Multitasking by the OC Lineage during Bone Infection: Bone Resorption, Immune Modulation, and Microbial Niche


Abstract
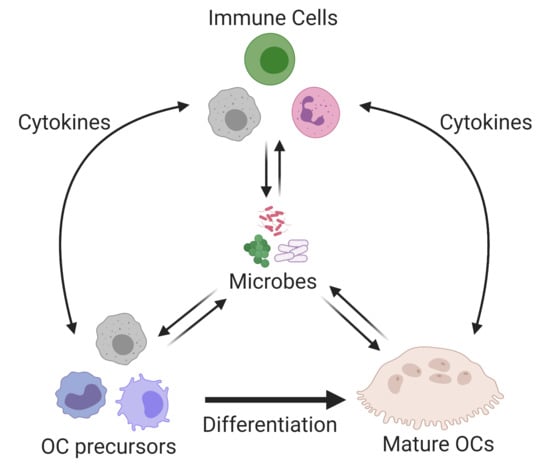
{kind=link}
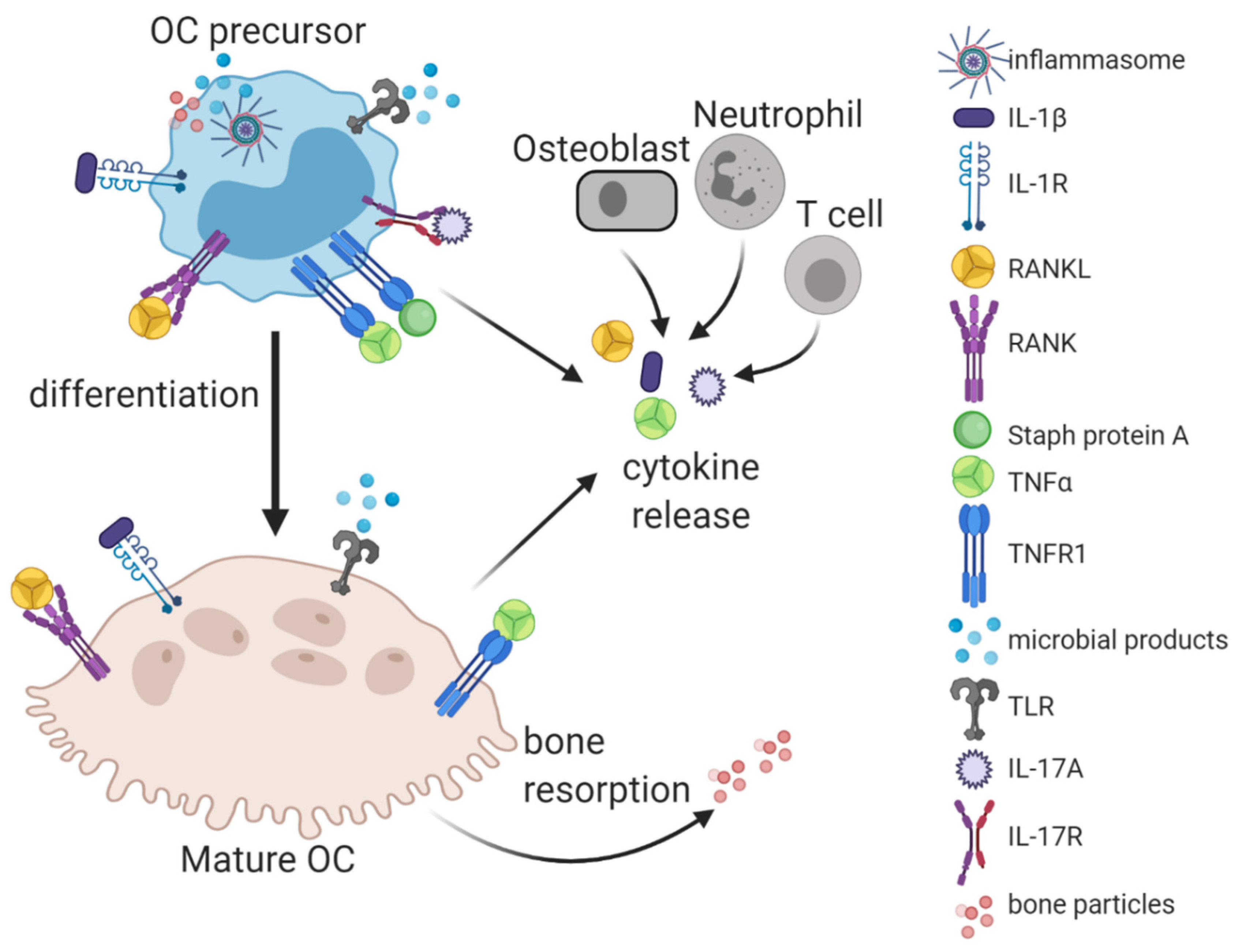{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Homeostatic Osteoclastogenesis
3. The Infectious Milieu, a Noxious Brew of Pathogen- and Host-Derived Factors Driving Osteoclastogenesis and Bone Resorption
3.1. Pathogen-Derived Factors and Pattern-Recognition Receptors
3.2. Cytokines and Other Host-Generated Osteoclastogenic Factors
4. Immune Modulation by OCs and Their Conventional and Unconventional Precursors
4.1. OC Precursors as Potential Immune Suppressors
4.2. Relationship between OCs, Dendritic Cells, and T Cells
5. Intracellular Infection of OCs Providing a Proliferative Niche
6. Clinical Implications
7. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Lew, D.P.; Waldvogel, F.A. Osteomyelitis. Lancet 2004, 364, 369–379. [Google Scholar] [CrossRef]
- Barth, K.; Remick, D.G.; Genco, C.A. Disruption of immune regulation by microbial pathogens and resulting chronic inflammation. J. Cell. Physiol. 2013, 228, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Kremers, H.M.; Nwojo, M.E.; Ransom, J.E.; Wood-Wentz, C.M.; Melton, L.J., 3rd; Huddleston, P.M., 3rd. Trends in the epidemiology of osteomyelitis: A population-based study, 1969 to 2009. J. Bone Jt. Surg. Am. 2015, 97, 837–845. [Google Scholar] [CrossRef] [PubMed]
- Goergens, E.; McEvoy, A.; Watson, M.; Barrett, I. Acute osteomyelitis and septic arthritis in children. J. Paediatr. Child Health 2005, 41, 59–62. [Google Scholar] [CrossRef]
- Sendi, P.; Kaempfen, A.; Uçkay, I.; Meier, R. Bone and joint infections of the hand. Clin. Microbiol. Infect. 2020, 26, 848–856. [Google Scholar] [CrossRef]
- Seebach, E.; Kubatzky, K.F. Chronic implant-related bone infections-can immune modulation be a therapeutic strategy? Front. Immunol. 2019, 10, 1724. [Google Scholar] [CrossRef]
- Novack, D.V.; Mbalaviele, G. Osteoclasts-key players in skeletal health and disease. Microbiol. Spectr. 2016, 4. [Google Scholar] [CrossRef]
- Kumar, G.; Roger, P.M. From crosstalk between immune and bone cells to bone erosion in infection. Int. J. Mol. Sci. 2019, 20, 5154. [Google Scholar] [CrossRef]
- Madel, M.B.; Ibáñez, L.; Wakkach, A.; de Vries, T.J.; Teti, A.; Apparailly, F.; Blin-Wakkach, C. Immune function and diversity of osteoclasts in normal and pathological conditions. Front. Immunol. 2019, 10, 1408. [Google Scholar] [CrossRef]
- Souza, P.P.C.; Lerner, U.H. Finding a toll on the route: The fate of osteoclast progenitors after toll-like receptor activation. Front. Immunol. 2019, 10, 1663. [Google Scholar] [CrossRef]
- Gu, Y.; Han, X. Toll-like receptor signaling and immune regulatory lymphocytes in periodontal disease. Int. J. Mol. Sci. 2020, 21, 3329. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Yang, J.; Park, O.J.; Kang, S.S.; Kim, W.S.; Kurokawa, K.; Yun, C.H.; Kim, H.H.; Lee, B.L.; Han, S.H. Lipoproteins are an important bacterial component responsible for bone destruction through the induction of osteoclast differentiation and activation. J. Bone Miner. Res. 2013, 28, 2381–2391. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Liu, J.; Xu, Q.; Harber, G.; Feng, X.; Michalek, S.M.; Katz, J. TLR2-dependent modulation of osteoclastogenesis by Porphyromonas gingivalis through differential induction of NFATc1 and NF-kappaB. J. Biol. Chem. 2011, 286, 24159–24169. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Sojar, H.; Genco, R.J.; DeNardin, E. Intracellular signaling and cytokine induction upon interactions of Porphyromonas gingivalis fimbriae with pattern-recognition receptors. Immunol. Investig. 2004, 33, 157–172. [Google Scholar] [CrossRef]
- Hienz, S.A.; Paliwal, S.; Ivanovski, S. Mechanisms of bone resorption in periodontitis. J. Immunol. Res. 2015, 2015, 615486. [Google Scholar] [CrossRef]
- Cao, F.; Zhou, W.; Liu, G.; Xia, T.; Liu, M.; Mi, B.; Liu, Y. Staphylococcus aureus peptidoglycan promotes osteoclastogenesis via TLR2-mediated activation of the NF-κB/NFATc1 signaling pathway. Am. J. Transl. Res. 2017, 9, 5022–5030. [Google Scholar]
- Ishida, M.; Kitaura, H.; Kimura, K.; Sugisawa, H.; Aonuma, T.; Takada, H.; Takano-Yamamoto, T. Muramyl dipeptide enhances lipopolysaccharide-induced osteoclast formation and bone resorption through increased RANKL expression in stromal cells. J. Immunol. Res. 2015, 2015, 132765. [Google Scholar] [CrossRef]
- Kishimoto, T.; Kaneko, T.; Ukai, T.; Yokoyama, M.; Haro, R.A.; Yoshinaga, Y.; Yoshimura, A.; Hara, Y. Peptidoglycan and lipopolysaccharide synergistically enhance bone resorption and osteoclastogenesis. J. Periodontal Res. 2012, 47, 446–454. [Google Scholar] [CrossRef]
- Zhu, X.; Zhao, Y.; Jiang, Y.; Qin, T.; Chen, J.; Chu, X.; Yi, Q.; Gao, S.; Wang, S. Dectin-1 signaling inhibits osteoclastogenesis via IL-33-induced inhibition of NFATc1. Oncotarget 2017, 8, 53366–53374. [Google Scholar] [CrossRef]
- Yamasaki, T.; Ariyoshi, W.; Okinaga, T.; Adachi, Y.; Hosokawa, R.; Mochizuki, S.; Sakurai, K.; Nishihara, T. The dectin 1 agonist curdlan regulates osteoclastogenesis by inhibiting nuclear factor of activated T cells cytoplasmic 1 (NFATc1) through Syk kinase. J. Biol. Chem. 2014, 289, 19191–19203. [Google Scholar] [CrossRef]
- Mbalaviele, G.; Novack, D.V.; Schett, G.; Teitelbaum, S.L. Inflammatory osteolysis: A conspiracy against bone. J. Clin. Investig. 2017, 127, 2030–2039. [Google Scholar] [CrossRef] [PubMed]
- Alippe, Y.; Mbalaviele, G. Omnipresence of inflammasome activities in inflammatory bone diseases. Semin. Immunopathol. 2019, 41, 607–618. [Google Scholar] [CrossRef]
- de Andrade, K.Q.; Almeida-da-Silva, C.L.C.; Coutinho-Silva, R. Immunological pathways triggered by porphyromonas gingivalis and fusobacterium nucleatum: Therapeutic possibilities? Mediat. Inflamm. 2019, 2019, 7241312. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Planillo, R.; Franchi, L.; Miller, L.S.; Nunez, G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J. Immunol. 2009, 183, 3942–3948. [Google Scholar] [CrossRef] [PubMed]
- Craven, R.R.; Gao, X.; Allen, I.C.; Gris, D.; Wardenburg, J.B.; McElvania-Tekippe, E.; Ting, J.P.; Duncan, J.A. Staphylococcus aureus alpha-hemolysin activates the NLRP3-inflammasome in human and mouse monocytic cells. PLoS ONE 2009, 4, e7446. [Google Scholar] [CrossRef]
- Hanamsagar, R.; Aldrich, A.; Kielian, T. Critical role for the AIM2 inflammasome during acute CNS bacterial infection. J. Neurochem. 2014, 129, 704–711. [Google Scholar] [CrossRef]
- Bonar, S.L.; Brydges, S.D.; Mueller, J.L.; McGeough, M.D.; Pena, C.; Chen, D.; Grimston, S.K.; Hickman-Brecks, C.L.; Ravindran, S.; McAlinden, A.; et al. Constitutively activated NLRP3 inflammasome causes inflammation and abnormal skeletal development in mice. PLoS ONE 2012, 7, e35979. [Google Scholar] [CrossRef]
- Qu, C.; Bonar, S.L.; Hickman-Brecks, C.L.; Abu-Amer, S.; McGeough, M.D.; Pena, C.A.; Broderick, L.; Yang, C.; Grimston, S.K.; Kading, J.; et al. NLRP3 mediates osteolysis through inflammation-dependent and -independent mechanisms. FASEB J. 2015, 29, 1269–1279. [Google Scholar] [CrossRef]
- Alippe, Y.; Wang, C.; Ricci, B.; Xiao, J.; Qu, C.; Zou, W.; Novack, D.V.; Abu-Amer, Y.; Civitelli, R.; Mbalaviele, G. Bone matrix components activate the NLRP3 inflammasome and promote osteoclast differentiation. Sci. Rep. 2017, 7, 6630. [Google Scholar] [CrossRef]
- Kim, H.; Walsh, M.C.; Takegahara, N.; Middleton, S.A.; Shin, H.I.; Kim, J.; Choi, Y. The purinergic receptor P2X5 regulates inflammasome activity and hyper-multinucleation of murine osteoclasts. Sci. Rep. 2017, 7, 196. [Google Scholar] [CrossRef]
- Lam, J.; Takeshita, S.; Barker, J.E.; Kanagawa, O.; Ross, F.P.; Teitelbaum, S.L. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J. Clin. Investig. 2000, 106, 1481–1488. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-H.; Heulsmann, A.; Tondravi, M.M.; Mukherjee, A.; Abu-Amer, Y. Tumor necrosis factor-a (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type 1 receptor and RANK signaling pathways. J. Biol. Chem. 2001, 276, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Kitaura, H.; Zhou, P.; Ross, F.P.; Teitelbaum, S.L. IL-1 mediates TNF-induced osteoclastogenesis. J. Clin. Investig. 2005, 115, 282–290. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Yamada, H.; Shibata, T.N.; Mitomi, H.; Nomoto, S.; Ozaki, S. Dual role of interleukin-17 in pannus growth and osteoclastogenesis in rheumatoid arthritis. Arthritis Res. Ther. 2011, 13, R14. [Google Scholar] [CrossRef]
- O’Brien, W.; Fissel, B.M.; Maeda, Y.; Yan, J.; Ge, X.; Gravallese, E.M.; Aliprantis, A.O.; Charles, J.F. RANK-independent osteoclast formation and bone erosion in inflammatory arthritis. Arthritis Rheumatol. 2016, 68, 2889–2900. [Google Scholar] [CrossRef]
- Marriott, I.; Hughes, F.M., Jr.; Bost, K.L. Bacterial infection of osteoblasts induces interleukin-1beta and interleukin-18 transcription but not protein synthesis. J. Interferon Cytokine Res. 2002, 22, 1049–1055. [Google Scholar] [CrossRef]
- Alexander, E.H.; Rivera, F.A.; Marriott, I.; Anguita, J.; Bost, K.L.; Hudson, M.C. Staphylococcus aureus-induced tumor necrosis factor related apoptosis-inducing ligand expression mediates apoptosis and caspase-8 activation in infected osteoblasts. BMC Microbiol. 2003, 3, 5. [Google Scholar] [CrossRef]
- McCall, S.H.; Sahraei, M.; Young, A.B.; Worley, C.S.; Duncan, J.A.; Ting, J.P.; Marriott, I. Osteoblasts express NLRP3, a nucleotide-binding domain and leucine-rich repeat region containing receptor implicated in bacterially induced cell death. J. Bone Miner. Res. 2008, 23, 30–40. [Google Scholar] [CrossRef]
- Kassem, A.; Lindholm, C.; Lerner, U.H. Toll-like receptor 2 stimulation of osteoblasts mediates staphylococcus aureus induced bone resorption and osteoclastogenesis through enhanced RANKL. PLoS ONE 2016, 11, e0156708. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, X.; Dou, C.; Cao, Z.; Liu, C.; Dong, S.; Fei, J. Staphylococcal protein A promotes osteoclastogenesis through MAPK signaling during bone infection. J. Cell. Physiol. 2017, 232, 2396–2406. [Google Scholar] [CrossRef]
- Ren, L.R.; Wang, H.; He, X.Q.; Song, M.G.; Chen, X.Q.; Xu, Y.Q. Staphylococcus aureus protein A induces osteoclastogenesis via the NF-κB signaling pathway. Mol. Med. Rep. 2017, 16, 6020–6028. [Google Scholar] [CrossRef] [PubMed]
- Claro, T.; Widaa, A.; McDonnell, C.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein A binding to osteoblast tumour necrosis factor receptor 1 results in activation of nuclear factor kappa B and release of interleukin-6 in bone infection. Microbiology 2013, 159, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, A.M.; Delpino, M.V.; Lattar, S.; Giai, C.; Llana, M.N.; Sanjuan, N.; Cassat, J.E.; Sordelli, D.; Gómez, M.I. Staphylococcus aureus protein A enhances osteoclastogenesis via TNFR1 and EGFR signaling. Biochim. Biophys. Acta 2016, 1862, 1975–1983. [Google Scholar] [CrossRef] [PubMed]
- Kamohara, A.; Hirata, H.; Xu, X.; Shiraki, M.; Yamada, S.; Zhang, J.Q.; Kukita, T.; Toyonaga, K.; Hara, H.; Urano, Y.; et al. IgG immune complexes with Staphylococcus aureus protein A enhance osteoclast differentiation and bone resorption by stimulating Fc receptors and TLR2. Int. Immunol. 2020, 32, 89–104. [Google Scholar] [CrossRef] [PubMed]
- Somayaji, S.N.; Ritchie, S.; Sahraei, M.; Marriott, I.; Hudson, M.C. Staphylococcus aureus induces expression of receptor activator of NF-kappaB ligand and prostaglandin E2 in infected murine osteoblasts. Infect. Immun. 2008, 76, 5120–5126. [Google Scholar] [CrossRef] [PubMed]
- Flammier, S.; Rasigade, J.P.; Badiou, C.; Henry, T.; Vandenesch, F.; Laurent, F.; Trouillet-Assant, S. Human monocyte-derived osteoclasts are targeted by staphylococcal pore-forming toxins and superantigens. PLoS ONE 2016, 11, e0150693. [Google Scholar] [CrossRef]
- Loughran, A.J.; Gaddy, D.; Beenken, K.E.; Meeker, D.G.; Morello, R.; Zhao, H.; Byrum, S.D.; Tackett, A.J.; Cassat, J.E.; Smeltzer, M.S. Impact of sarA and phenol-soluble modulins on the pathogenesis of osteomyelitis in diverse clinical isolates of staphylococcus aureus. Infect. Immun. 2016, 84, 2586–2594. [Google Scholar] [CrossRef]
- Cassat, J.E.; Hammer, N.D.; Campbell, J.P.; Benson, M.A.; Perrien, D.S.; Mrak, L.N.; Smeltzer, M.S.; Torres, V.J.; Skaar, E.P. A secreted bacterial protease tailors the Staphylococcus aureus virulence repertoire to modulate bone remodeling during osteomyelitis. Cell Host Microbe 2013, 13, 759–772. [Google Scholar] [CrossRef]
- Cai, X.; Li, Z.; Zhao, Y.; Katz, J.; Michalek, S.M.; Feng, X.; Li, Y.; Zhang, P. Enhanced dual function of osteoclast precursors following calvarial Porphyromonas gingivalis infection. J. Periodontal Res. 2020, 55, 410–425. [Google Scholar] [CrossRef]
- Yasuhara, R.; Miyamoto, Y.; Takami, M.; Imamura, T.; Potempa, J.; Yoshimura, K.; Kamijo, R. Lysine-specific gingipain promotes lipopolysaccharide- and active-vitamin D3-induced osteoclast differentiation by degrading osteoprotegerin. Biochem. J. 2009, 419, 159–166. [Google Scholar] [CrossRef]
- Mo, W.; Luo, H.; Wu, J.; Xu, N.; Zhang, F.; Qiu, Q.; Zhu, W.; Liang, M. Gingipains promote RANKL-induced osteoclastogenesis through the enhancement of integrin β3 in RAW264.7 cells. J. Mol. Histol. 2020, 51, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Kanzaki, H.; Movila, A.; Kayal, R.; Napimoga, M.H.; Egashira, K.; Dewhirst, F.; Sasaki, H.; Howait, M.; Al-Dharrab, A.; Mira, A.; et al. Phosphoglycerol dihydroceramide, a distinctive ceramide produced by Porphyromonas gingivalis, promotes RANKL-induced osteoclastogenesis by acting on non-muscle myosin II-A (Myh9), an osteoclast cell fusion regulatory factor. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 452–462. [Google Scholar] [CrossRef] [PubMed]
- Yokota, K.; Sato, K.; Miyazaki, T.; Kitaura, H.; Kayama, H.; Miyoshi, F.; Araki, Y.; Akiyama, Y.; Takeda, K.; Mimura, T. Combination of tumor necrosis factor α and interleukin-6 induces mouse osteoclast-like cells with bone resorption activity both in vitro and in vivo. Arthritis Rheumatol. 2014, 66, 121–129. [Google Scholar] [CrossRef]
- Zhao, B.; Grimes, S.N.; Li, S.; Hu, X.; Ivashkiv, L.B. TNF-induced osteoclastogenesis and inflammatory bone resorption are inhibited by transcription factor RBP-J. J. Exp. Med. 2012, 209, 319–334. [Google Scholar] [CrossRef]
- De Vries, T.J.; Schoenmaker, T.; Aerts, D.; Grevers, L.C.; Souza, P.P.; Nazmi, K.; van de Wiel, M.; Ylstra, B.; Lent, P.L.; Leenen, P.J.; et al. M-CSF priming of osteoclast precursors can cause osteoclastogenesis-insensitivity, which can be prevented and overcome on bone. J. Cell. Physiol. 2015, 230, 210–225. [Google Scholar] [CrossRef]
- Kotake, S.; Udagawa, N.; Takahashi, N.; Matsuzaki, K.; Itoh, K.; Ishiyama, S.; Saito, S.; Inoue, K.; Kamatani, N.; Gillespie, M.T.; et al. IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J. Clin. Investig. 1999, 103, 1345–1352. [Google Scholar] [CrossRef]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef]
- Shen, F.; Ruddy, M.J.; Plamondon, P.; Gaffen, S.L. Cytokines link osteoblasts and inflammation: Microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J. Leukoc. Biol. 2005, 77, 388–399. [Google Scholar] [CrossRef]
- Komatsu, N.; Takayanagi, H. Autoimmune arthritis: The interface between the immune system and joints. Adv. Immunol. 2012, 115, 45–71. [Google Scholar] [CrossRef]
- Dey, I.; Bishayi, B. Impact of simultaneous neutralization of IL-17A and treatment with recombinant IL-2 on Th17-Treg cell population in S.aureus induced septic arthritis. Microb. Pathog. 2020, 139, 103903. [Google Scholar] [CrossRef]
- Infante-Duarte, C.; Horton, H.F.; Byrne, M.C.; Kamradt, T. Microbial lipopeptides induce the production of IL-17 in Th cells. J. Immunol. 2000, 165, 6107–6115. [Google Scholar] [CrossRef]
- Putnam, N.E.; Fulbright, L.E.; Curry, J.M.; Ford, C.A.; Petronglo, J.R.; Hendrix, A.S.; Cassat, J.E. MyD88 and IL-1R signaling drive antibacterial immunity and osteoclast-driven bone loss during Staphylococcus aureus osteomyelitis. PLoS Pathog. 2019, 15, e1007744. [Google Scholar] [CrossRef] [PubMed]
- Adamopoulos, I.E.; Chao, C.C.; Geissler, R.; Laface, D.; Blumenschein, W.; Iwakura, Y.; McClanahan, T.; Bowman, E.P. Interleukin-17A upregulates receptor activator of NF-kappaB on osteoclast precursors. Arthritis Res. Ther. 2010, 12, R29. [Google Scholar] [CrossRef] [PubMed]
- Kitaura, H.; Zhou, P.; Kim, H.J.; Novack, D.V.; Ross, F.P.; Teitelbaum, S.L. M-CSF mediates TNF-induced inflammatory osteolysis. J. Clin. Investig. 2005, 115, 3418–3427. [Google Scholar] [CrossRef]
- Yao, Z.; Xing, L.; Boyce, B.F. NF-kappaB p100 limits TNF-induced bone resorption in mice by a TRAF3-dependent mechanism. J. Clin. Investig. 2009, 119, 3024–3034. [Google Scholar] [CrossRef]
- Andreev, D.; Liu, M.; Weidner, D.; Kachler, K.; Faas, M.; Grüneboom, A.; Schlötzer-Schrehardt, U.; Muñoz, L.E.; Steffen, U.; Grötsch, B.; et al. Osteocyte necrosis triggers osteoclast-mediated bone loss through macrophage-inducible C-type lectin. J. Clin. Investig. 2020. [Google Scholar] [CrossRef]
- Komori, T. Cell death in chondrocytes, osteoblasts, and osteocytes. Int. J. Mol. Sci. 2016, 17, 2045. [Google Scholar] [CrossRef]
- Su, X.; Floyd, D.H.; Hughes, A.; Xiang, J.; Schneider, J.G.; Uluckan, O.; Heller, E.; Deng, H.; Zou, W.; Craft, C.S.; et al. The ADP receptor P2RY12 regulates osteoclast function and pathologic bone remodeling. J. Clin. Investig. 2012, 122, 3579–3592. [Google Scholar] [CrossRef]
- Dong, Y.; Chen, Y.; Zhang, L.; Tian, Z.; Dong, S. P2X7 receptor acts as an efficient drug target in regulating bone metabolism system. Biomed. Pharmacother. 2020, 125, 110010. [Google Scholar] [CrossRef]
- Korcok, J.; Raimundo, L.N.; Du, X.; Sims, S.M.; Dixon, S.J. P2Y6 nucleotide receptors activate NF-kappaB and increase survival of osteoclasts. J. Biol. Chem. 2005, 280, 16909–16915. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef]
- Kirkwood, K.L.; Zhang, L.; Thiyagarajan, R.; Seldeen, K.L.; Troen, B.R. Myeloid-derived suppressor cells at the intersection of inflammaging and bone fragility. Immunol. Investig. 2018, 47, 844–854. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Huang, Y.; Wang, S.; Fu, R.; Guo, C.; Wang, H.; Zhao, J.; Gaskin, F.; Chen, J.; Yang, N.; et al. Myeloid-derived suppressor cells contribute to bone erosion in collagen-induced arthritis by differentiating to osteoclasts. J. Autoimmun. 2015, 65, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Charles, J.F.; Hsu, L.Y.; Niemi, E.C.; Weiss, A.; Aliprantis, A.O.; Nakamura, M.C. Inflammatory arthritis increases mouse osteoclast precursors with myeloid suppressor function. J. Clin. Investig. 2012, 122, 4592–4605. [Google Scholar] [CrossRef] [PubMed]
- Sawant, A.; Ponnazhagan, S. Myeloid-derived suppressor cells as osteoclast progenitors: A novel target for controlling osteolytic bone metastasis. Cancer Res. 2013, 73, 4606–4610. [Google Scholar] [CrossRef] [PubMed]
- Heim, C.E.; Vidlak, D.; Scherr, T.D.; Kozel, J.A.; Holzapfel, M.; Muirhead, D.E.; Kielian, T. Myeloid-derived suppressor cells contribute to Staphylococcus aureus orthopedic biofilm infection. J. Immunol. 2014, 192, 3778–3792. [Google Scholar] [CrossRef]
- Heim, C.E.; Vidlak, D.; Scherr, T.D.; Hartman, C.W.; Garvin, K.L.; Kielian, T. IL-12 promotes myeloid-derived suppressor cell recruitment and bacterial persistence during Staphylococcus aureus orthopedic implant infection. J. Immunol. 2015, 194, 3861–3872. [Google Scholar] [CrossRef]
- Heim, C.E.; Vidlak, D.; Kielian, T. Interleukin-10 production by myeloid-derived suppressor cells contributes to bacterial persistence during Staphylococcus aureus orthopedic biofilm infection. J. Leukoc. Biol. 2015, 98, 1003–1013. [Google Scholar] [CrossRef]
- Yamada, K.J.; Heim, C.E.; Aldrich, A.L.; Gries, C.M.; Staudacher, A.G.; Kielian, T. Arginase-1 Expression in Myeloid Cells Regulates Staphylococcus aureus Planktonic but Not Biofilm Infection. Infect. Immun. 2018, 86. [Google Scholar] [CrossRef]
- Stoll, H.; Ost, M.; Singh, A.; Mehling, R.; Neri, D.; Schäfer, I.; Velic, A.; Macek, B.; Kretschmer, D.; Weidenmaier, C.; et al. Staphylococcal enterotoxins dose-dependently modulate the generation of myeloid-derived suppressor cells. Front. Cell. Infect. Microbiol. 2018, 8, 321. [Google Scholar] [CrossRef]
- Su, L.; Xu, Q.; Zhang, P.; Michalek, S.M.; Katz, J. Phenotype and function of myeloid-derived suppressor cells induced by porphyromonas gingivalis infection. Infect. Immun. 2017, 85. [Google Scholar] [CrossRef]
- Gallois, A.; Lachuer, J.; Yvert, G.; Wierinckx, A.; Brunet, F.; Rabourdin-Combe, C.; Delprat, C.; Jurdic, P.; Mazzorana, M. Genome-wide expression analyses establish dendritic cells as a new osteoclast precursor able to generate bone-resorbing cells more efficiently than monocytes. J. Bone Miner. Res. 2010, 25, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Kiesel, J.; Miller, C.; Abu-Amer, Y.; Aurora, R. Systems level analysis of osteoclastogenesis reveals intrinsic and extrinsic regulatory interactions. Dev. Dyn. 2007, 236, 2181–2197. [Google Scholar] [CrossRef]
- Ibáñez, L.; Abou-Ezzi, G.; Ciucci, T.; Amiot, V.; Belaïd, N.; Obino, D.; Mansour, A.; Rouleau, M.; Wakkach, A.; Blin-Wakkach, C. Inflammatory osteoclasts prime TNFα-producing CD4(+) T cells and express CX(3) CR1. J. Bone Miner. Res. 2016, 31, 1899–1908. [Google Scholar] [CrossRef] [PubMed]
- Grassi, F.; Manferdini, C.; Cattini, L.; Piacentini, A.; Gabusi, E.; Facchini, A.; Lisignoli, G. T cell suppression by osteoclasts in vitro. J. Cell. Physiol. 2011, 226, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Kikuta, J.; Wada, Y.; Kowada, T.; Wang, Z.; Sun-Wada, G.H.; Nishiyama, I.; Mizukami, S.; Maiya, N.; Yasuda, H.; Kumanogoh, A.; et al. Dynamic visualization of RANKL and Th17-mediated osteoclast function. J. Clin. Investig. 2013, 123, 866–873. [Google Scholar] [CrossRef] [PubMed]
- Pöllinger, B.; Junt, T.; Metzler, B.; Walker, U.A.; Tyndall, A.; Allard, C.; Bay, S.; Keller, R.; Raulf, F.; Di Padova, F.; et al. Th17 cells, not IL-17+ γδ T cells, drive arthritic bone destruction in mice and humans. J. Immunol. 2011, 186, 2602–2612. [Google Scholar] [CrossRef]
- Kiesel, J.R.; Buchwald, Z.S.; Aurora, R. Cross-presentation by osteoclasts induces FoxP3 in CD8+ T cells. J. Immunol. 2009, 182, 5477–5487. [Google Scholar] [CrossRef]
- Buchwald, Z.S.; Kiesel, J.R.; DiPaolo, R.; Pagadala, M.S.; Aurora, R. Osteoclast activated FoxP3+ CD8+ T-cells suppress bone resorption in vitro. PLoS ONE 2012, 7, e38199. [Google Scholar] [CrossRef]
- Li, H.; Hong, S.; Qian, J.; Zheng, Y.; Yang, J.; Yi, Q. Cross talk between the bone and immune systems: Osteoclasts function as antigen-presenting cells and activate CD4+ and CD8+ T cells. Blood 2010, 116, 210–217. [Google Scholar] [CrossRef]
- Fischer, L.; Herkner, C.; Kitte, R.; Dohnke, S.; Riewaldt, J.; Kretschmer, K.; Garbe, A.I. Foxp3(+) Regulatory T Cells in Bone and Hematopoietic Homeostasis. Front. Endocrinol. 2019, 10, 578. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Zhu, G.; Lu, Y.; Wang, M.; Jules, J.; Zhou, X.; Chen, W. Deficiency of cathepsin K prevents inflammation and bone erosion in rheumatoid arthritis and periodontitis and reveals its shared osteoimmune role. FEBS Lett. 2015, 589, 1331–1339. [Google Scholar] [CrossRef] [PubMed]
- Hao, L.; Chen, J.; Zhu, Z.; Reddy, M.S.; Mountz, J.D.; Chen, W.; Li, Y.P. Odanacatib, A cathepsin K-specific inhibitor, inhibits inflammation and bone loss caused by periodontal diseases. J. Periodontol. 2015, 86, 972–983. [Google Scholar] [CrossRef] [PubMed]
- Josse, J.; Velard, F.; Gangloff, S.C. Staphylococcus aureus vs. osteoblast: Relationship and consequences in osteomyelitis. Front. Cell. Infect. Microbiol. 2015, 5, 85. [Google Scholar] [CrossRef] [PubMed]
- de Bentley, K.L.M.; Trombetta, R.; Nishitani, K.; Bello-Irizarry, S.N.; Ninomiya, M.; Zhang, L.; Chung, H.L.; McGrath, J.L.; Daiss, J.L.; Awad, H.A.; et al. Evidence of staphylococcus aureus deformation, proliferation, and migration in canaliculi of live cortical bone in murine models of osteomyelitis. J. Bone Miner. Res. 2017, 32, 985–990. [Google Scholar] [CrossRef]
- Yang, D.; Wijenayaka, A.R.; Solomon, L.B.; Pederson, S.M.; Findlay, D.M.; Kidd, S.P.; Atkins, G.J. Novel insights into staphylococcus aureus deep bone infections: The involvement of osteocytes. mBio 2018, 9. [Google Scholar] [CrossRef]
- Krauss, J.L.; Roper, P.M.; Ballard, A.; Shih, C.C.; Fitzpatrick, J.A.J.; Cassat, J.E.; Ng, P.Y.; Pavlos, N.J.; Veis, D.J. Staphylococcus aureus infects osteoclasts and replicates intracellularly. mBio 2019, 10. [Google Scholar] [CrossRef]
- Hoshino, A.; Hanada, S.; Yamada, H.; Mii, S.; Takahashi, M.; Mitarai, S.; Yamamoto, K.; Manome, Y. Mycobacterium tuberculosis escapes from the phagosomes of infected human osteoclasts reprograms osteoclast development via dysregulation of cytokines and chemokines. Pathog. Dis. 2014, 70, 28–39. [Google Scholar] [CrossRef]
- Kukita, A.; Ichigi, Y.; Takigawa, I.; Watanabe, T.; Kukita, T.; Miyamoto, H. Infection of RANKL-primed RAW-D macrophages with Porphyromonas gingivalis promotes osteoclastogenesis in a TNF-α-independent manner. PLoS ONE 2012, 7, e38500. [Google Scholar] [CrossRef][Green Version]
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature 2019, 568, 541–545. [Google Scholar] [CrossRef]
- Gaida, M.M.; Mayer, B.; Stegmaier, S.; Schirmacher, P.; Wagner, C.; Hansch, G.M. Polymorphonuclear neutrophils in osteomyelitis: Link to osteoclast generation and bone resorption. Eur. J. Inflamm. 2012, 413–426. [Google Scholar] [CrossRef]
- Masters, E.A.; Trombetta, R.P.; de Mesy Bentley, K.L.; Boyce, B.F.; Gill, A.L.; Gill, S.R.; Nishitani, K.; Ishikawa, M.; Morita, Y.; Ito, H.; et al. Evolving concepts in bone infection: Redefining “biofilm”, “acute vs. chronic osteomyelitis”, “the immune proteome” and “local antibiotic therapy”. Bone Res. 2019, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Gross, C.; Weber, M.; Creutzburg, K.; Möbius, P.; Preidl, R.; Amann, K.; Wehrhan, F. Osteoclast profile of medication-related osteonecrosis of the jaw secondary to bisphosphonate therapy: A comparison with osteoradionecrosis and osteomyelitis. J. Transl. Med. 2017, 15, 128. [Google Scholar] [CrossRef]
- Wan, J.T.; Sheeley, D.M.; Somerman, M.J.; Lee, J.S. Mitigating osteonecrosis of the jaw (ONJ) through preventive dental care and understanding of risk factors. Bone Res. 2020, 8, 14. [Google Scholar] [CrossRef] [PubMed]
- AlDhalaan, N.A.; BaQais, A.; Al-Omar, A. Medication-related osteonecrosis of the jaw: A review. Cureus 2020, 12, e6944. [Google Scholar] [CrossRef] [PubMed]
- Ro, D.H.; Jin, H.; Park, J.Y.; Lee, M.C.; Won, S.; Han, H.S. The use of bisphosphonates after joint arthroplasty is associated with lower implant revision rate. Knee Surg. Sports Traumatol. Arthrosc. 2019, 27, 2082–2089. [Google Scholar] [CrossRef]
- Kalyan, S.; Wang, J.; Quabius, E.S.; Huck, J.; Wiltfang, J.; Baines, J.F.; Kabelitz, D. Systemic immunity shapes the oral microbiome and susceptibility to bisphosphonate-associated osteonecrosis of the jaw. J. Transl. Med. 2015, 13, 212. [Google Scholar] [CrossRef]
- Hansen, T.; Kunkel, M.; Weber, A.; Kirkpatrick, C.J. Osteonecrosis of the jaws in patients treated with bisphosphonates–histomorphologic analysis in comparison with infected osteoradionecrosis. J. Oral Pathol. Med. 2006, 35, 155–160. [Google Scholar] [CrossRef]
- Rosen, H.N. Risks of Bisphosphonate Therapy in Patients with Osteoporosis; Post, T.W., Ed.; UpToDate: Waltham, MA, USA, 2020. [Google Scholar]
- Kim, J.; Kim, S.W.; Lee, S.Y.; Kim, T.H.; Jung, J.H. Bone mineral density in osteoporotic patients with pyogenic vertebral osteomyelitis: Effect of early versus late treatment for osteoporosis. Osteoporos. Int. 2018, 29, 2761–2770. [Google Scholar] [CrossRef]
- Hallmer, F.; Korduner, M.; Møystad, A.; Bjørnland, T. Treatment of diffuse sclerosing osteomyelitis of the jaw with denosumab shows remarkable results-A report of two cases. Clin. Case Rep. 2018, 6, 2434–2437. [Google Scholar] [CrossRef]
- Otto, S.; Burian, E.; Troeltzsch, M.; Kaeppler, G.; Ehrenfeld, M. Denosumab as a potential treatment alternative for patients suffering from diffuse sclerosing osteomyelitis of the mandible-A rapid communication. J. Craniomaxillofac. Surg. 2018, 46, 534–537. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Ferguson, P.J. Chronic nonbacterial osteomyelitis and chronic recurrent multifocal osteomyelitis in children. Pediatr. Clin. N. Am. 2018, 65, 783–800. [Google Scholar] [CrossRef] [PubMed]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roper, P.M.; Shao, C.; Veis, D.J. Multitasking by the OC Lineage during Bone Infection: Bone Resorption, Immune Modulation, and Microbial Niche. Cells 2020, 9, 2157. https://doi.org/10.3390/cells9102157
Roper PM, Shao C, Veis DJ. Multitasking by the OC Lineage during Bone Infection: Bone Resorption, Immune Modulation, and Microbial Niche. Cells. 2020; 9(10):2157. https://doi.org/10.3390/cells9102157
Chicago/Turabian StyleRoper, Philip M., Christine Shao, and Deborah J. Veis. 2020. "Multitasking by the OC Lineage during Bone Infection: Bone Resorption, Immune Modulation, and Microbial Niche" Cells 9, no. 10: 2157. https://doi.org/10.3390/cells9102157
APA StyleRoper, P. M., Shao, C., & Veis, D. J. (2020). Multitasking by the OC Lineage during Bone Infection: Bone Resorption, Immune Modulation, and Microbial Niche. Cells, 9(10), 2157. https://doi.org/10.3390/cells9102157