T3 Critically Affects the Mhrt/Brg1 Axis to Regulate the Cardiac MHC Switch: Role of an Epigenetic Cross-Talk



 ,
,  ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Design and Animal Models
2.2. Neonatal Rat Cardiomyocyte Cell Culture
2.3. In Silico Analyses
2.3.1. Identification of the Mhrt Coding Sequence in Rattus norvegicus
2.3.2. miRNA Target Prediction
2.3.3. Identification of Putative Thyroid Hormone Response Elements
2.4. RNA Extraction and RT-qPCR
2.5. Mhrt Silencing and miR-Mimic Over-Expression
2.6. Plasmid Construction and Reporter Assays
2.7. Statistical Analysis
3. Results
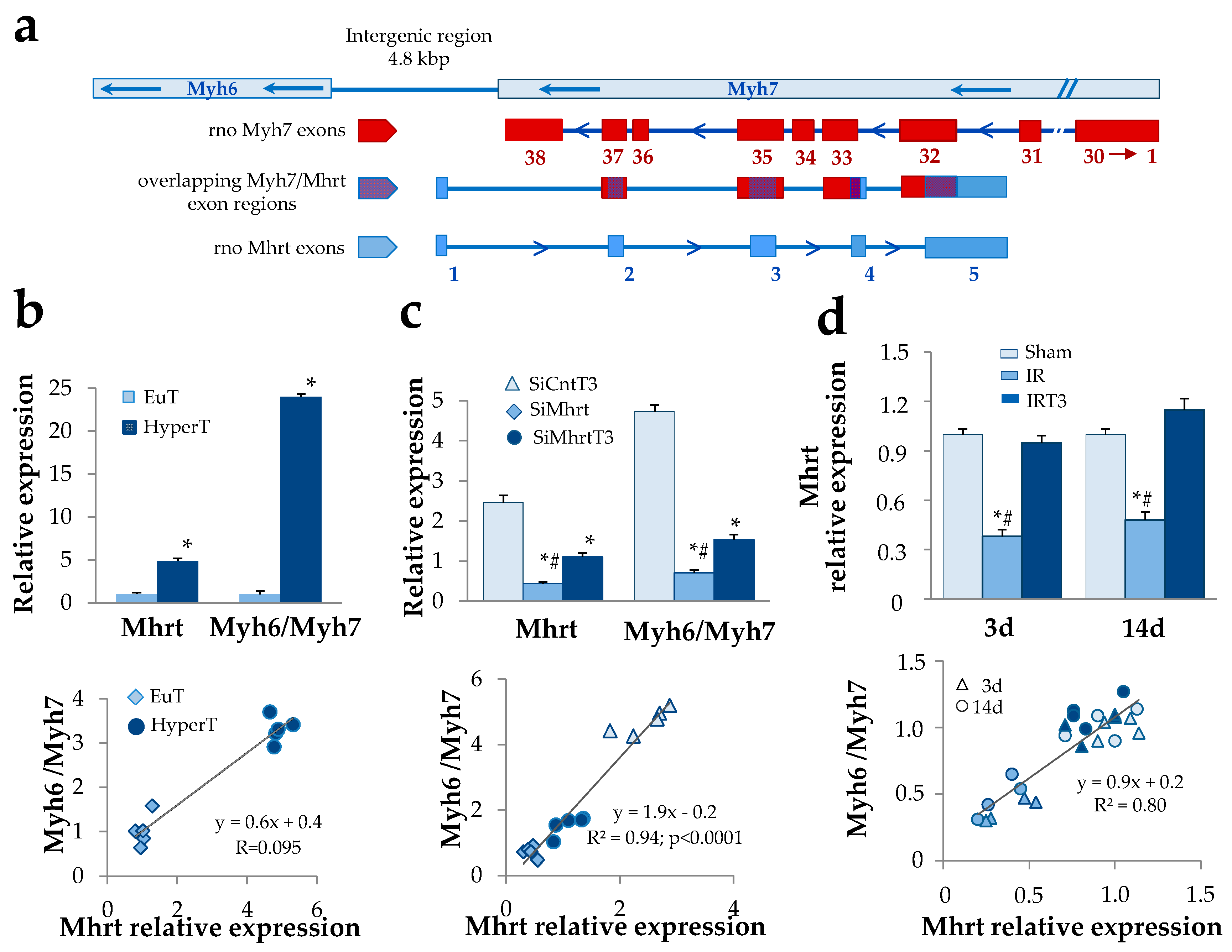
3.1. T3-Dependent Up-Regulation of Mhrt Greatly Contributes to the Cardiac Increase of the Myh6/Myh7 Expression Ratio
3.2. T3 Modulation of Mhrt Promoter and Brg1 Activity Relies on Chromatin Remodeling
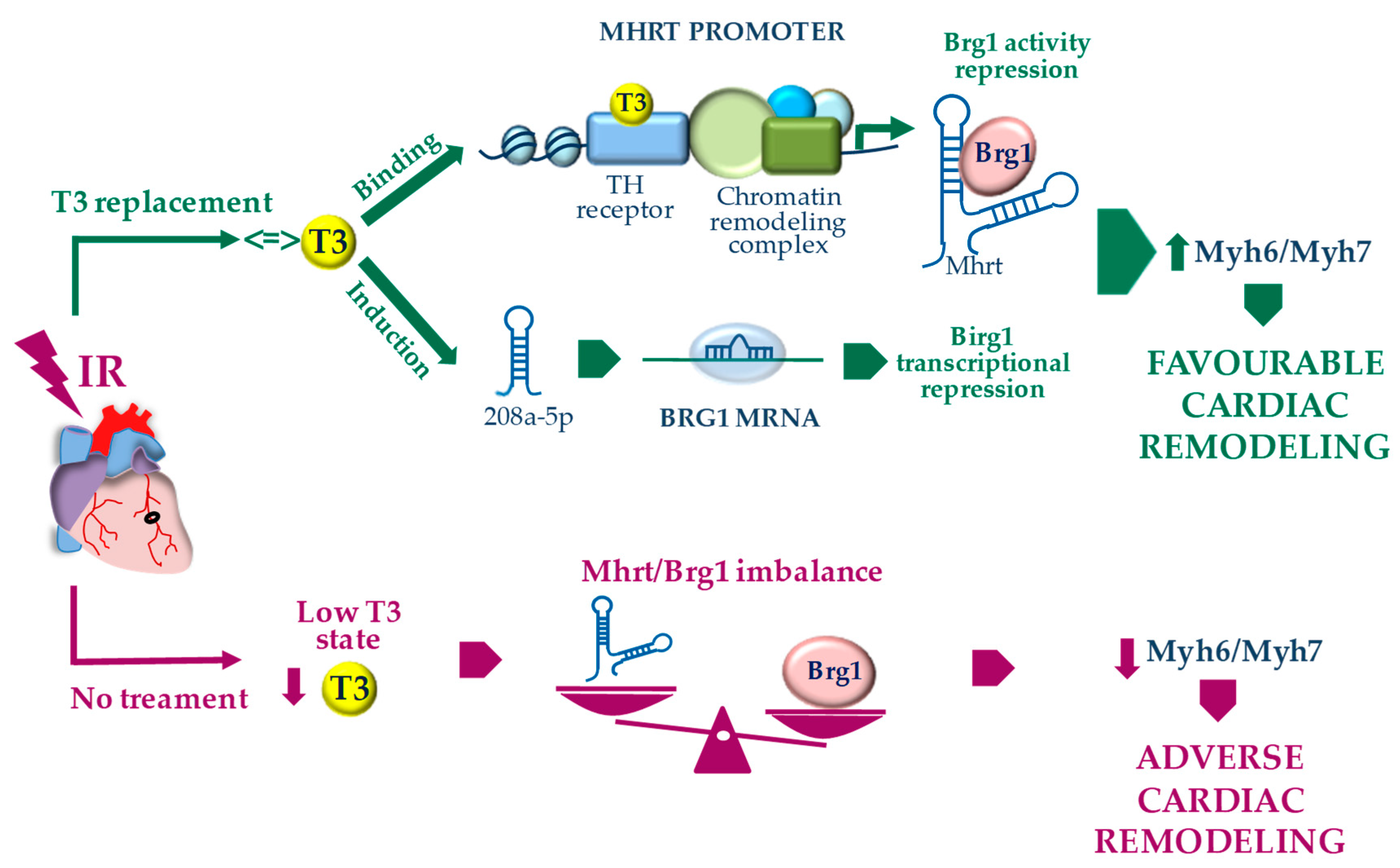
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Velagaleti, R.S.; Gona, P.; Pencina, M.J.; Aragam, J.; Wang, T.J.; Levy, D.; D’Agostino, R.B.; Lee, D.S.; Kannel, W.B.; Benjamin, E.J.; et al. Left ventricular hypertrophy patterns and incidence of heart failure with preserved versus reduced ejection fraction. Am. J. Cardiol. 2014, 113, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Dirkx, E.; da Costa Martins, P.A.; De Windt, L.J. Regulation of fetal gene expression in heart failure. Biochim. Biophys. Acta 2013, 1832, 2414–2424. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, W.; Lin, C.H.; Yang, J.; Shang, C.; Nuernberg, S.T.; Jin, K.K.; Xu, W.; Lin, C.Y.; Lin, C.J.; et al. A long noncoding RNA protects the heart from pathological hypertrophy. Nature 2014, 514, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Hang, C.T.; Yang, J.; Han, P.; Cheng, H.L.; Shang, C.; Ashley, E.; Zhou, B.; Chang, C.P. Chromatin regulation by Brg1 underlies heart muscle development and disease. Nature 2000, 466, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Han, P.; Li, W.; Yang, J.; Shang, C.; Lin, C.H.; Cheng, W.; Hang, C.T.; Cheng, H.L.; Chen, C.H.; Wong, J.; et al. Epigenetic response to environmental stress: Assembly of BRG1-G9a/GLP-DNMT3 repressive chromatin complex on Myh6 promoter in pathologically stressed hearts. Biochim. Biophys. Acta 2016, 1863, 1772–1781. [Google Scholar] [CrossRef]
- Moog, N.K.; Entringer, S.; Heim, C.; Wadhwa, P.D.; Kathmann, N.; Buss, C. Influence of maternal thyroid hormones during gestation on fetal brain development. Neuroscience 2017, 342, 68–100. [Google Scholar] [CrossRef]
- Gerdes, A.M. Restoration of thyroid hormone balance: A game changer in the treatment of heart failure? Am. J. Physiol. Heart Circ. Physiol. 2015, 308, 1–10. [Google Scholar] [CrossRef][Green Version]
- de Vries, E.M.; Fliers, E.; Boelen, A. The molecular basis of the non-thyroidal illness syndrome. J. Endocrinol. 2015, 225, 67–81. [Google Scholar] [CrossRef]
- Janssen, R.; Muller, A.; Simonides, W.S. Cardiac thyroid hormone metabolism and heart failure. Eur. ThyroidJ. 2017, 6, 130–137. [Google Scholar] [CrossRef]
- Nakamura, M.; Sadoshima, J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat. Rev. Cardiol. 2018, 15, 387–407. [Google Scholar] [CrossRef]
- Pingitore, A.; Nicolini, G.; Kusmic, C.; Iervasi, G.; Grigolini, P.; Forini, F. Cardioprotection and thyroid hormones. Heart Fail. Rev. 2016, 21, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Cokkinos, D.V.; Chryssanthopoulos, S. Thyroid hormones and cardiac remodeling. Heart Fail. Rev. 2016, 21, 365–372. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, A.M.; Iervasi, G. Thyroid replacement therapy and heart failure. Circulation 2010, 122, 385–393. [Google Scholar] [CrossRef] [PubMed]
- Jabbar, A.; Pingitore, A.; Pearce, S.H.S.; Zaman, A.; Iervasi, G.; Razvi, S. Thyroid hormones and cardiovascular disease. Nat. Rev. Cardiol. 2017, 14, 39–55. [Google Scholar] [CrossRef]
- Wang, K.; Ojamaa, K.; Samuels, A.; Gilani, N.; Zhang, K.; An, S.; Zhang, Y.; Tang, Y.D.; Askari, B.; Gerdes, A.M. BNP as a New Biomarker of Cardiac Thyroid Hormone Function. Front. Physiol. 2020, 11, 729. [Google Scholar] [CrossRef]
- Donzelli, R.; Colligiani, D.; Kusmic, C.; Sabatini, M.; Lorenzini, L.; Accorroni, A.; Nannipieri, M.; Saba, A.; Iervasi, G.; Zucchi, R. Effect of Hypothyroidism and Hyperthyroidism on Tissue Thyroid Hormone Concentrations in Rat. Eur. Thyroid J. 2016, 5, 27–34. [Google Scholar] [CrossRef][Green Version]
- Forini, F.; Nicolini, G.; Kusmic, C.; D’Aurizio, R.; Rizzo, M.; Baumgart, M.; Groth, M.; Doccini, S.; Iervasi, G.; Pitto, L. Integrative analysis of differentially expressed genes and miRNAs predicts complex T3-mediated protective circuits in a rat model of cardiac ischemia reperfusion. Sci. Rep. 2018, 8, 13870. [Google Scholar] [CrossRef]
- Nicolini, G.; Forini, F.; Kusmic, C.; Pitto, L.; Mariani, L.; Iervasi, G. Early and Short-term Triiodothyronine Supplementation Prevents Adverse Postischemic Cardiac Remodeling: Role of Transforming Growth Factor-β1 and Antifibrotic miRNA Signaling. Mol. Med. 2016, 21, 900–911. [Google Scholar] [CrossRef]
- Olivares, E.L.; Marassi, M.P.; Fortunato, R.S.; da Silva, A.C.; Costa-e-Sousa, R.H.; Araujo, I.G.; Mattos, E.C.; Masuda, M.O.; Mulcahey, M.A.; Huang, S.A.; et al. Thyroid function disturbance and type 3 iodothyronine deiodinase induction after myocardial infarction in rats a time course study. Endocrinology 2007, 148, 4786–4792. [Google Scholar] [CrossRef]
- Poliseno, L.; Evangelista, M.; Mercatanti, A.; Mariani, L.; Citti, L.; Rainaldi, G. The energy profiling of short interfering RNAs is highly predictive of their activity. Oligonucleotides 2004, 14, 227–232. [Google Scholar] [CrossRef]
- Janssen, R.; Zuidwijk, M.J.; Kuster, D.W.; Muller, A.; Simonides, W.S. Thyroid Hormone-Regulated Cardiac microRNAs are Predicted to Suppress Pathological Hypertrophic Signaling. Front. Endocrinol. 2014, 5, 171. [Google Scholar] [CrossRef] [PubMed]
- Forini, F.; Nicolini, G.; Pitto, L.; Iervasi, G. Novel Insight into the Epigenetic and Post-transcriptional Control of Cardiac Gene Expression by Thyroid Hormone. Front. Endocrinol. 2019, 29, 601. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Bodell, P.W.; Qin, A.X.; Giger, J.M.; Baldwin, K.M. Role of antisense RNA in coordinating cardiac myosin heavy chain gene switching. J. Biol. Chem. 2003, 278, 37132–37138. [Google Scholar] [CrossRef]
- Giger, J.; Qin, A.X.; Bodell, P.W.; Baldwin, K.M.; Haddad, F. Activity of the beta-myosin heavy chain antisense promoter responds to diabetes and hypothyroidism. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, 3065–3071. [Google Scholar] [CrossRef] [PubMed]
- Danzi, S.; Klein, S.; Klein, I. Differential regulation of the myosin heavy chain genes alpha and beta in rat atria and ventricles: Role of antisense RNA. Thyroid 2008, 18, 761–768. [Google Scholar] [CrossRef]
- Heimeier, R.A.; Hsia, V.S.; Shi, Y.B. Participation of Brahma-related gene 1 (BRG1)-associated factor 57 and BRG1-containing chromatin remodeling complexes in thyroid hormone-dependent gene activation during vertebrate development. Mol. Endocrinol. 2008, 22, 1065–1077. [Google Scholar] [CrossRef]
- Bedadala, G.R.; Pinnoji, R.C.; Palem, J.R.; Hsia, S.C. Thyroid hormone controls the gene expression of HSV-1 LAT and ICP0 in neuronal cells. Cell Res. 2010, 20, 587–598. [Google Scholar] [CrossRef]
- Gillis, N.E.; Taber, T.H.; Bolf, E.L.; Beaudet, C.M.; Tomczak, J.A.; White, J.H.; Stein, J.L.; Stein, G.S.; Lian, J.B.; Frietze, S.; et al. Thyroid Hormone Receptor β Suppression of RUNX2 Is Mediated by Brahma-Related Gene 1-Dependent Chromatin Remodeling. Endocrinology 2018, 159, 2484–2494. [Google Scholar] [CrossRef]
- Haddad, F.; Jiang, W.; Bodell, P.W.; Qin, A.X.; Baldwin, K.M. Cardiac myosin heavy chain gene regulation by thyroid hormone involves altered histone modifications. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, 1968–1980. [Google Scholar] [CrossRef][Green Version]
- Morrison, E.A.; Sanchez, J.C.; Ronan, J.L.; Farrell, D.P.; Varzavand, K.; Johnson, J.K.; Gu, B.X.; Crabtree, G.R.; Musselman, C.A. DNA binding drives the association of BRG1/hBRM bromodomains with nucleosomes. Nat. Commun. 2017, 8, 16080. [Google Scholar] [CrossRef]
- Pol, C.J.; Muller, A.; Simonides, W.S. Cardiomyocyte-specific inactivation of thyroid hormone in pathologic ventricular hypertrophy: An adaptative response or part of the problem? Heart Fail. Rev. 2010, 15, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Iervasi, G.; Pingitore, A.; Landi, P.; Raciti, M.; Ripoli, A.; Scarlattini, M.; L’Abbate, A.; Donato, L. Low-T3 syndrome: A strong prognostic predictor of death in patients with heart disease. Circulation 2003, 107, 708–713. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Forini, F.; Nicolini, G.; Kusmic, C.; D’Aurizio, R.; Mercatanti, A.; Iervasi, G.; Pitto, L. T3 Critically Affects the Mhrt/Brg1 Axis to Regulate the Cardiac MHC Switch: Role of an Epigenetic Cross-Talk. Cells 2020, 9, 2155. https://doi.org/10.3390/cells9102155
Forini F, Nicolini G, Kusmic C, D’Aurizio R, Mercatanti A, Iervasi G, Pitto L. T3 Critically Affects the Mhrt/Brg1 Axis to Regulate the Cardiac MHC Switch: Role of an Epigenetic Cross-Talk. Cells. 2020; 9(10):2155. https://doi.org/10.3390/cells9102155
Chicago/Turabian StyleForini, Francesca, Giuseppina Nicolini, Claudia Kusmic, Romina D’Aurizio, Alberto Mercatanti, Giorgio Iervasi, and Letizia Pitto. 2020. "T3 Critically Affects the Mhrt/Brg1 Axis to Regulate the Cardiac MHC Switch: Role of an Epigenetic Cross-Talk" Cells 9, no. 10: 2155. https://doi.org/10.3390/cells9102155
APA StyleForini, F., Nicolini, G., Kusmic, C., D’Aurizio, R., Mercatanti, A., Iervasi, G., & Pitto, L. (2020). T3 Critically Affects the Mhrt/Brg1 Axis to Regulate the Cardiac MHC Switch: Role of an Epigenetic Cross-Talk. Cells, 9(10), 2155. https://doi.org/10.3390/cells9102155