018, 115, E8395–E8402. [Google Scholar] [CrossRef] [Green Version]
- Blaumueller, C.M.; Qi, H.; Zagouras, P.; Artavanis-Tsakonas, S. Intracellular cleavage of Notch leads to a heterodimeric receptor on the plasma membrane. Cell 1997, 90, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Rand, M.D.; Grimm, L.M.; Artavanis-Tsakonas, S.; Patriub, V.; Blacklow, S.C.; Sklar, J.; Aster, J.C. Calcium depletion dissociates and activates heterodimeric notch receptors. Mol. Cell Biol. 2000, 20, 1825–1835. [Google Scholar] [CrossRef] [Green Version]
- Logeat, F.; Bessia, C.; Brou, C.; LeBail, O.; Jarriault, S.; Seidah, N.G.; Israel, A. The Notch1 receptor is cleaved constitutively by a furin-like convertase. Proc. Natl. Acad. Sci. USA 1998, 95, 8108–8112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.M. Nodal signaling: Developmental roles and regulation. Development 2007, 134, 1023–1034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schier, A.F.; Shen, M.M. Nodal signalling in vertebrate development. Nature 2000, 403, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, K.; Nagaoka, T.; Lee, J.M.; Bianco, C.; Gonzales, M.; Castro, N.P.; Rangel, M.C.; Sakamoto, K.; Sun, Y.; Callahan, R.; et al. Enhancement of Notch receptor maturation and signaling sensitivity by Cripto-1. J. Cell Biol. 2009, 187, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Ntziachristos, P.; Lim, J.S.; Sage, J.; Aifantis, I. From fly wings to targeted cancer therapies: A centennial for notch signaling. Cancer Cell 2014, 25, 318–334. [Google Scholar] [CrossRef] [Green Version]
- Brou, C.; Logeat, F.; Gupta, N.; Bessia, C.; LeBail, O.; Doedens, J.R.; Cumano, A.; Roux, P.; Black, R.A.; Israël, A. A novel proteolytic cleavage involved in Notch signaling: The role of the disintegrin-metalloprotease TACE. Mol. Cell 2000, 5, 207–216. [Google Scholar] [CrossRef]
- Mumm, J.S.; Schroeter, E.H.; Saxena, M.T.; Griesemer, A.; Tian, X.; Pan, D.J.; Ray, W.J.; Kopan, R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol. Cell 2000, 5, 197–206. [Google Scholar] [CrossRef]
- Parks, A.L.; Klueg, K.M.; Stout, J.R.; Muskavitch, M.A. Ligand endocytosis drives receptor dissociation and activation in the Notch pathway. Development 2000, 127, 1373–1385. [Google Scholar]
- van Tetering, G.; van Diest, P.; Verlaan, I.; van der Wall, E.; Kopan, R.; Vooijs, M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J.Biol. Chem. 2009, 284, 31018–31027. [Google Scholar] [CrossRef] [Green Version]
- Bozkulak, E.C.; Weinmaster, G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol. Cell Biol. 2009, 29, 5679–5695. [Google Scholar] [CrossRef] [Green Version]
- Schroeter, E.H.; Kisslinger, J.A.; Kopan, R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 1998, 393, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Struhl, G.; Greenwald, I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 1999, 398, 522–525. [Google Scholar] [CrossRef] [PubMed]
- De Strooper, B. Aph-1, Pen-2, and Nicastrin with Presenilin generate an active gamma-Secretase complex. Neuron 2003, 38, 9–12. [Google Scholar] [CrossRef] [Green Version]
- Haapasalo, A.; Kovacs, D.M. The many substrates of presenilin/γ-secretase. J. Alzheimers Dis 2011, 25, 3–28. [Google Scholar] [CrossRef] [PubMed]
- Kimberly, W.T.; LaVoie, M.J.; Ostaszewski, B.L.; Ye, W.; Wolfe, M.S.; Selkoe, D.J. Gamma-secretase is a membrane protein complex comprised of presenilin, nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. USA 2003, 100, 6382–6387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Aster, J.C.; Blacklow, S.C.; Lake, R.; Artavanis-Tsakonas, S.; Griffin, J.D. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 2000, 26, 484–489. [Google Scholar] [CrossRef]
- Jarriault, S.; Brou, C.; Logeat, F.; Schroeter, E.H.; Kopan, R.; Israel, A. Signalling downstream of activated mammalian Notch. Nature 1995, 377, 355–358. [Google Scholar] [CrossRef]
- Gerhardt, D.M.; Pajcini, K.V.; D’Altri, T.; Tu, L.; Jain, R.; Xu, L.; Chen, M.J.; Rentschler, S.; Shestova, O.; Wertheim, G.B.; et al. The Notch1 transcriptional activation domain is required for development and reveals a novel role for Notch1 signaling in fetal hematopoietic stem cells. Genes Dev. 2014, 28, 576–593. [Google Scholar] [CrossRef] [Green Version]
- Seugnet, L.; Simpson, P.; Haenlin, M. Requirement for dynamin during Notch signaling in Drosophila neurogenesis. Dev. Biol. 1997, 192, 585–598. [Google Scholar] [CrossRef] [Green Version]
- Kandachar, V.; Roegiers, F. Endocytosis and control of Notch signaling. Curr. Opin. Cell Biol. 2012, 24, 534–540. [Google Scholar] [CrossRef] [Green Version]
- Schnute, B.; Troost, T.; Klein, T. Endocytic Trafficking of the Notch Receptor. Adv. Exp. Med. Biol. 2018, 1066, 99–122. [Google Scholar] [CrossRef] [PubMed]
- Gupta-Rossi, N.; Six, E.; LeBail, O.; Logeat, F.; Chastagner, P.; Olry, A.; Israël, A.; Brou, C. Monoubiquitination and endocytosis direct gamma-secretase cleavage of activated Notch receptor. J. Cell Biol. 2004, 166, 73–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorensen, E.B.; Conner, S.D. gamma-secretase-dependent cleavage initiates notch signaling from the plasma membrane. Traffic 2010, 11, 1234–1245. [Google Scholar] [CrossRef] [Green Version]
- Windler, S.L.; Bilder, D. Endocytic internalization routes required for delta/notch signaling. Curr Biol. 2010, 20, 538–543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerial, M.; McBride, H. Rab proteins as membrane organizers. Nat. Rev. Mol. Cell Biol. 2001, 2, 107–117. [Google Scholar] [CrossRef]
- Chastagner, P.; Israël, A.; Brou, C. AIP4/Itch regulates Notch receptor degradation in the absence of ligand. PLoS ONE 2008, 3, e2735. [Google Scholar] [CrossRef] [Green Version]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Huotari, J.; Helenius, A. Endosome maturation. EMBO J. 2011, 30, 3481–3500. [Google Scholar] [CrossRef]
- Yeh, C.-H.; Bellon, M.; Nicot, C. FBXW7: A critical tumor suppressor of human cancers. Mol. Cancer 2018, 17, 115. [Google Scholar] [CrossRef]
- Pasternak, S.H.; Bagshaw, R.D.; Guiral, M.; Zhang, S.; Ackerley, C.A.; Pak, B.J.; Callahan, J.W.; Mahuran, D.J. Presenilin-1, nicastrin, amyloid precursor protein, and γ-secretase activity are co-localized in the lysosomal membrane. J. Biol. Chem. 2003, 278, 26687–26694. [Google Scholar] [CrossRef] [Green Version]
- Sakata, T.; Sakaguchi, H.; Tsuda, L.; Higashitani, A.; Aigaki, T.; Matsuno, K.; Hayashi, S. Drosophila Nedd4 regulates endocytosis of notch and suppresses its ligand-independent activation. Curr. Biol. 2004, 14, 2228–2236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busseau, I.; Diederich, R.J.; Xu, T.; Artavanis-Tsakonas, S. A member of the Notch group of interacting loci, deltex encodes a cytoplasmic basic protein. Genetics 1994, 136, 585–596. [Google Scholar] [PubMed]
- Wilkin, M.; Tongngok, P.; Gensch, N.; Clemence, S.; Motoki, M.; Yamada, K.; Hori, K.; Taniguchi-Kanai, M.; Franklin, E.; Matsuno, K. Drosophila HOPS and AP-3 complex genes are required for a Deltex-regulated activation of notch in the endosomal trafficking pathway. Dev. Cell 2008, 15, 762–772. [Google Scholar] [CrossRef]
- Dell’Angelica, E.C. AP-3-dependent trafficking and disease: The first decade. Curr. Opin. Cell Biol. 2009, 21, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Fortini, M.E. Notch signaling: The core pathway and its posttranslational regulation. Dev. Cell 2009, 16, 633–647. [Google Scholar] [CrossRef] [Green Version]
- Fortini, M.E.; Bilder, D. Endocytic regulation of Notch signaling. Curr. Opin. Genet. Dev. 2009, 19, 323–328. [Google Scholar] [CrossRef] [Green Version]
- Yamamoto, S.; Charng, W.-L.; Bellen, H.J. Endocytosis and intracellular trafficking of Notch and its ligands. In Current Topics in Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2010; pp. 165–200. [Google Scholar]
- Troost, T.; Jaeckel, S.; Ohlenhard, N.; Klein, T. The tumour suppressor Lethal (2) giant discs is required for the function of the ESCRT-III component Shrub/CHMP4. J. Cell Sci. 2012, 125, 763–776. [Google Scholar] [CrossRef] [Green Version]
- Yan, Y.; Denef, N.; Schüpbach, T. The vacuolar proton pump, V-ATPase, is required for notch signaling and endosomal trafficking in Drosophila. Dev. Cell 2009, 17, 387–402. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.M.; Petrucelli, L.; Fisher, G.; Ramdath, S.; Castillo, J.; Di Fiore, M.M.; D’Aniello, A. Evidence for D-aspartyl-β-amyloid secretase activity in human brain. J. Neuropathol. Exp. Neurol. 2002, 61, 125–131. [Google Scholar] [CrossRef]
- Zhang, L.; Song, L.; Terracina, G.; Liu, Y.; Pramanik, B.; Parker, E. Biochemical characterization of the γ-secretase activity that produces β-amyloid peptides. Biochemistry 2001, 40, 5049–5055. [Google Scholar] [CrossRef]
- Tagami, S.; Okochi, M.; Yanagida, K.; Ikuta, A.; Fukumori, A.; Matsumoto, N.; Ishizuka-Katsura, Y.; Nakayama, T.; Itoh, N.; Jiang, J. Regulation of Notch signaling by dynamic changes in the precision of S3 cleavage of Notch-1. Mol. Cellular Biol. 2008, 28, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artavanis-Tsakonas, S. The molecular biology of the Notch locus and the fine tuning of differentiation in Drosophila. Trends Genet. 1988, 4, 95–100. [Google Scholar] [CrossRef]
- Pajcini, K.V.; Speck, N.A.; Pear, W.S. Notch signaling in mammalian hematopoietic stem cells. Leukemia 2011, 25, 1525–1532. [Google Scholar] [CrossRef] [Green Version]
- Gering, M.; Patient, R. Notch signalling and haematopoietic stem cell formation during embryogenesis. J. Cell. Physiol. 2010, 222, 11–16. [Google Scholar] [CrossRef]
- Bugeon, L.; Taylor, H.B.; Progatzky, F.; Lin, M.I.; Ellis, C.D.; Welsh, N.; Smith, E.; Vargesson, N.; Gray, C.; Renshaw, S.A. The NOTCH pathway contributes to cell fate decision in myelopoiesis. Haematologica 2011, 96, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The varied roles of Notch in cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef] [Green Version]
- Khosla, R.; Vyas, A.K.; Trehanpati, N. Dichotomy of Notch signalling in regulating tumour immune surveillance. Scand. J. Immunol. 2019, 89, e12744. [Google Scholar]
- Shang, Y.; Smith, S.; Hu, X. Role of Notch signaling in regulating innate immunity and inflammation in health and disease. Protein Cell 2016, 7, 159–174. [Google Scholar] [CrossRef] [Green Version]
- Meurette, O.; Mehlen, P. Notch signaling in the tumor microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Delgado-Calle, J.; Anderson, J.; Cregor, M.D.; Hiasa, M.; Chirgwin, J.M.; Carlesso, N.; Yoneda, T.; Mohammad, K.S.; Plotkin, L.I.; Roodman, G.D. Bidirectional Notch signaling and osteocyte-derived factors in the bone marrow microenvironment promote tumor cell proliferation and bone destruction in multiple myeloma. Cancer Res. 2016, 76, 1089–1100. [Google Scholar] [CrossRef] [Green Version]
- Demehri, S.; Turkoz, A.; Kopan, R. Epidermal Notch1 loss promotes skin tumorigenesis by impacting the stromal microenvironment. Cancer Cell 2009, 16, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Stoeck, A.; Lejnine, S.; Truong, A.; Pan, L.; Wang, H.; Zang, C.; Yuan, J.; Ware, C.; MacLean, J.; Garrett-Engele, P.W. Discovery of biomarkers predictive of GSI response in triple-negative breast cancer and adenoid cystic carcinoma. Cancer Discov. 2014, 4, 1154–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldoni, S.; Del Papa, B.; Dorillo, E.; Aureli, P.; De Falco, F.; Rompietti, C.; Sorcini, D.; Varasano, E.; Cecchini, D.; Zei, T.; et al. Bepridil exhibits anti-leukemic activity associated with NOTCH1 pathway inhibition in chronic lymphocytic leukemia. Int. J. Cancer 2018, 143, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Aifantis, I.; Raetz, E.; Buonamici, S. Molecular pathogenesis of T-cell leukaemia and lymphoma. Nat. Rev. Immunol. 2008, 8, 380–390. [Google Scholar] [CrossRef] [PubMed]
- Ellisen, L.W.; Bird, J.; West, D.C.; Soreng, A.L.; Reynolds, T.C.; Smith, S.D.; Sklar, J. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 1991, 66, 649–661. [Google Scholar] [CrossRef]
- Capobianco, A.J.; Zagouras, P.; Blaumueller, C.M.; Artavanis-Tsakonas, S.; Bishop, J.M. Neoplastic transformation by truncated alleles of human NOTCH1/TAN1 and NOTCH2. Mol. Cell. Biol. 1997, 17, 6265–6273. [Google Scholar] [CrossRef] [Green Version]
- Pear, W.S.; Aster, J.C.; Scott, M.L.; Hasserjian, R.P.; Soffer, B.; Sklar, J.; Baltimore, D. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J. Exp. Med. 1996, 183, 2283–2291. [Google Scholar] [CrossRef]
- Weng, A.P.; Ferrando, A.A.; Lee, W.; Morris, J.P.; Silverman, L.B.; Sanchez-Irizarry, C.; Blacklow, S.C.; Look, A.T.; Aster, J.C. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 2004, 306, 269–271. [Google Scholar] [CrossRef] [Green Version]
- Malecki, M.J.; Sanchez-Irizarry, C.; Mitchell, J.L.; Histen, G.; Xu, M.L.; Aster, J.C.; Blacklow, S.C. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol. Cell. Biol. 2006, 26, 4642–4651. [Google Scholar] [CrossRef] [Green Version]
- Sulis, M.L.; Williams, O.; Palomero, T.; Tosello, V.; Pallikuppam, S.; Real, P.J.; Barnes, K.; Zuurbier, L.; Meijerink, J.P.; Ferrando, A.A. NOTCH1 extracellular juxtamembrane expansion mutations in T-ALL. Blood J. Am. Soc. Hematol. 2008, 112, 733–740. [Google Scholar] [CrossRef] [Green Version]
- Gupta-Rossi, N.; Le Bail, O.; Gonen, H.; Brou, C.; Logeat, F.; Six, E.; Ciechanover, A.; Israël, A. Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J. Biol. Chem. 2001, 276, 34371–34378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, B.J.; Buonamici, S.; Sulis, M.L.; Palomero, T.; Vilimas, T.; Basso, G.; Ferrando, A.; Aifantis, I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J. Exp. Med. 2007, 204, 1825–1835. [Google Scholar] [CrossRef] [PubMed]
- O’Neil, J.; Grim, J.; Strack, P.; Rao, S.; Tibbitts, D.; Winter, C.; Hardwick, J.; Welcker, M.; Meijerink, J.P.; Pieters, R.; et al. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J. Exp. Med. 2007, 204, 1813–1824. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Masiero, M.; Banham, A.H. Notch signaling: Its roles and therapeutic potential in hematological malignancies. Oncotarget 2016, 7, 29804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Breit, S.; Stanulla, M.; Flohr, T.; Schrappe, M.; Ludwig, W.-D.; Tolle, G.; Happich, M.; Muckenthaler, M.U.; Kulozik, A.E. Activating NOTCH1 mutations predict favorable early treatment response and long-term outcome in childhood precursor T-cell lymphoblastic leukemia. Blood 2006, 108, 1151–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.J.; Taki, T.; Oda, M.; Watanabe, T.; Yumura-Yagi, K.; Kobayashi, R.; Suzuki, N.; Hara, J.; Horibe, K.; Hayashi, Y. FBXW7 and NOTCH1 mutations in childhood T cell acute lymphoblastic leukaemia and T cell non-Hodgkin lymphoma. Br. J. Haematol. 2009, 145, 198–206. [Google Scholar] [CrossRef]
- Mansour, M.R.; Sulis, M.L.; Duke, V.; Foroni, L.; Jenkinson, S.; Koo, K.; Allen, C.G.; Gale, R.E.; Buck, G.; Richards, S. Prognostic implications of NOTCH1 and FBXW7 mutations in adults with T-cell acute lymphoblastic leukemia treated on the MRC UKALLXII/ECOG E2993 protocol. J. Clin. Oncol. 2009, 27, 4352. [Google Scholar] [CrossRef] [Green Version]
- Ben Abdelali, R.; Asnafi, V.; Leguay, T.; Boissel, N.; Buzyn, A.; Chevallier, P.; Thomas, X.; Lepretre, S.; Huguet, F.; Vey, N. Pediatric-inspired intensified therapy of adult T-ALL reveals the favorable outcome of NOTCH1/FBXW7 mutations, but not of low ERG/BAALC expression: A GRAALL study. Blood J. Am. Soc. Hematol. 2011, 118, 5099–5107. [Google Scholar] [CrossRef] [Green Version]
- Pui, J.C.; Allman, D.; Xu, L.; DeRocco, S.; Karnell, F.G.; Bakkour, S.; Lee, J.Y.; Kadesch, T.; Hardy, R.R.; Aster, J.C. Notch1 expression in early lymphopoiesis influences B versus T lineage determination. Immunity 1999, 11, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Saito, T.; Chiba, S.; Ichikawa, M.; Kunisato, A.; Asai, T.; Shimizu, K.; Yamaguchi, T.; Yamamoto, G.; Seo, S.; Kumano, K. Notch2 is preferentially expressed in mature B cells and indispensable for marginal zone B lineage development. Immunity 2003, 18, 675–685. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Trifonov, V.; Fangazio, M.; Bruscaggin, A.; Rasi, S.; Spina, V.; Monti, S.; Vaisitti, T.; Arruga, F.; Famà, R.; et al. The coding genome of splenic marginal zone lymphoma: Activation of NOTCH2 and other pathways regulating marginal zone development. J. Exp. Med. 2012, 209, 1537–1551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiel, M.J.; Velusamy, T.; Betz, B.L.; Zhao, L.; Weigelin, H.G.; Chiang, M.Y.; Huebner-Chan, D.R.; Bailey, N.G.; Yang, D.T.; Bhagat, G.; et al. Whole-genome sequencing identifies recurrent somatic NOTCH2 mutations in splenic marginal zone lymphoma. J. Exp. Med. 2012, 209, 1553–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nadeu, F.; Clot, G.; Delgado, J.; Martin-Garcia, D.; Baumann, T.; Salaverria, I.; Beà, S.; Pinyol, M.; Jares, P.; Navarro, A. Clinical impact of the subclonal architecture and mutational complexity in chronic lymphocytic leukemia. Leukemia 2018, 32, 645–653. [Google Scholar] [CrossRef] [PubMed]
- Sportoletti, P.; Baldoni, S.; Cavalli, L.; Del Papa, B.; Bonifacio, E.; Ciurnelli, R.; Bell, A.S.; Di Tommaso, A.; Rosati, E.; Crescenzi, B.; et al. NOTCH1 PEST domain mutation is an adverse prognostic factor in B-CLL. Br. J. Haematol 2010, 151, 404–406. [Google Scholar] [CrossRef] [PubMed]
- Kridel, R.; Meissner, B.; Rogic, S.; Boyle, M.; Telenius, A.; Woolcock, B.; Gunawardana, J.; Jenkins, C.; Cochrane, C.; Ben-Neriah, S.; et al. Whole transcriptome sequencing reveals recurrent NOTCH1 mutations in mantle cell lymphoma. Blood 2012, 119, 1963–1971. [Google Scholar] [CrossRef]
- Fabbri, G.; Rasi, S.; Rossi, D.; Trifonov, V.; Khiabanian, H.; Ma, J.; Grunn, A.; Fangazio, M.; Capello, D.; Monti, S. Analysis of the chronic lymphocytic leukemia coding genome: Role of NOTCH1 mutational activation. J. Exp. Med. 2011, 208, 1389–1401. [Google Scholar] [CrossRef] [Green Version]
- Puente, X.S.; Pinyol, M.; Quesada, V.; Conde, L.; Ordóñez, G.R.; Villamor, N.; Escaramis, G.; Jares, P.; Beà, S.; González-Díaz, M. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature 2011, 475, 101–105. [Google Scholar] [CrossRef] [Green Version]
- South, A.P.; Cho, R.J.; Aster, J.C. The double-edged sword of notch signaling in cancer. In Seminars in Cell and Developmental Biology; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Rossi, D.; Rasi, S.; Fabbri, G.; Spina, V.; Fangazio, M.; Forconi, F.; Marasca, R.; Laurenti, L.; Bruscaggin, A.; Cerri, M. Mutations of NOTCH1 are an independent predictor of survival in chronic lymphocytic leukemia. Blood J. Am. Soc. Hematol. 2012, 119, 521–529. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.y.; Kumano, K.; Nakazaki, K.; Sanada, M.; Matsumoto, A.; Yamamoto, G.; Nannya, Y.; Suzuki, R.; Ota, S.; Ota, Y. Gain-of-function mutations and copy number increases of Notch2 in diffuse large B-cell lymphoma. Cancer Sci. 2009, 100, 920–926. [Google Scholar] [CrossRef] [Green Version]
- Schmitz, R.; Wright, G.W.; Huang, D.W.; Johnson, C.A.; Phelan, J.D.; Wang, J.Q.; Roulland, S.; Kasbekar, M.; Young, R.M.; Shaffer, A.L. Genetics and pathogenesis of diffuse large B-cell lymphoma. N. Engl. J. Med. 2018, 378, 1396–1407. [Google Scholar] [CrossRef]
- Boissard, F.; Tosolini, M.; Ligat, L.; Quillet-Mary, A.; Lopez, F.; Fournié, J.-J.; Ysebaert, L.; Poupot, M. Nurse-like cells promote CLL survival through LFA-3/CD2 interactions. Oncotarget 2016, 8, 52225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, X.S.; Jares, P.; Campo, E. Chronic lymphocytic leukemia and mantle cell lymphoma: Crossroads of genetic and microenvironment interactions. Blood 2018, 131, 2283–2296. [Google Scholar] [CrossRef] [PubMed]
- van Attekum, M.H.; Eldering, E.; Kater, A.P. Chronic lymphocytic leukemia cells are active participants in microenvironmental cross-talk. Haematologica 2017, 102, 1469–1476. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Yang, J.; Qian, J.; Li, H.; Romaguera, J.E.; Kwak, L.W.; Wang, M.; Yi, Q. Role of the microenvironment in mantle cell lymphoma: IL-6 is an important survival factor for the tumor cells. Blood 2012, 120, 3783–3792. [Google Scholar] [CrossRef] [Green Version]
- Silkenstedt, E.; Arenas, F.; Colom-Sanmartí, B.; Xargay-Torrent, S.; Higashi, M.; Giró, A.; Rodriguez, V.; Fuentes, P.; Aulitzky, W.E.; van der Kuip, H.; et al. Notch1 signaling in NOTCH1-mutated mantle cell lymphoma depends on Delta-Like ligand 4 and is a potential target for specific antibody therapy. J. Exp. Clin. Cancer Res. 2019, 38, 446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Indraccolo, S.; Minuzzo, S.; Masiero, M.; Pusceddu, I.; Persano, L.; Moserle, L.; Reboldi, A.; Favaro, E.; Mecarozzi, M.; Di Mario, G.; et al. Cross-talk between tumor and endothelial cells involving the Notch3-Dll4 interaction marks escape from tumor dormancy. Cancer Res. 2009, 69, 1314–1323. [Google Scholar] [CrossRef] [Green Version]
- Westhoff, B.; Colaluca, I.N.; D’Ario, G.; Donzelli, M.; Tosoni, D.; Volorio, S.; Pelosi, G.; Spaggiari, L.; Mazzarol, G.; Viale, G. Alterations of the Notch pathway in lung cancer. Proc. Natl. Acad. Sci. USA 2009, 106, 22293–22298. [Google Scholar] [CrossRef] [Green Version]
- Northcott, P.A.; Buchhalter, I.; Morrissy, A.S.; Hovestadt, V.; Weischenfeldt, J.; Ehrenberger, T.; Gröbner, S.; Segura-Wang, M.; Zichner, T.; Rudneva, V.A. The whole-genome landscape of medulloblastoma subtypes. Nature 2017, 547, 311–317. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Messer, A.R.; Amitai-Lange, A.; Melamed, Z.e.; Ohana, R.; Bell, R.E.; Kapitansky, O.; Lerman, G.; Greenberger, S.; Khaled, M. Interactions of melanoma cells with distal keratinocytes trigger metastasis via notch signaling inhibition of MITF. Mol. Cell 2015, 59, 664–676. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, B.; Ji, Z.Z.; Zheng, P.S. Notch1 regulates the growth of human colon cancers. Cancer 2010, 116, 5207–5218. [Google Scholar] [CrossRef]
- Reedijk, M.; Odorcic, S.; Zhang, H.; Chetty, R.; Tennert, C.; Dickson, B.C.; Lockwood, G.; Gallinger, S.; Egan, S.E. Activation of Notch signaling in human colon adenocarcinoma. Int. J. Oncol. 2008, 33, 1223–1229. [Google Scholar] [PubMed] [Green Version]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.R.; Kalyana-Sundaram, S.; Wu, Y.-M.; Shankar, S.; Cao, X.; Ateeq, B.; Asangani, I.A.; Iyer, M.; Maher, C.A.; Grasso, C.S. Functionally recurrent rearrangements of the MAST kinase and Notch gene families in breast cancer. Nat. Med. 2011, 17, 1646. [Google Scholar] [CrossRef] [Green Version]
- Gallahan, D.; Callahan, R. The mouse mammary tumor associated gene INT3 is a unique member of the NOTCH gene family (NOTCH4). Oncogene 1997, 14, 1883–1890. [Google Scholar] [CrossRef] [Green Version]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of breast cancer stem cell activity by signaling through the Notch4 receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reedijk, M.; Odorcic, S.; Chang, L.; Zhang, H.; Miller, N.; McCready, D.R.; Lockwood, G.; Egan, S.E. High-level coexpression of JAG1 and NOTCH1 is observed in human breast cancer and is associated with poor overall survival. Cancer Res. 2005, 65, 8530–8537. [Google Scholar] [CrossRef] [Green Version]
- Ayyanan, A.; Civenni, G.; Ciarloni, L.; Morel, C.; Mueller, N.; Lefort, K.; Mandinova, A.; Raffoul, W.; Fiche, M.; Dotto, G.P. Increased Wnt signaling triggers oncogenic conversion of human breast epithelial cells by a Notch-dependent mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 3799–3804. [Google Scholar] [CrossRef] [Green Version]
- Weijzen, S.; Rizzo, P.; Braid, M.; Vaishnav, R.; Jonkheer, S.M.; Zlobin, A.; Osborne, B.A.; Gottipati, S.; Aster, J.C.; Hahn, W.C. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat. Med. 2002, 8, 979–986. [Google Scholar] [CrossRef]
- Fitzgerald, K.; Harrington, A.; Leder, P. Ras pathway signals are required for notch-mediated oncogenesis. Oncogene 2000, 19, 4191–4198. [Google Scholar] [CrossRef] [Green Version]
- Pickering, C.R.; Zhou, J.H.; Lee, J.J.; Drummond, J.A.; Peng, S.A.; Saade, R.E.; Tsai, K.Y.; Curry, J.L.; Tetzlaff, M.T.; Lai, S.Y.; et al. Mutational landscape of aggressive cutaneous squamous cell carcinoma. Clin. Cancer Res. 2014, 20, 6582–6592. [Google Scholar] [CrossRef] [Green Version]
- South, A.P.; Purdie, K.J.; Watt, S.A.; Haldenby, S.; Den Breems, N.Y.; Dimon, M.; Arron, S.T.; Kluk, M.J.; Aster, J.C.; McHugh, A. NOTCH1 mutations occur early during cutaneous squamous cell carcinogenesis. J. Investig. Dermatol. 2014, 134, 2630–2638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.-X.; Zhang, J.; Wang, J. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Li, L.; Ou, Y.; Gao, Z.; Li, E.; Li, X.; Zhang, W.; Wang, J.; Xu, L.; Zhou, Y. Identification of genomic alterations in oesophageal squamous cell cancer. Nature 2014, 509, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Rampias, T.; Vgenopoulou, P.; Avgeris, M.; Polyzos, A.; Stravodimos, K.; Valavanis, C.; Scorilas, A.; Klinakis, A. A new tumor suppressor role for the Notch pathway in bladder cancer. Nat. Med. 2014, 20, 1199–1205. [Google Scholar] [CrossRef]
- Nowell, C.S.; Radtke, F. Notch as a tumour suppressor. Nat. Rev. Cancer 2017, 17, 145. [Google Scholar] [CrossRef]
- Wang, N.J.; Sanborn, Z.; Arnett, K.L.; Bayston, L.J.; Liao, W.; Proby, C.M.; Leigh, I.M.; Collisson, E.A.; Gordon, P.B.; Jakkula, L.; et al. Loss-of-function mutations in Notch receptors in cutaneous and lung squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2011, 108, 17761–17766. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, M.; Wolfer, A.; Raj, K.; Kummer, J.A.; Mill, P.; van Noort, M.; Hui, C.-C.; Clevers, H.; Dotto, G.P.; Radtke, F. Notch1 functions as a tumor suppressor in mouse skin. Nat. Genet. 2003, 33, 416. [Google Scholar] [CrossRef]
- Nguyen, B.-C.; Lefort, K.; Mandinova, A.; Antonini, D.; Devgan, V.; Della Gatta, G.; Koster, M.I.; Zhang, Z.; Wang, J.; Di Vignano, A.T. Cross-regulation between Notch and p63 in keratinocyte commitment to differentiation. Genes Dev. 2006, 20, 1028–1042. [Google Scholar] [CrossRef] [Green Version]
- Rangarajan, A.; Talora, C.; Okuyama, R.; Nicolas, M.; Mammucari, C.; Oh, H.; Aster, J.C.; Krishna, S.; Metzger, D.; Chambon, P.; et al. Notch signaling is a direct determinant of keratinocyte growth arrest and entry into differentiation. EMBO J. 2001, 20, 3427–3436. [Google Scholar] [CrossRef] [Green Version]
- Extance, A. Alzheimer’s Failure Raises Questions about Disease-Modifying Strategies; Nature Publishing Group: Berlin, Germany, 2010. [Google Scholar]
- Doody, R.S.; Raman, R.; Sperling, R.A.; Seimers, E.; Sethuraman, G.; Mohs, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Sun, X. Peripheral and central effects of γ-secretase inhibition by semagacestat in Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henley, D.B.; Sundell, K.L.; Sethuraman, G.; Dowsett, S.A.; May, P.C. Safety profile of semagacestat, a gamma-secretase inhibitor: IDENTITY trial findings. Curr. Med. Res. Opin. 2014, 30, 2021–2032. [Google Scholar] [CrossRef] [PubMed]
- Restivo, G.; Nguyen, B.C.; Dziunycz, P.; Ristorcelli, E.; Ryan, R.J.; Özuysal, Ö.Y.; Di Piazza, M.; Radtke, F.; Dixon, M.J.; Hofbauer, G.F. IRF6 is a mediator of Notch pro-differentiation and tumour suppressive function in keratinocytes. EMBO J. 2011, 30, 4571–4585. [Google Scholar] [CrossRef] [PubMed]
- Nowell, C.S.; Odermatt, P.D.; Azzolin, L.; Hohnel, S.; Wagner, E.F.; Fantner, G.E.; Lutolf, M.P.; Barrandon, Y.; Piccolo, S.; Radtke, F. Chronic inflammation imposes aberrant cell fate in regenerating epithelia through mechanotransduction. Nat. Cell Biol. 2016, 18, 168–180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demehri, S.; Liu, Z.; Lee, J.; Lin, M.-H.; Crosby, S.D.; Roberts, C.J.; Grigsby, P.W.; Miner, J.H.; Farr, A.G.; Kopan, R. Notch-deficient skin induces a lethal systemic B-lymphoproliferative disorder by secreting TSLP, a sentinel for epidermal integrity. PLoS Biol. 2008, 6, e123. [Google Scholar] [CrossRef]
- Di Piazza, M.; Nowell, C.S.; Koch, U.; Durham, A.-D.; Radtke, F. Loss of cutaneous TSLP-dependent immune responses skews the balance of inflammation from tumor protective to tumor promoting. Cancer Cell 2012, 22, 479–493. [Google Scholar] [CrossRef] [Green Version]
- Viatour, P.; Ehmer, U.; Saddic, L.A.; Dorrell, C.; Andersen, J.B.; Lin, C.; Zmoos, A.-F.; Mazur, P.K.; Schaffer, B.E.; Ostermeier, A. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J. Exp. Med. 2011, 208, 1963–1976. [Google Scholar] [CrossRef] [Green Version]
- Giachino, C.; Boulay, J.-L.; Ivanek, R.; Alvarado, A.; Tostado, C.; Lugert, S.; Tchorz, J.; Coban, M.; Mariani, L.; Bettler, B. A tumor suppressor function for notch signaling in forebrain tumor subtypes. Cancer Cell 2015, 28, 730–742. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-S.; Chu, I.-S.; Mikaelyan, A.; Calvisi, D.F.; Heo, J.; Reddy, J.K.; Thorgeirsson, S.S. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat. Genet. 2004, 36, 1306–1311. [Google Scholar] [CrossRef]
- Kawaguchi, K.; Honda, M.; Yamashita, T.; Okada, H.; Shirasaki, T.; Nishikawa, M.; Nio, K.; Arai, K.; Sakai, Y.; Yamashita, T. Jagged1 DNA copy number variation is associated with poor outcome in liver cancer. Am. J. Pathol. 2016, 186, 2055–2067. [Google Scholar] [CrossRef] [Green Version]
- Ma, L.; Dong, P.; Liu, L.; Gao, Q.; Duan, M.; Zhang, S.; Chen, S.; Xue, R.; Wang, X. Overexpression of protein O-fucosyltransferase 1 accelerates hepatocellular carcinoma progression via the Notch signaling pathway. Biochem. Biophys. Res. Commun. 2016, 473, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Koch, U.; Lehal, R.; Radtke, F. Stem cells living with a Notch. Development 2013, 140, 689–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giachino, C.; Basak, O.; Lugert, S.; Knuckles, P.; Obernier, K.; Fiorelli, R.; Frank, S.; Raineteau, O.; Alvarez-Buylla, A.; Taylor, V. Molecular diversity subdivides the adult forebrain neural stem cell population. Stem Cells 2014, 32, 70–84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Aoki, K.; Chiba, K.; Sato, Y.; Shiozawa, Y.; Shiraishi, Y.; Shimamura, T.; Niida, A.; Motomura, K.; Ohka, F. Mutational landscape and clonal architecture in grade II and III gliomas. Nat. Genet. 2015, 47, 458. [Google Scholar] [CrossRef]
- Network, C.G.A.R. Comprehensive, integrative genomic analysis of diffuse lower-grade gliomas. New Engl. J. Med. 2015, 372, 2481–2498. [Google Scholar]
- Klinakis, A.; Lobry, C.; Abdel-Wahab, O.; Oh, P.; Haeno, H.; Buonamici, S.; van De Walle, I.; Cathelin, S.; Trimarchi, T.; Araldi, E.; et al. A novel tumour-suppressor function for the Notch pathway in myeloid leukaemia. Nature 2011, 473, 230–233. [Google Scholar] [CrossRef]
- Lobry, C.; Ntziachristos, P.; Ndiaye-Lobry, D.; Oh, P.; Cimmino, L.; Zhu, N.; Araldi, E.; Hu, W.; Freund, J.; Abdel-Wahab, O. Notch pathway activation targets AML-initiating cell homeostasis and differentiation. J. Exp. Med. 2013, 210, 301–319. [Google Scholar] [CrossRef]
- Kannan, S.; Sutphin, R.M.; Hall, M.G.; Golfman, L.S.; Fang, W.; Nolo, R.M.; Akers, L.J.; Hammitt, R.A.; McMurray, J.S.; Kornblau, S.M. Notch activation inhibits AML growth and survival: A potential therapeutic approach. J. Exp. Med. 2013, 210, 321–337. [Google Scholar] [CrossRef]
- Kode, A.; Manavalan, J.S.; Mosialou, I.; Bhagat, G.; Rathinam, C.V.; Luo, N.; Khiabanian, H.; Lee, A.; Murty, V.V.; Friedman, R. Leukaemogenesis induced by an activating β-catenin mutation in osteoblasts. Nature 2014, 506, 240–244. [Google Scholar] [CrossRef] [Green Version]
- Sorrentino, C.; Cuneo, A.; Roti, G. Therapeutic Targeting of Notch Signaling Pathway in Hematological Malignancies. Mediterr. J. Hematol. Infect. 2019, 11, e2019037. [Google Scholar] [CrossRef]
- Nickoloff, B.J.; Osborne, B.A.; Miele, L. Notch signaling as a therapeutic target in cancer: A new approach to the development of cell fate modifying agents. Oncogene 2003, 22, 6598–6608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rizzo, P.; Osipo, C.; Foreman, K.; Golde, T.; Osborne, B.; Miele, L. Rational targeting of Notch signaling in cancer. Oncogene 2008, 27, 5124–5131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hounjet, J.; Habets, R.; Schaaf, M.B.; Hendrickx, T.C.; Barbeau, L.M.O.; Yahyanejad, S.; Rouschop, K.M.; Groot, A.J.; Vooijs, M. The anti-malarial drug chloroquine sensitizes oncogenic NOTCH1 driven human T-ALL to γ-secretase inhibition. Oncogene 2019, 38, 5457–5468. [Google Scholar] [CrossRef] [PubMed]
- Pinazza, M.; Ghisi, M.; Minuzzo, S.; Agnusdei, V.; Fossati, G.; Ciminale, V.; Pezzè, L.; Ciribilli, Y.; Pilotto, G.; Venturoli, C.; et al. Histone deacetylase 6 controls Notch3 trafficking and degradation in T-cell acute lymphoblastic leukemia cells. Oncogene 2018, 37, 3839–3851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, M.; Troost, T.; Grawe, F.; Martinez-Arias, A.; Klein, T. Activation of Notch in lgd mutant cells requires the fusion of late endosomes with the lysosome. J. Cell Sci. 2013, 126, 645–656. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Habets, R.A.; de Bock, C.E.; Serneels, L.; Lodewijckx, I.; Verbeke, D.; Nittner, D.; Narlawar, R.; Demeyer, S.; Dooley, J.; Liston, A.; et al. Safe targeting of T cell acute lymphoblastic leukemia by pathology-specific NOTCH inhibition. Sci. Transl. Med. 2019, 11, eaau6246. [Google Scholar] [CrossRef] [Green Version]
- Roti, G.; Carlton, A.; Ross, K.N.; Markstein, M.; Pajcini, K.; Su, A.H.; Perrimon, N.; Pear, W.S.; Kung, A.L.; Blacklow, S.C.; et al. Complementary genomic screens identify SERCA as a therapeutic target in NOTCH1 mutated cancer. Cancer Cell 2013, 23, 390–405. [Google Scholar] [CrossRef] [Green Version]
- Nolin, E.; Gans, S.; Llamas, L.; Bandyopadhyay, S.; Brittain, S.M.; Bernasconi-Elias, P.; Carter, K.P.; Loureiro, J.J.; Thomas, J.R.; Schirle, M.; et al. Discovery of a ZIP7 inhibitor from a Notch pathway screen. Nat. Chem. Biol. 2019, 15, 179–188. [Google Scholar] [CrossRef]
- Periz, G.; Fortini, M.E. Ca(2+)-ATPase function is required for intracellular trafficking of the Notch receptor in Drosophila. EMBO J. 1999, 18, 5983–5993. [Google Scholar] [CrossRef] [Green Version]
- Doan, N.T.; Paulsen, E.S.; Sehgal, P.; Moller, J.V.; Nissen, P.; Denmeade, S.R.; Isaacs, J.T.; Dionne, C.A.; Christensen, S.B. Targeting thapsigargin towards tumors. Steroids 2015, 97, 2–7. [Google Scholar] [CrossRef]
- Sagara, Y.; Wade, J.; Inesi, G. A conformational mechanism for formation of a dead-end complex by the sarcoplasmic reticulum ATPase with thapsigargin. J. Biol. Chem. 1992, 267, 1286–1292. [Google Scholar] [PubMed]
- Toyoshima, C.; Nomura, H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature 2002, 418, 605–611. [Google Scholar] [CrossRef] [PubMed]
- Wong, G.T.; Manfra, D.; Poulet, F.M.; Zhang, Q.; Josien, H.; Bara, T.; Engstrom, L.; Pinzon-Ortiz, M.; Fine, J.S.; Lee, H.J.; et al. Chronic treatment with the gamma-secretase inhibitor LY-411,575 inhibits beta-amyloid peptide production and alters lymphopoiesis and intestinal cell differentiation. J. Biol. Chem. 2004, 279, 12876–12882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lytton, J.; Westlin, M.; Hanley, M.R. Thapsigargin inhibits the sarcoplasmic or endoplasmic reticulum Ca-ATPase family of calcium pumps. J. Biol. Chem. 1991, 266, 17067–17071. [Google Scholar] [PubMed]
- Denmeade, S.R.; Jakobsen, C.M.; Janssen, S.; Khan, S.R.; Garrett, E.S.; Lilja, H.; Christensen, S.B.; Isaacs, J.T. Prostate-specific antigen-activated thapsigargin prodrug as targeted therapy for prostate cancer. J. Nat. Cancer Inst. 2003, 95, 990–1000. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A Phase II, Multicenter, Single-Arm Study of Mipsagargin (G-202) as a Second-Line Therapy Following Sorafenib for Adult Patients with Progressive Advanced Hepatocellular Carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denmeade, S.R.; Mhaka, A.M.; Rosen, D.M.; Brennen, W.N.; Dalrymple, S.; Dach, I.; Olesen, C.; Gurel, B.; Demarzo, A.M.; Wilding, G.; et al. Engineering a prostate-specific membrane antigen-activated tumor endothelial cell prodrug for cancer therapy. Sci. Transl. Med. 2012, 4, 140ra186. [Google Scholar] [CrossRef] [Green Version]
- Roti, G.; Qi, J.; Kitara, S.; Sanchez-Martin, M.; Saur Conway, A.; Varca, A.C.; Su, A.; Wu, L.; Kung, A.L.; Ferrando, A.A.; et al. Leukemia-specific delivery of mutant NOTCH1 targeted therapy. J. Exp. Med. 2018, 215, 197–216. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.-C.; Yang, Z.-X.; Zhou, J.-S.; Zhang, H.-T.; Huang, Q.-K.; Dang, L.-L.; Liu, G.-X.; Tao, K.-S. Curcumin regulates hepatoma cell proliferation and apoptosis through the Notch signaling pathway. Int. J. Clin. Exp. Med. 2014, 7, 714. [Google Scholar]
- Subramaniam, D.; Ponnurangam, S.; Ramamoorthy, P.; Standing, D.; Battafarano, R.J.; Anant, S.; Sharma, P. Curcumin induces cell death in esophageal cancer cells through modulating Notch signaling. PLoS ONE 2012, 7, e30590. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, J.; Ma, D.; Zhang, L.; Si, M.; Yin, H.; Li, J. Curcumin inhibits proliferation and invasion of osteosarcoma cells through inactivation of Notch-1 signaling. FEBS J. 2012, 279, 2247–2259. [Google Scholar] [CrossRef] [PubMed]
- Aridoss, G.; Zhou, B.; Hermanson, D.L.; Bleeker, N.P.; Xing, C. Structure-activity relationship (SAR) study of ethyl 2-amino-6-(3,5-dimethoxyphenyl)-4-(2-ethoxy-2-oxoethyl)-4H-chromene-3-carboxylate (CXL017) and the potential of the lead against multidrug resistance in cancer treatment. J. Med. Chem. 2012, 55, 5566–5581. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, N.P.; Cornea, R.L.; Thomas, D.D.; Xing, C. A novel SERCA inhibitor demonstrates synergy with classic SERCA inhibitors and targets multidrug-resistant AML. Mol. Pharm. 2013, 10, 4358–4366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Ford, C.; Calderón, C.; Sehgal, P.; Fedosova, N.U.; Murillo, R.; Olesen, C.; Nissen, P.; Møller, J.V.; Merfort, I. Discovery of Tricyclic Clerodane Diterpenes as Sarco/Endoplasmic Reticulum Ca2+-ATPase Inhibitors and Structure–Activity Relationships. J. Nat. Prod. 2015, 78, 1262–1270. [Google Scholar] [CrossRef] [PubMed]
- De Ford, C.; Heidersdorf, B.; Haun, F.; Murillo, R.; Friedrich, T.; Borner, C.; Merfort, I. The clerodane diterpene casearin J induces apoptosis of T-ALL cells through SERCA inhibition, oxidative stress, and interference with Notch1 signaling. Cell Death Dis. 2016, 7, e2070. [Google Scholar] [CrossRef] [PubMed]
- Marchesini, M.; Gherli, A.; Montanaro, A.; Patrizi, L.; Sorrentino, C.; Pagliaro, L.; Rompietti, C.; Kitara, S.; Heit, S.; Olesen, C.E.; et al. Blockade of Oncogenic NOTCH1 with the SERCA Inhibitor CAD204520 in T Cell Acute Lymphoblastic Leukemia. Cell Chem. Biol. 2020, 27, 678–697. [Google Scholar] [CrossRef]
- Sehgal, P.; Szalai, P.; Olesen, C.; Praetorius, H.A.; Nissen, P.; Christensen, S.B.; Engedal, N.; Møller, J.V. Inhibition of the sarco/endoplasmic reticulum (ER) Ca2+-ATPase by thapsigargin analogs induces cell death via ER Ca2+ depletion and the unfolded protein response. J. Biol. Chem. 2017, 292, 19656–19673. [Google Scholar] [CrossRef] [Green Version]
- Hollingshead, L.M.; Faulds, D.; Fitton, A. Bepridil. A review of its pharmacological properties and therapeutic use in stable angina pectoris. Drugs 1992, 44, 835–857. [Google Scholar] [CrossRef]
- Roti, G.; Ross, K.; Ferrando, A.; Blacklow, S.; Aster, J.; Stegmaier, K. Expression-Based Screen Identifies the Calcium Channel Antagonist Bepridil as a Notch1 Modulator in T-ALL. Blood 2009, 114, 366. [Google Scholar] [CrossRef]
- Arruga, F.; Gizdic, B.; Serra, S.; Vaisitti, T.; Ciardullo, C.; Coscia, M.; Laurenti, L.; D’Arena, G.; Jaksic, O.; Inghirami, G. Functional impact of NOTCH1 mutations in chronic lymphocytic leukemia. Leukemia 2014, 28, 1060. [Google Scholar] [CrossRef]
- Groth, C.; Sasamura, T.; Khanna, M.R.; Whitley, M.; Fortini, M.E. Protein trafficking abnormalities in Drosophila tissues with impaired activity of the ZIP7 zinc transporter Catsup. Development 2013, 140, 3018–3027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, I.; Gagner, J.P.; Law, M.; Newcomb, E.W.; Zagzag, D. Angiogenesis in gliomas: Biology and molecular pathophysiology. Brain Pathol. 2005, 15, 297–310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Chen, T.; Zhang, J.; Mao, Q.; Li, S.; Xiong, W.; Qiu, Y.; Xie, Q.; Ge, J. Notch1 promotes glioma cell migration and invasion by stimulating β-catenin and NF-κB signaling via AKT activation. Cancer Sci. 2012, 103, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S. Molecular mechanisms of glioma invasiveness: The role of proteases. Nat. Rev. Cancer 2003, 3, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Sidenius, N.; Blasi, F. The urokinase plasminogen activator system in cancer: Recent advances and implication for prognosis and therapy. Cancer Metastasis Rev. 2003, 22, 205–222. [Google Scholar] [CrossRef]
- Raghu, H.; Gondi, C.S.; Dinh, D.H.; Gujrati, M.; Rao, J.S. Specific knockdown of uPA/uPAR attenuates invasion in glioblastoma cells and xenografts by inhibition of cleavage and trafficking of Notch -1 receptor. Mol. Cancer 2011, 10, 130. [Google Scholar] [CrossRef] [Green Version]
- Kanwar, R.; Fortini, M.E. Notch signaling: A different sort makes the cut. Curr. Biol. 2004, 14, R1043–R1045. [Google Scholar] [CrossRef] [Green Version]
- Weber, U.; Eroglu, C.; Mlodzik, M. Phospholipid membrane composition affects EGF receptor and Notch signaling through effects on endocytosis during Drosophila development. Dev. Cell 2003, 5, 559–570. [Google Scholar] [CrossRef] [Green Version]
- Sakamoto, K.; Aldana-Masangkay, G. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2011, 2011, 10. [Google Scholar]
- Gao, Y.-s.; Hubbert, C.C.; Yao, T.-P. The microtubule-associated histone deacetylase 6 (HDAC6) regulates epidermal growth factor receptor (EGFR) endocytic trafficking and degradation. J. Biol. Chem. 2010, 285, 11219–11226. [Google Scholar] [CrossRef] [Green Version]
- Bolden, J.E.; Peart, M.J.; Johnstone, R.W. Anticancer activities of histone deacetylase inhibitors. Nat. Rev. Drug Dis. 2006, 5, 769–784. [Google Scholar] [CrossRef] [PubMed]
- Mosquera, J.M.; Sboner, A.; Zhang, L.; Chen, C.L.; Sung, Y.S.; Chen, H.W.; Agaram, N.P.; Briskin, D.; Basha, B.M.; Singer, S. Novel MIR143-NOTCH fusions in benign and malignant glomus tumors. Genes Chromosomes Cancer 2013, 52, 1075–1087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tzoneva, G.; Ferrando, A.A. Recent advances on NOTCH signaling in T-ALL. Curr. Top. Microbiol. Immunol. 2012, 360, 163–182. [Google Scholar] [CrossRef] [PubMed]
- Kaether, C.; Haass, C.; Steiner, H. Assembly, trafficking and function of γ-secretase. Neurodegener. Dis. 2006, 3, 275–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Green, K.N.; Demuro, A.; Akbari, Y.; Hitt, B.D.; Smith, I.F.; Parker, I.; LaFerla, F.M. SERCA pump activity is physiologically regulated by presenilin and regulates amyloid beta production. J. Cell. Biol. 2008, 181, 1107–1116. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Easton, J.; Shao, Y.; Maciaszek, J.; Wang, Z.; Wilkinson, M.R.; McCastlain, K.; Edmonson, M.; Pounds, S.B.; Shi, L. The genomic landscape of pediatric and young adult T-lineage acute lymphoblastic leukemia. Nat. Genet. 2017, 49, 1211. [Google Scholar] [CrossRef] [Green Version]
- Atak, Z.K.; Gianfelici, V.; Hulselmans, G.; De Keersmaecker, K.; Devasia, A.G.; Geerdens, E.; Mentens, N.; Chiaretti, S.; Durinck, K.; Uyttebroeck, A. Comprehensive analysis of transcriptome variation uncovers known and novel driver events in T-cell acute lymphoblastic leukemia. PLoS Genet. 2013, 9, e1003997. [Google Scholar] [CrossRef]
- Milano, J.; McKay, J.; Dagenais, C.; Foster-Brown, L.; Pognan, F.; Gadient, R.; Jacobs, R.T.; Zacco, A.; Greenberg, B.; Ciaccio, P.J. Modulation of notch processing by γ-secretase inhibitors causes intestinal goblet cell metaplasia and induction of genes known to specify gut secretory lineage differentiation. Toxicol. Sci. 2004, 82, 341–358. [Google Scholar] [CrossRef]
- Deangelo, D.J.; Stone, R.M.; Silverman, L.B.; Stock, W.; Attar, E.C.; Fearen, I.; Dallob, A.; Matthews, C.; Stone, J.; Freedman, S.J.; et al. A phase I clinical trial of the notch inhibitor MK-0752 in patients with T-cell acute lymphoblastic leukemia/lymphoma (T-ALL) and other leukemias. J. Clin. Oncol. 2006, 24, 6585. [Google Scholar] [CrossRef]
- Mukherjee, N.; Almeida, A.; Partyka, K.A.; Lu, Y.; Schwan, J.V.; Lambert, K.; Rogers, M.; Robinson, W.A.; Robinson, S.E.; Applegate, A.J. Combining a GSI and BCL-2 inhibitor to overcome melanoma’s resistance to current treatments. Oncotarget 2016, 7, 84594. [Google Scholar] [CrossRef] [Green Version]
- Real, P.J.; Ferrando, A.A. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia 2009, 23, 1374–1377. [Google Scholar] [PubMed] [Green Version]
- Shi, T.-T.; Yu, X.-X.; Yan, L.-J.; Xiao, H.-T. Research progress of hydroxychloroquine and autophagy inhibitors on cancer. Cancer Chemother. Pharmacol. 2017, 79, 287–294. [Google Scholar] [PubMed]
- De Strooper, B.; Vassar, R.; Golde, T. The secretases: Enzymes with therapeutic potential in Alzheimer disease. Nat. Rev. Neurol 2010, 6, 99–107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cullion, K.; Draheim, K.M.; Hermance, N.; Tammam, J.; Sharma, V.M.; Ware, C.; Nikov, G.; Krishnamoorthy, V.; Majumder, P.K.; Kelliher, M.A. Targeting the Notch1 and mTOR pathways in a mouse T-ALL model. Blood 2009, 113, 6172–6181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papayannidis, C.; DeAngelo, D.J.; Stock, W.; Huang, B.; Shaik, M.N.; Cesari, R.; Zheng, X.; Reynolds, J.M.; English, P.A.; Ozeck, M.; et al. A Phase 1 study of the novel gamma-secretase inhibitor PF-03084014 in patients with T-cell acute lymphoblastic leukemia and T-cell lymphoblastic lymphoma. Blood Cancer J. 2015, 5, e350. [Google Scholar] [CrossRef] [Green Version]
- Zweidler-McKay, P.A.; DeAngelo, D.J.; Douer, D.; Dombret, H.; Ottmann, O.G.; Vey, N.; Thomas, D.A.; Zhu, L.; Huang, F.; Bajaj, G. The safety and activity of BMS-906024, a gamma secretase inhibitor (GSI) with anti-notch activity. In Patients with Relapsed T-Cell Acute Lymphoblastic Leukemia (T-ALL): Initial Results of a Phase 1 Trial; American Society of Hematology: Washington, DC, USA, 2014. [Google Scholar]
- Knoechel, B.; Bhatt, A.; Pan, L.; Pedamallu, C.S.; Severson, E.; Gutierrez, A.; Dorfman, D.M.; Kuo, F.C.; Kluk, M.; Kung, A.L.; et al. Complete hematologic response of early T-cell progenitor acute lymphoblastic leukemia to the γ-secretase inhibitor BMS-906024: Genetic and epigenetic findings in an outlier case. Cold Spring Harb. Mol. Case Stud. 2015, 1, a000539. [Google Scholar] [CrossRef] [Green Version]
- Riccio, O.; van Gijn, M.E.; Bezdek, A.C.; Pellegrinet, L.; van Es, J.H.; Zimber-Strobl, U.; Strobl, L.J.; Honjo, T.; Clevers, H.; Radtke, F. Loss of intestinal crypt progenitor cells owing to inactivation of both Notch1 and Notch2 is accompanied by derepression of CDK inhibitors p27Kip1 and p57Kip2. EMBO Rep. 2008, 9, 377–383. [Google Scholar] [CrossRef] [Green Version]
- Filipović, A.; Lombardo, Y.; Faronato, M.; Abrahams, J.; Aboagye, E.; Nguyen, Q.D.; d’Aqua, B.B.; Ridley, A.; Green, A.; Rahka, E.; et al. Anti-nicastrin monoclonal antibodies elicit pleiotropic anti-tumour pharmacological effects in invasive breast cancer cells. Breast Cancer Res. Treat. 2014, 148, 455–462. [Google Scholar] [CrossRef] [Green Version]
- Arai, M.A.; Akamine, R.; Tsuchiya, A.; Yoneyama, T.; Koyano, T.; Kowithayakorn, T.; Ishibashi, M. The Notch inhibitor cowanin accelerates nicastrin degradation. Sci. Rep. 2018, 8, 5376. [Google Scholar] [CrossRef]
- Suisse, A.; Treisman, J.E. Reduced SERCA Function Preferentially Affects Wnt Signaling by Retaining E-Cadherin in the Endoplasmic Reticulum. Cell Rep. 2019, 26, 322–329. [Google Scholar] [CrossRef] [Green Version]
- Wu, R.; Yang, H.; Wan, J.; Deng, X.; Chen, L.; Hao, S.; Ma, L. Knockdown of the Hippo transducer YAP reduces proliferation and promotes apoptosis in the Jurkat leukemia cell. Mol. Med. Rep. 2018, 18, 5379–5388. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
| Chemical Structure | Class | Compound | Activity on Notch Trafficking | Ref. |
|---|---|---|---|---|
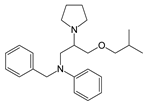 | Antiarrhythmic class IV | Bepridil | Ion flux modulation | [84] |
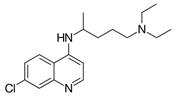 | Antimalarial | Chloroquine | Endosomal trafficking impairment | [166] |
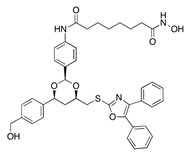 | HDAC6 inhibitors | Tubacin | Endosomal trafficking impairment | [167] |
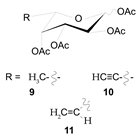 | POFUT1 analogs | Compounds 9, 10, and 11 | EGF-fucosylation inhibition | [168] |
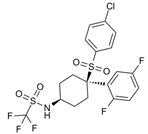 | PSEN1 inhibitors | MRK-560 | GS complex inhibition | [169] |
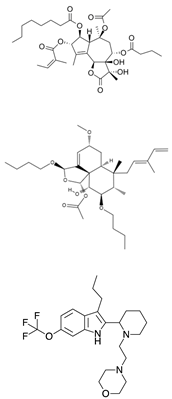 | SERCA inhibitors | Thapsigargin Casearin J CAD204520 | Ion flux modulation | [170] |
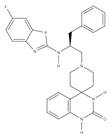 | ZIP7 inhibitors | NVS-ZP7-4 | Ion flux modulation | [171] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pagliaro, L.; Sorrentino, C.; Roti, G. Targeting Notch Trafficking and Processing in Cancers. Cells 2020, 9, 2212. https://doi.org/10.3390/cells9102212
Pagliaro L, Sorrentino C, Roti G. Targeting Notch Trafficking and Processing in Cancers. Cells. 2020; 9(10):2212. https://doi.org/10.3390/cells9102212
Chicago/Turabian StylePagliaro, Luca, Claudia Sorrentino, and Giovanni Roti. 2020. "Targeting Notch Trafficking and Processing in Cancers" Cells 9, no. 10: 2212. https://doi.org/10.3390/cells9102212
APA StylePagliaro, L., Sorrentino, C., & Roti, G. (2020). Targeting Notch Trafficking and Processing in Cancers. Cells, 9(10), 2212. https://doi.org/10.3390/cells9102212