The Role of Cyclic AMP Signaling in Cardiac Fibrosis


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Cellular and Molecular Mechanisms Controlling Cardiac Fibrosis
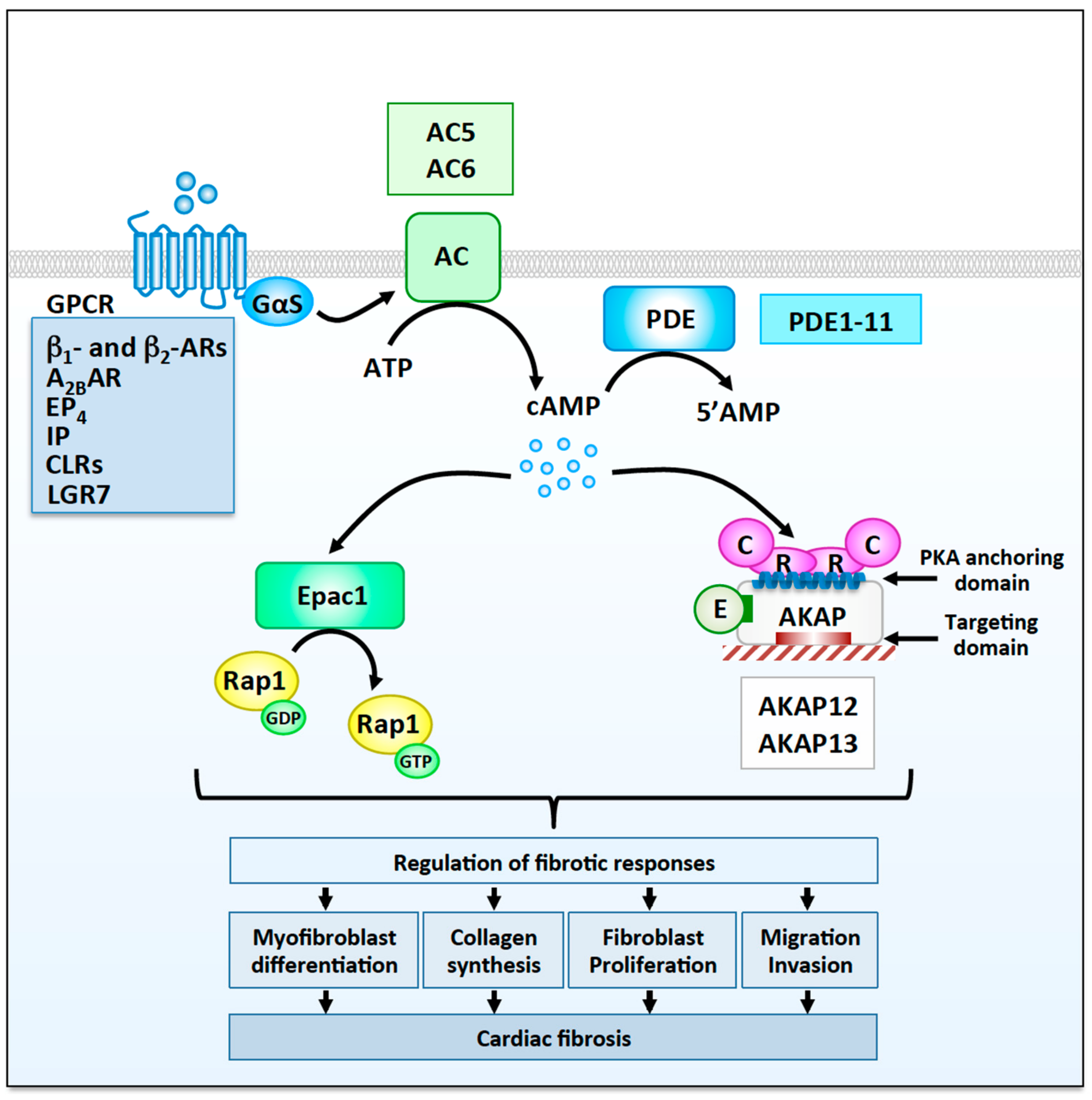
3. The Functional Role of cAMP Signaling in Cardiac Fibroblasts
3.1. G Protein-Coupled Receptors
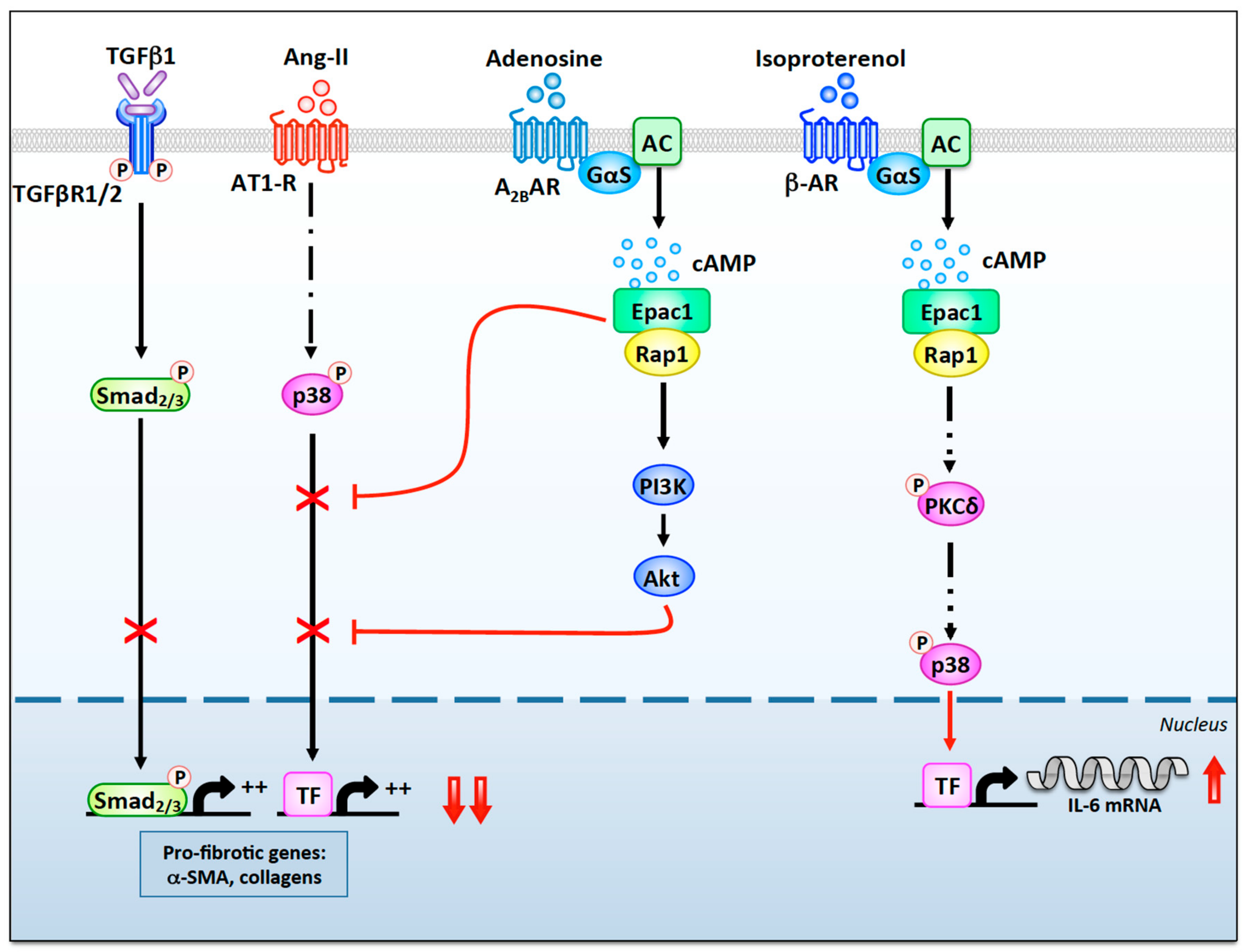
3.1.1. β-Adrenergic Receptors
3.1.2. A2B Adenosine Receptors
3.1.3. Additional Gs-Coupled GPCR Regulating Fibrotic Responses
3.2. cAMP Regulators
3.2.1. Adenylyl Cyclases
3.2.2. Phosphodiesterases
3.3. cAMP Effectors
3.3.1. Exchange Protein Activated by cAMP
3.3.2. Protein Kinase A
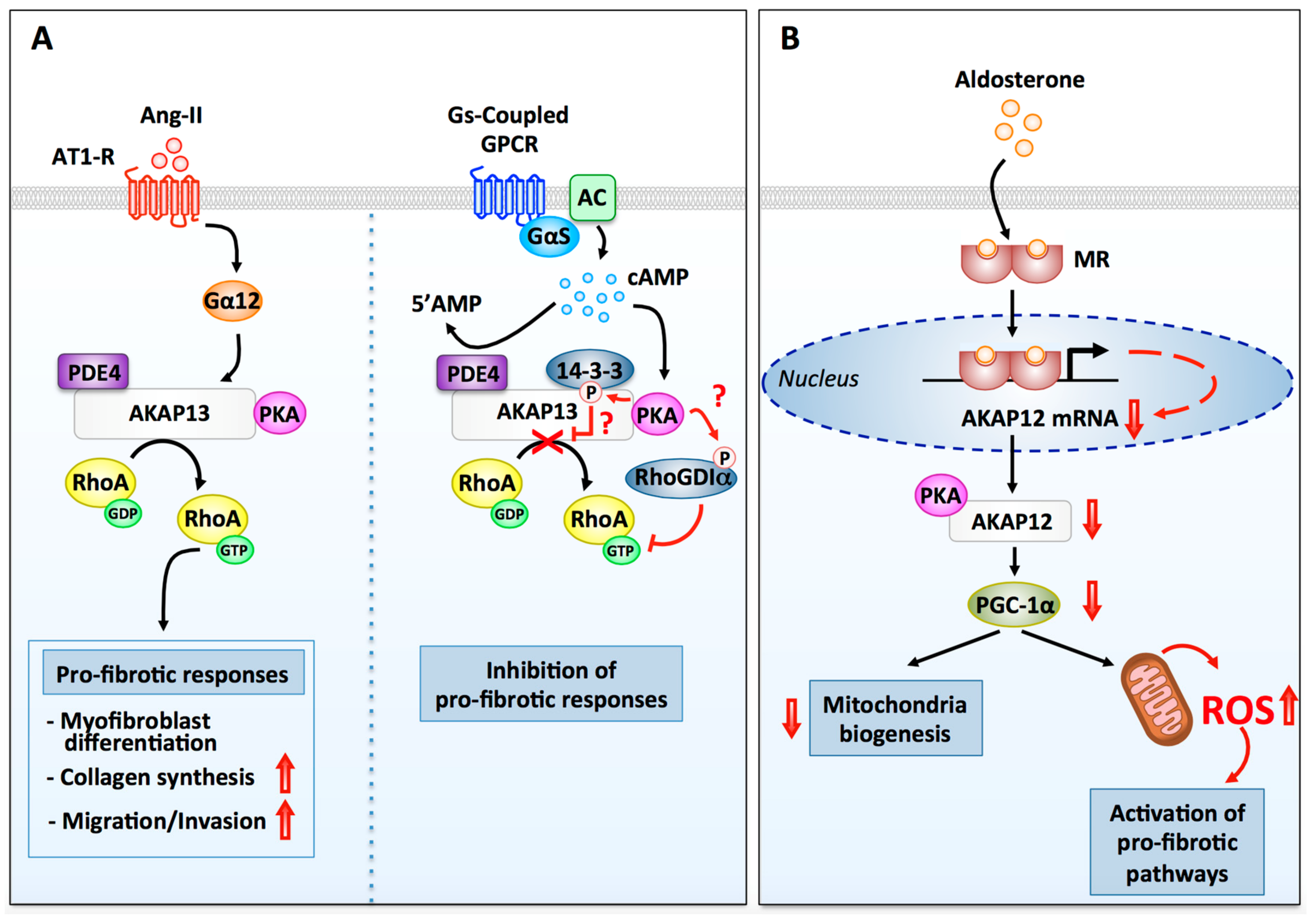
3.3.3. A-Kinase Anchoring Proteins
AKAP13
AKAP12
4. Conclusions and Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Towbin, J.A.; Bowles, N.E. The failing heart. Nature 2002, 415, 227–233. [Google Scholar] [CrossRef] [PubMed]
- Xin, M.; Olson, E.N.; Bassel-Duby, R. Mending broken hearts: Cardiac development as a basis for adult heart regeneration and repair. Nat. Rev. Mol. Cell Biol. 2013, 14, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Hill, J.A.; Olson, E.N. Cardiac plasticity. N. Engl. J. Med. 2008, 358, 1370–1380. [Google Scholar] [CrossRef] [PubMed]
- Barry, S.P.; Townsend, P.A. What causes a broken heart—molecular insights into heart failure. Int. Rev. Cell Mol. Biol. 2010, 284, 113–179. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y. Myocardial repair/remodelling following infarction: Roles of local factors. Cardiovasc. Res. 2009, 81, 482–490. [Google Scholar] [CrossRef]
- Burchfield, J.S.; Xie, M.; Hill, J.A. Pathological ventricular remodeling: Mechanisms: Part 1 of 2. Circulation 2013, 128, 388–400. [Google Scholar] [CrossRef]
- Diviani, D.; Reggi, E.; Arambasic, M.; Caso, S.; Maric, D. Emerging roles of A-kinase anchoring proteins in cardiovascular pathophysiology. Biochim. Biophys. Acta 2016, 1863, 1926–1936. [Google Scholar] [CrossRef]
- Sharma, K.; Kass, D.A. Heart failure with preserved ejection fraction: Mechanisms, clinical features, and therapies. Circ. Res. 2014, 115, 79–96. [Google Scholar] [CrossRef]
- Morissette, M.R.; Rosenzweig, A. Targeting survival signaling in heart failure. Curr. Opin. Pharmacol. 2005, 5, 165–170. [Google Scholar] [CrossRef]
- Frangogiannis, N.G. Cardiac fibrosis: Cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Asp. Med. 2019, 65, 70–99. [Google Scholar] [CrossRef]
- Xie, M.; Burchfield, J.S.; Hill, J.A. Pathological ventricular remodeling: Therapies: Part 2 of 2. Circulation 2013, 128, 1021–1030. [Google Scholar] [CrossRef] [PubMed]
- Aghajanian, H.; Kimura, T.; Rurik, J.G.; Hancock, A.S.; Leibowitz, M.S.; Li, L.; Scholler, J.; Monslow, J.; Lo, A.; Han, W.; et al. Targeting cardiac fibrosis with engineered T cells. Nature 2019, 573, 430–433. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Roth, D.M.; Olson, E.R.; Naugle, J.E.; Meszaros, J.G.; Insel, P.A. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc. Natl. Acad. Sci. USA 2005, 102, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Surinkaew, S.; Aflaki, M.; Takawale, A.; Chen, Y.; Qi, X.Y.; Gillis, M.A.; Shi, Y.F.; Tardif, J.C.; Chattipakorn, N.; Nattel, S. Exchange protein activated by cyclic-adenosine monophosphate (Epac) regulates atrial fibroblast function and controls cardiac remodelling. Cardiovasc. Res. 2019, 115, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Souders, C.A.; Bowers, S.L.; Baudino, T.A. Cardiac fibroblast: The renaissance cell. Circ. Res. 2009, 105, 1164–1176. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Porter, K.E. Function and fate of myofibroblasts after myocardial infarction. Fibrogenesis Tissue Repair 2013, 6, 5. [Google Scholar] [CrossRef] [PubMed]
- Kong, P.; Christia, P.; Frangogiannis, N.G. The pathogenesis of cardiac fibrosis. Cell. Mol. Life Sci. 2014, 71, 549–574. [Google Scholar] [CrossRef] [PubMed]
- Travers, J.G.; Kamal, F.A.; Robbins, J.; Yutzey, K.E.; Blaxall, B.C. Cardiac Fibrosis: The Fibroblast Awakens. Circ. Res. 2016, 118, 1021–1040. [Google Scholar] [CrossRef]
- Davis, J.; Molkentin, J.D. Myofibroblasts: Trust your heart and let fate decide. J. Mol. Cell. Cardiol. 2014, 70, 9–18. [Google Scholar] [CrossRef]
- Kanisicak, O.; Khalil, H.; Ivey, M.J.; Karch, J.; Maliken, B.D.; Correll, R.N.; Brody, M.J.; SC, J.L.; Aronow, B.J.; Tallquist, M.D.; et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 2016, 7, 12260. [Google Scholar] [CrossRef]
- Gonzalez, A.; Lopez, B.; Ravassa, S.; San Jose, G.; Diez, J. The complex dynamics of myocardial interstitial fibrosis in heart failure. Focus on collagen cross-linking. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1421–1432. [Google Scholar] [CrossRef] [PubMed]
- Khalil, H.; Kanisicak, O.; Prasad, V.; Correll, R.N.; Fu, X.; Schips, T.; Vagnozzi, R.J.; Liu, R.; Huynh, T.; Lee, S.J.; et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J. Clin. Investig. 2017, 127, 3770–3783. [Google Scholar] [CrossRef] [PubMed]
- Leask, A. TGFbeta, cardiac fibroblasts, and the fibrotic response. Cardiovasc. Res. 2007, 74, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Molkentin, J.D.; Bugg, D.; Ghearing, N.; Dorn, L.E.; Kim, P.; Sargent, M.A.; Gunaje, J.; Otsu, K.; Davis, J.M. Fibroblast-Specific Genetic Manipulation of p38 MAPK in vivo Reveals its Central Regulatory Role in Fibrosis. Circulation 2017, 136, 549–561. [Google Scholar] [CrossRef]
- Francis, S.H.; Corbin, J.D. Structure and function of cyclic nuleotide-dependent protein kinases. Ann. Rev. Physiol. 1994, 56, 237–272. [Google Scholar] [CrossRef]
- Turnham, R.E.; Scott, J.D. Protein kinase A catalytic subunit isoform PRKACA; History, function and physiology. Gene 2016, 577, 101–108. [Google Scholar] [CrossRef]
- De Rooij, J.; Zwartkruis, F.J.T.; Verheijen, M.H.G.; Cool, R.H.; Nijman, S.M.B.; Wittinghofer, A.; Bos, J.L. Epac is a Rap1 guanine-nucleotide-exchange factor directly activated by cyclic AMP. Nature 1998, 396, 474–477. [Google Scholar] [CrossRef]
- Brand, T. POPDC proteins and cardiac function. Biochem. Soc. Trans. 2019, 47, 1393–1404. [Google Scholar] [CrossRef]
- Sartiani, L.; Romanelli, M.N.; Mugelli, A.; Cerbai, E. Updates on HCN Channels in the Heart: Function, Dysfunction and Pharmacology. Curr. Drug Targets 2015, 16, 868–876. [Google Scholar] [CrossRef]
- Chao, Y.C.; Surdo, N.C.; Pantano, S.; Zaccolo, M. Imaging cAMP nanodomains in the heart. Biochem. Soc. Trans. 2019, 47, 1383–1392. [Google Scholar] [CrossRef]
- Baldwin, T.A.; Dessauer, C.W. Function of Adenylyl Cyclase in Heart: The AKAP Connection. J. Cardiovasc. Dev. Dis. 2018, 5, 2. [Google Scholar] [CrossRef] [PubMed]
- Lomas, O.; Zaccolo, M. Phosphodiesterases Maintain Signaling Fidelity via Compartmentalization of Cyclic Nucleotides. Physiology 2014, 29, 141–149. [Google Scholar] [CrossRef] [PubMed]
- Ercu, M.; Klussmann, E. Roles of A-Kinase Anchoring Proteins and Phosphodiesterases in the Cardiovascular System. J. Cardiovasc. Dev. Dis. 2018, 5, 14. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Dessauer, C.W.; Tasken, K. Creating order from chaos: Cellular regulation by kinase anchoring. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 187–210. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Sun, S.Q.; Hassid, A.; Ostrom, R.S. cAMP inhibits transforming growth factor-beta-stimulated collagen synthesis via inhibition of extracellular signal-regulated kinase 1/2 and Smad signaling in cardiac fibroblasts. Mol. Pharmacol. 2006, 70, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- Horio, T.; Nishikimi, T.; Yoshihara, F.; Matsuo, H.; Takishita, S.; Kangawa, K. Effects of adrenomedullin on cultured rat cardiac myocytes and fibroblasts. Eur. J. Pharmacol. 1999, 382, 1–9. [Google Scholar] [CrossRef]
- Woo, A.Y.H.; Xiao, R.P. beta-Adrenergic receptor subtype signaling in heart: From bench to bedside. Acta Pharmacol. Sin. 2012, 33, 335–341. [Google Scholar] [CrossRef]
- Wang, J.L.; Gareri, C.; Rockman, H.A. G-Protein-Coupled Receptors in Heart Disease. Circ. Res. 2018, 123, 716–735. [Google Scholar] [CrossRef] [PubMed]
- Daaka, Y.; Luttrell, L.M.; Lefkowitz, R.J. Switching of the coupling of the beta2-adrenergic receptor to different G proteins by protein kinase A. Nature 1997, 390, 88–91. [Google Scholar] [CrossRef]
- Fraser, I.; Cong, M.; Kim, J.; Rollins, E.; Daaka, Y.; Lefkowitz, R.; Scott, J. Assembly of an AKAP/beta2-adrenergic receptor signaling complex facilitates receptor phosphorylation and signaling. Curr. Biol. 2000, 10, 409–412. [Google Scholar] [CrossRef]
- Leicht, M.; Greipel, N.; Zimmer, H. Comitogenic effect of catecholamines on rat cardiac fibroblasts in culture. Cardiovasc. Res. 2000, 48, 274–284. [Google Scholar] [CrossRef]
- Kim, J.; Eckhart, A.D.; Eguchi, S.; Koch, W.J. Beta-adrenergic receptor-mediated DNA synthesis in cardiac fibroblasts is dependent on transactivation of the epidermal growth factor receptor and subsequent activation of extracellular signal-regulated kinases. J. Biol. Chem. 2002, 277, 32116–32123. [Google Scholar] [CrossRef] [PubMed]
- Turner, N.A.; Porter, K.E.; Smith, W.H.; White, H.L.; Ball, S.G.; Balmforth, A.J. Chronic beta2-adrenergic receptor stimulation increases proliferation of human cardiac fibroblasts via an autocrine mechanism. Cardiovasc. Res. 2003, 57, 784–792. [Google Scholar] [CrossRef]
- Lv, T.T.; Du, Y.H.; Cao, N.; Zhang, S.L.; Gong, Y.L.; Bai, Y.; Wang, W.; Liu, H.R. Proliferation in cardiac fibroblasts induced by beta(1)-adrenoceptor autoantibody and the underlying mechanisms. Sci. Rep. 2016, 6, 32430. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.; Benicke, M.; Deten, A.; Zimmer, H.G. Catecholamines stimulate interleukin-6 synthesis in rat cardiac fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2001, 281, H14–H21. [Google Scholar] [CrossRef]
- Yin, F.; Wang, Y.Y.; Du, J.H.; Li, C.; Lu, Z.Z.; Han, C.; Zhang, Y.Y. Noncanonical cAMP pathway and p38 MAPK mediate beta2-adrenergic receptor-induced IL-6 production in neonatal mouse cardiac fibroblasts. J. Mol. Cell. Cardiol. 2006, 40, 384–393. [Google Scholar] [CrossRef]
- Aranguiz-Urroz, P.; Canales, J.; Copaja, M.; Troncoso, R.; Vicencio, J.M.; Carrillo, C.; Lara, H.; Lavandero, S.; Diaz-Araya, G. Beta(2)-adrenergic receptor regulates cardiac fibroblast autophagy and collagen degradation. Biochim. Biophys. Acta Mol. Basis Dis. 2011, 1812, 23–31. [Google Scholar] [CrossRef]
- Patterson, A.J.; Zhu, W.; Chow, A.; Agrawal, R.; Kosek, J.; Xiao, R.P.; Kobilka, B. Protecting the myocardium: A role for the beta2 adrenergic receptor in the heart. Crit. Care Med. 2004, 32, 1041–1048. [Google Scholar] [CrossRef]
- Communal, C.; Singh, K.; Sawyer, D.B.; Colucci, W.S. Opposing effects of beta(1)- and beta(2)-adrenergic receptors on cardiac myocyte apoptosis: Role of a pertussis toxin-sensitive G protein. Circulation 1999, 100, 2210–2212. [Google Scholar] [CrossRef]
- Engelhardt, S.; Hein, L.; Wiesmann, F.; Lohse, M.J. Progressive hypertrophy and heart failure in beta1-adrenergic receptor transgenic mice. Proc. Natl. Acad. Sci. USA 1999, 96, 7059–7064. [Google Scholar] [CrossRef]
- Nakaya, M.; Nishida, M.; Kurose, H. Induction of cardiac fibrosis by beta-blocker in G protein-independent but GRK5/beta-arrestin2-dependent signaling pathways. J. Pharmacol. Sci. 2011, 115, 35669–35677. [Google Scholar]
- Brilla, C.G. Regression of myocardial fibrosis in hypertensive heart disease: Diverse effects of various antihypertensive drugs. Cardiovasc. Res. 2000, 46, 324–331. [Google Scholar] [CrossRef][Green Version]
- Vecchio, E.A.; White, P.J.; May, L.T. Targeting Adenosine Receptors for the Treatment of Cardiac Fibrosis. Front. Pharmacol. 2017, 8, 243. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K.; Gillespie, D.G.; Jackson, E.K. Adenosine inhibits collagen and protein synthesis in cardiac fibroblasts: Role of A2B receptors. Hypertension 1998, 31, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K.; Gillespie, D.G.; Mi, Z.; Jackson, E.K. Exogenous and endogenous adenosine inhibits fetal calf serum-induced growth of rat cardiac fibroblasts: Role of A2B receptors. Circulation 1997, 96, 2656–2666. [Google Scholar] [CrossRef] [PubMed]
- Dubey, R.K.; Gillespie, D.G.; Mi, Z.; Jackson, E.K. Endogenous cyclic AMP-adenosine pathway regulates cardiac fibroblast growth. Hypertension 2001, 37, 1095–1100. [Google Scholar] [CrossRef]
- Vecchio, E.A.; Chuo, C.H.; Baltos, J.A.; Ford, L.; Scammells, P.J.; Wang, B.H.; Christopoulos, A.; White, P.J.; May, L.T. The hybrid molecule, VCP746, is a potent adenosine A2B receptor agonist that stimulates anti-fibrotic signalling. Biochem. Pharmacol. 2016, 117, 46–56. [Google Scholar] [CrossRef]
- Villarreal, F.; Epperson, S.A.; Ramirez-Sanchez, I.; Yamazaki, K.G.; Brunton, L.L. Regulation of cardiac fibroblast collagen synthesis by adenosine: Roles for Epac and PI3K. Am. J. Physiol. Cell Physiol. 2009, 296, C1178–C1184. [Google Scholar] [CrossRef]
- Phosri, S.; Arieyawong, A.; Bunrukchai, K.; Parichatikanond, W.; Nishimura, A.; Nishida, M.; Mangmool, S. Stimulation of Adenosine A(2B) Receptor Inhibits Endothelin-1-Induced Cardiac Fibroblast Proliferation and alpha-Smooth Muscle Actin Synthesis Through the cAMP/Epac/PI3K/Akt-Signaling Pathway. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef]
- Phosri, S.; Bunrukchai, K.; Parichatikanond, W.; Sato, V.H.; Mangmool, S. Epac is required for exogenous and endogenous stimulation of adenosine A(2B) receptor for inhibition of angiotensin II-induced collagen synthesis and myofibroblast differentiation. Purinergic Signal. 2018, 14, 141–156. [Google Scholar] [CrossRef]
- Wakeno, M.; Minamino, T.; Seguchi, O.; Okazaki, H.; Tsukamoto, O.; Okada, K.; Hirata, A.; Fujita, M.; Asanuma, H.; Kim, J.; et al. Long-term stimulation of adenosine A2b receptors begun after myocardial infarction prevents cardiac remodeling in rats. Circulation 2006, 114, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Sassi, Y.; Ahles, A.; Truong, D.J.J.; Baqi, Y.; Lee, S.Y.; Husse, B.; Hulot, J.S.; Foinquinos, A.; Thum, T.; Muller, C.E.; et al. Cardiac myocyte-secreted cAMP exerts paracrine action via adenosine receptor activation. J. Clin. Investig. 2014, 124, 5385–5397. [Google Scholar] [CrossRef] [PubMed]
- Toldo, S.; Zhong, H.; Mezzaroma, E.; Van Tassell, B.W.; Kannan, H.; Zeng, D.; Belardinelli, L.; Voelkel, N.F.; Abbate, A. GS-6201, a selective blocker of the A2B adenosine receptor, attenuates cardiac remodeling after acute myocardial infarction in the mouse. J. Pharmacol. Exp. Ther. 2012, 343, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhong, H.; Everett, T.H.t.; Wilson, E.; Chang, R.; Zeng, D.; Belardinelli, L.; Olgin, J.E. Blockade of A2B adenosine receptor reduces left ventricular dysfunction and ventricular arrhythmias 1 week after myocardial infarction in the rat model. Heart Rhythm 2014, 11, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Chan, E.C.; Dusting, G.J.; Guo, N.; Peshavariya, H.M.; Taylor, C.J.; Dilley, R.; Narumiya, S.; Jiang, F. Prostacyclin receptor suppresses cardiac fibrosis: Role of CREB phosphorylation. J. Mol. Cell. Cardiol. 2010, 49, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Oka, T.; Yamagami, K.; Lee, J.K.; Akazawa, H.; Naito, A.T.; Yasui, T.; Ishizu, T.; Nakaoka, Y.; Sakata, Y.; et al. An EP4 Receptor Agonist Inhibits Cardiac Fibrosis Through Activation of PKA Signaling in Hypertrophied Heart. Int. Heart J. 2017, 58, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yang, S.J.; Yao, W.J.; Zhu, H.Y.; Xu, X.L.; Meng, G.L.; Zhang, W. Prostacyclin Analogue Beraprost Inhibits Cardiac Fibroblast Proliferation Depending on Prostacyclin Receptor Activation through a TGF beta-Smad Signal Pathway. PLoS ONE 2014, 9, e98483. [Google Scholar] [CrossRef]
- Shindo, T.; Tanaka, M.; Kamiyoshi, A.; Ichikawa-Shindo, Y.; Kawate, H.; Yamauchi, A.; Sakurai, T. Regulation of cardiovascular development and homeostasis by the adrenomedullin-RAMP system. Peptides 2019, 111, 55–61. [Google Scholar] [CrossRef]
- Yang, J.H.; Cai, Y.; Duan, X.H.; Ma, C.G.; Wang, X.; Tang, C.S.; Qi, Y.F. Intermedin 1-53 inhibits rat cardiac fibroblast activation induced by angiotensin II. Regul. Pept. 2009, 158, 19–25. [Google Scholar] [CrossRef]
- Nishikimi, T.; Tadokoro, K.; Akimoto, K.; Mori, Y.; Ishikawa, Y.; Ishimura, K.; Horio, T.; Kangawa, K.; Matsuoka, H. Response of adrenomedullin system to cytokine in cardiac fibroblasts-role of adrenomedullin as an antifibrotic factor. Cardiovasc. Res. 2005, 66, 104–113. [Google Scholar] [CrossRef][Green Version]
- Samuel, C.S.; Unemori, E.N.; Mookerjee, I.; Bathgate, R.A.; Layfield, S.L.; Mak, J.; Tregear, G.W.; Du, X.J. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology 2004, 145, 4125–4133. [Google Scholar] [CrossRef] [PubMed]
- Sadana, R.; Dessauer, C.W. Physiological Roles for G Protein-Regulated Adenylyl Cyclase Isoforms: Insights from Knockout and Overexpression Studies. Neurosignals 2009, 17, 5–22. [Google Scholar] [CrossRef] [PubMed]
- Schmid, A.; Meili, D.; Salathe, M. Soluble adenylyl cyclase in health and disease. Biochim. Biophys. Acta 2014, 1842, 2584–2592. [Google Scholar] [CrossRef] [PubMed]
- Efendiev, R.; Dessauer, C.W. AKAPs and Adenylyl Cyclase in Cardiovascular Physiology and Pathology. J. Cardiovasc. Pharmacol. 2011, 58, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, R.S.; Naugle, J.E.; Hase, M.; Gregorian, C.; Swaney, J.S.; Insel, P.A.; Brunton, L.L.; Meszaros, J.G. Angiotensin II enhances adenylyl cyclase signaling via Ca2+/calmodulin. Gq-Gs cross-talk regulates collagen production in cardiac fibroblasts. J. Biol. Chem. 2003, 278, 24461–24468. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Kawabe, J.-I.; Yatani, A.; Takagi, G.; Lee, M.-C.; Hong, C.; Liu, J.; Takagi, I.; Sadoshima, J.; Vatner, D.E.; et al. Type 5 adenylyl cyclase disruption alters not only sympathetic but also parasympathetic and calcium-mediated cardiac regulation. Circ. Res. 2003, 93, 364–371. [Google Scholar] [CrossRef]
- Swaney, J.S.; Patel, H.H.; Yokoyama, U.; Head, B.P.; Roth, D.M.; Insel, P.A. Focal adhesions in (myo)fibroblasts scaffold adenylyl cyclase with phosphorylated caveolin. J. Biol. Chem. 2006, 281, 17173–17179. [Google Scholar] [CrossRef]
- Swaney, J.S.; Patel, H.H.; Yokoyama, U.; Lai, N.C.; Spellman, M.; Insel, P.A.; Roth, D.M. Adenylyl cyclase activity and function are decreased in rat cardiac fibroblasts after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3216–H3220. [Google Scholar] [CrossRef]
- Takahashi, T.; Tang, T.; Lai, N.C.; Roth, D.M.; Rebodello, B.; Saito, M.; Lew, W.Y.W.; Clopton, P.; Hammond, H.K. Increased cardiac adenylyl cyclase expression is associated with increased survival after myocardial infarction. Circulation 2006, 114, 388–396. [Google Scholar] [CrossRef]
- Guellich, A.; Gao, S.; Hong, C.; Yan, L.; Wagner, T.E.; Dhar, S.K.; Ghaleh, B.; Hittinger, L.; Iwatsubo, K.; Ishikawa, Y.; et al. Effects of cardiac overexpression of type 6 adenylyl cyclase affects on the response to chronic pressure overload. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H707–H712. [Google Scholar] [CrossRef]
- Hammond, H.K.; Penny, W.F.; Traverse, J.H.; Henry, T.D.; Watkins, M.W.; Yancy, C.W.; Sweis, R.N.; Adler, E.D.; Patel, A.N.; Murray, D.R.; et al. Intracoronary Gene Transfer of Adenylyl Cyclase 6 in Patients With Heart Failure. JAMA Cardiol. 2016, 1, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Takagi, G.; Kawabe, J.-i.; Yang, G.; Lee, M.-C.; Hong, C.; Liu, J.; Vatner, D.E.; Sadoshima, J.; Vatner, S.F.; et al. Disruption of type 5 adenylyl cyclase gene preserves cardiac function against pressure overload. Proc. Natl. Acad. Sci. USA 2003, 100, 9986–9990. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Vatner, D.E.; Kurotani, R.; Bai, Y.; Gao, S.; Yuan, Z.; Iwatsubo, K.; Ulucan, C.; Kawabe, J.-i.; Ghosh, K.; et al. Disruption of type 5 adenylyl cyclase enhances desensitization of cyclic adenosine monophosphate signal and increases Akt signal with chronic catecholamine stress. Circulation 2007, 116, 1776–1783. [Google Scholar] [CrossRef] [PubMed]
- Iwatsubo, K.; Minamisawa, S.; Tsunematsu, T.; Nakagome, M.; Toya, Y.; Tomlinson, J.E.; Umemura, S.; Scarborough, R.M.; Levy, D.E.; Ishikawa, Y. Direct inhibition of type 5 adenylyl cyclase prevents myocardial apoptosis without functional deterioration. J. Biol. Chem. 2004, 279, 40938–40945. [Google Scholar] [CrossRef]
- Pavan, B.; Biondi, C.; Dalpiaz, A. Adenylyl cyclases as innovative therapeutic goals. Drug Discov. Today 2009, 14, 982–991. [Google Scholar] [CrossRef]
- Pierre, S.; Eschenhagen, T.; Geisslinger, G.; Scholich, K. Capturing adenylyl cyclases as potential drug targets. Nat. Rev. Drug Discov. 2009, 8, 321–335. [Google Scholar] [CrossRef]
- Ho, D.; Yan, L.; Iwatsubo, K.; Vatner, D.E.; Vatner, S.F. Modulation of beta-adrenergic receptor signaling in heart failure and longevity: Targeting adenylyl cyclase type 5. Heart Fail. Rev. 2010, 15, 495–512. [Google Scholar] [CrossRef]
- Zhang, J.; Levy, D.; Oydanich, M.; Bravo, C.A.; Yoon, S.; Vatner, D.E.; Vatner, S.F. A novel adenylyl cyclase type 5 inhibitor that reduces myocardial infarct size even when administered after coronary artery reperfusion. J. Mol. Cell. Cardiol. 2018, 121, 13–15. [Google Scholar] [CrossRef]
- Bender, A.T.; Beavo, J.A. Cyclic nucleotide phosphodiesterases: Molecular regulation to clinical use. Pharmacol. Rev. 2006, 58, 488–520. [Google Scholar] [CrossRef]
- Lugnier, C. Cyclic nucleotide phosphodiesterase (PDE) superfamily: A new target for the development of specific therapeutic agents. Pharmacol. Ther. 2006, 109, 366–398. [Google Scholar] [CrossRef]
- Miller, C.L.; Cai, Y.; Oikawa, M.; Thomas, T.; Dostmann, W.R.; Zaccolo, M.; Fujiwara, K.; Yan, C. Cyclic nucleotide phosphodiesterase 1A: A key regulator of cardiac fibroblast activation and extracellular matrix remodeling in the heart. Basic Res. Cardiol. 2011, 106, 1023–1039. [Google Scholar] [CrossRef] [PubMed]
- Brescia, M.; Zaccolo, M. Modulation of Compartmentalised Cyclic Nucleotide Signalling via Local Inhibition of Phosphodiesterase Activity. Int. J. Mol. Sci. 2016, 17, 1672. [Google Scholar] [CrossRef] [PubMed]
- Vettel, C.; Lämmle, S.; Ewens, S.; Cervirgen, C.; Emons, J.; Ongherth, A.; Dewenter, M.; Lindner, D.; Westermann, D.; Nikolaev, V.O.; et al. PDE2-mediated cAMP hydrolysis accelerates cardiac fibroblast to myofibroblast conversion and is antagonized by exogenous activation of cGMP signaling pathways. Am. J. Physiol. Heart Circ. Physiol. 2014, 306, H1246–H1252. [Google Scholar] [CrossRef] [PubMed]
- Knight, W.E.; Chen, S.; Zhang, Y.; Oikawa, M.; Wu, M.; Zhou, Q.; Miller, C.L.; Cai, Y.; Mickelsen, D.M.; Moravec, C.; et al. PDE1C deficiency antagonizes pathological cardiac remodeling and dysfunction. Proc. Natl. Acad. Sci. USA 2016, 113, E7116–E7125. [Google Scholar] [CrossRef]
- Pandit, J.; Forman, M.D.; Fennell, K.F.; Dillman, K.S.; Menniti, F.S. Mechanism for the allosteric regulation of phosphodiesterase 2A deduced from the X-ray structure of a near full-length construct. Proc. Natl. Acad. Sci. USA 2009, 106, 18225–18230. [Google Scholar] [CrossRef]
- Mehel, H.; Emons, J.; Vettel, C.; Wittkopper, K.; Seppelt, D.; Dewenter, M.; Lutz, S.; Sossalla, S.; Maier, L.S.; Lechene, P.; et al. Phosphodiesterase-2 is up-regulated in human failing hearts and blunts beta-adrenergic responses in cardiomyocytes. J. Am. Coll. Cardiol. 2013, 62, 1596–1606. [Google Scholar] [CrossRef]
- Baliga, R.S.; Preedy, M.E.J.; Dukinfield, M.S.; Chu, S.M.; Aubdool, A.A.; Bubb, K.J.; Moyes, A.J.; Tones, M.A.; Hobbs, A.J. Phosphodiesterase 2 inhibition preferentially promotes NO/guanylyl cyclase/cGMP signaling to reverse the development of heart failure. Proc. Natl. Acad. Sci. USA 2018, 115, E7428–E7437. [Google Scholar] [CrossRef]
- Chen, S.; Zhang, Y.; Lighthouse, J.K.; Mickelsen, D.M.; Wu, J.; Yao, P.; Small, E.M.; Yan, C. A Novel Role of Cyclic Nucleotide Phosphodiesterase 10A in Pathological Cardiac Remodeling and Dysfunction. Circulation 2019. [Google Scholar] [CrossRef]
- Patrucco, E.; Domes, K.; Sbroggio, M.; Blaich, A.; Schlossmann, J.; Desch, M.; Rybalkin, S.D.; Beavo, J.A.; Lukowski, R.; Hofmann, F. Roles of cGMP-dependent protein kinase I (cGKI) and PDE5 in the regulation of Ang II-induced cardiac hypertrophy and fibrosis. Proc. Natl. Acad. Sci. USA 2014, 111, 12925–12929. [Google Scholar] [CrossRef]
- Nakamura, T.; Zhu, G.; Ranek, M.J.; Kokkonen-Simon, K.; Zhang, M.; Kim, G.E.; Tsujita, K.; Kass, D.A. Prevention of PKG-1alpha Oxidation Suppresses Antihypertrophic/Antifibrotic Effects From PDE5 Inhibition but not sGC Stimulation. Circ. Heart Fail. 2018, 11, e004740. [Google Scholar] [CrossRef]
- Kawasaki, H.; Springett, G.M.; Mochizuki, N.; Toki, S.; Nakaya, M.; Matsuda, M.; Housman, D.E.; Graybiel, A.M. A family of cAMP-binding proteins that directly activate Rap1. Science 1998, 282, 2275–2279. [Google Scholar] [CrossRef] [PubMed]
- De Rooij, J.; Rehmann, H.; van Triest, M.; Cool, R.H.; Wittinghofer, A.; Bos, J.L. Mechanism of regulation of the Epac family of cAMP-dependent RapGEFs. J. Biol. Chem. 2000, 275, 20829–20836. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Epac proteins: Multi-purpose cAMP targets. Trends Biochem. Sci. 2006, 31, 680–686. [Google Scholar] [CrossRef] [PubMed]
- Robichaux, W.G.; Cheng, X. Intracellular cAMP Sensor EPAC: Physiology, Pathophysiology, and Therapeutics Development. Physiol. Rev. 2018, 98, 919–1053. [Google Scholar] [CrossRef] [PubMed]
- Metrich, M.; Lucas, A.; Gastineau, M.; Samuel, J.L.; Heymes, C.; Morel, E.; Lezoualc’h, F. Epac mediates beta-adrenergic receptor-induced cardiomyocyte hypertrophy. Circ. Res. 2008, 102, 959–965. [Google Scholar] [CrossRef]
- Insel, P.A.; Murray, F.; Yokoyama, U.; Romano, S.; Yun, H.; Brown, L.; Snead, A.; Lu, D.; Aroonsakool, N. cAMP and Epac in the regulation of tissue fibrosis. Br. J. Pharmacol. 2012, 166, 447–456. [Google Scholar] [CrossRef]
- Barker, G.; Parnell, E.; van Basten, B.; Buist, H.; Adams, D.R.; Yarwood, S.J. The Potential of a Novel Class of EPAC-Selective Agonists to Combat Cardiovascular Inflammation. J. Cardiovasc. Dev. Dis. 2017, 4, 22. [Google Scholar] [CrossRef]
- Brette, F.; Blandin, E.; Simard, C.; Guinamard, R.; Salle, L. Epac activator critically regulates action potential duration by decreasing potassium current in rat adult ventricle. J. Mol. Cell. Cardiol. 2013, 57, 96–105. [Google Scholar] [CrossRef]
- Lezoualc’h, F.; Fazal, L.; Laudette, M.; Conte, C. Cyclic AMP Sensor EPAC Proteins and Their Role in Cardiovascular Function and Disease. Circ. Res. 2016, 118, 881–897. [Google Scholar] [CrossRef]
- Yokoyama, U.; Patel, H.H.; Lai, N.C.; Aroonsakool, N.; Roth, D.M.; Insel, P.A. The cyclic AMP effector Epac integrates pro- and anti-fibrotic signals. Proc. Natl. Acad. Sci. USA 2008, 105, 6386–6391. [Google Scholar] [CrossRef]
- Chen, C.; Du, J.H.; Feng, W.; Song, Y.; Lu, Z.Z.; Xu, M.; Li, Z.J.; Zhang, Y.Y. Beta-Adrenergic receptors stimulate interleukin-6 production through Epac-dependent activation of PKCd/p38 MAPK signalling in neonatal mouse cardiac fibroblasts. Br. J. Pharmacol. 2012, 166, 676–688. [Google Scholar] [CrossRef] [PubMed]
- Okumura, S.; Fujita, T.; Cai, W.Q.; Jin, M.H.; Namekata, I.; Mototani, Y.; Jin, H.L.; Ohnuki, Y.; Tsuneoka, Y.; Kurotani, R.; et al. Epac1-dependent phospholamban phosphorylation mediates the cardiac response to stresses. J. Clin. Investig. 2014, 124, 2785–2801. [Google Scholar] [CrossRef] [PubMed]
- Laudette, M.; Coluccia, A.; Sainte-Marie, Y.; Solari, A.; Fazal, L.; Sicard, P.; Silvestri, R.; Mialet-Perez, J.; Pons, S.; Ghaleh, B.; et al. Identification of a pharmacological inhibitor of Epac1 that protects the heart against acute and chronic models of cardiac stress. Cardiovasc. Res. 2019, 115, 1766–1777. [Google Scholar] [CrossRef] [PubMed]
- Poppe, H.; Rybalkin, S.D.; Rehmann, H.; Hinds, T.R.; Tang, X.B.; Christensen, A.E.; Schwede, F.; Genieser, H.G.; Bos, J.L.; Doskeland, S.O.; et al. Cyclic nucleotide analogs as probes of signaling pathways. Nat. Methods 2008, 5, 277–278. [Google Scholar] [CrossRef]
- Enserink, J.M.; Christensen, A.E.; de Rooij, J.; van Triest, M.; Schwede, F.; Genieser, H.G.; Doskeland, S.O.; Blank, J.L.; Bos, J.L. A novel Epac-specific cAMP analogue demonstrates independent regulation of Rap1 and ERK. Nat. Cell Biol. 2002, 4, 901–906. [Google Scholar] [CrossRef]
- Vliem, M.J.; Ponsioen, B.; Schwede, F.; Pannekoek, W.J.; Riedl, J.; Kooistra, M.R.H.; Jalink, K.; Genieser, H.G.; Bos, J.L.; Rehmann, H. 8-pCPT-2’-O-Me-cAMP-AM: An improved Epac-selective cAMP analogue. Chembiochem 2008, 9, 2052–2054. [Google Scholar] [CrossRef]
- Wiejak, J.; van Basten, B.; Luchowska-Stanska, U.; Hamilton, G.; Yarwood, S.J. The novel exchange protein activated by cyclic AMP 1 (EPAC1) agonist, I942, regulates inflammatory gene expression in human umbilical vascular endothelial cells (HUVECs). Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 264–276. [Google Scholar] [CrossRef]
- Beck, E.M.; Parnell, E.; Cowley, A.; Porter, A.; Gillespie, J.; Robinson, J.; Robinson, L.; Pannifer, A.D.; Hamon, V.; Jones, P.; et al. Identification of A Novel Class of Benzofuran Oxoacetic Acid-Derived Ligands that Selectively Activate Cellular EPAC1. Cells 2019, 8, 1425. [Google Scholar] [CrossRef]
- Dema, A.; Perets, E.; Schulz, M.S.; Deak, V.A.; Klussmann, E. Pharmacological targeting of AKAP-directed compartmentalized cAMP signalling. Cell. Signal. 2015, 27, 2474–2487. [Google Scholar] [CrossRef]
- Kemp, B.E.; Benjamini, E.; Krebs, E.G. Synthetic hexapeptide substrates and inhibitors of 3’:5’-cyclic AMP-dependent protein kinase. Proc. Natl. Acad. Sci. USA 1976, 73, 1038–1042. [Google Scholar] [CrossRef]
- Smith, F.D.; Esseltine, J.L.; Nygren, P.J.; Veesler, D.; Byrne, D.P.; Vonderach, M.; Strashnov, I.; Eyers, C.E.; Eyers, P.A.; Langeberg, L.K.; et al. Local protein kinase A action proceeds through intact holoenzymes. Science 2017, 356, 1288–1293. [Google Scholar] [CrossRef] [PubMed]
- Torres-Quesada, O.; Mayrhofer, J.E.; Stefan, E. The many faces of compartmentalized PKA signalosomes. Cell. Signal. 2017, 37, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Dodge-Kafka, K.L.; Li, J.; Kapiloff, M.S. A-kinase anchoring proteins: Scaffolding proteins in the heart. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1742–H1753. [Google Scholar] [CrossRef] [PubMed]
- Ostrom, R.S.; Gregorian, C.; Drenan, R.M.; Xiang, Y.; Regan, J.W.; Insel, P.A. Receptor number and caveolar co-localization determine receptor coupling efficiency to adenylyl cyclase. J. Biol. Chem. 2001, 276, 42063–42069. [Google Scholar] [CrossRef] [PubMed]
- Patel, H.H.; Murray, F.; Insel, P.A. G-protein-coupled receptor-signaling components in membrane raft and caveolae microdomains. Handb. Exp. Pharmacol. 2008, 167–184. [Google Scholar] [CrossRef]
- Zaccolo, M. cAMP signal transduction in the heart: Understanding spatial control for the development of novel therapeutic strategies. Br. J. Pharmacol. 2009, 158, 50–60. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Li, Y.; Kim, S.; Fu, Q.; Parikh, D.; Sridhar, B.; Shi, Q.; Zhang, X.; Guan, Y.; Chen, X.; et al. Phosphodiesterases coordinate cAMP propagation induced by two stimulatory G protein-coupled receptors in hearts. Proc. Natl. Acad. Sci. USA 2012, 109, 6578–6583. [Google Scholar] [CrossRef]
- Oishi, A.; Makita, N.; Sato, J.; Iiri, T. Regulation of RhoA Signaling by the cAMP-dependent Phosphorylation of RhoGDI alpha. J. Biol. Chem. 2012, 287, 38705–38715. [Google Scholar] [CrossRef]
- Esseltine, J.L.; Scott, J.D. AKAP signaling complexes: Pointing towards the next generation of therapeutic targets? Trends Pharmacol. Sci. 2013, 34, 648–655. [Google Scholar] [CrossRef]
- Langeberg, L.K.; Scott, J.D. Signalling scaffolds and local organization of cellular behaviour. Nat. Rev. Mol. Cell Biol. 2015, 16, 232–244. [Google Scholar] [CrossRef]
- Klussmann, E. Protein-protein interactions of PDE4 family members—Functions, interactions and therapeutic value. Cell. Signal. 2016, 28, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Dessauer, C.W. Adenylyl cyclase—A-kinase anchoring protein complexes: The next dimension in cAMP signaling. Mol. Pharmacol. 2009, 76, 935–941. [Google Scholar] [CrossRef] [PubMed]
- Kapiloff, M.S.; Piggott, L.A.; Sadana, R.; Li, J.; Heredia, L.A.; Henson, E.; Efendiev, R.; Dessauer, C.W. An adenylyl cyclase-mAKAPbeta signaling complex regulates cAMP levels in cardiac myocytes. J. Biol. Chem. 2009, 284, 23540–23546. [Google Scholar] [CrossRef] [PubMed]
- Carr, D.W.; Stofko-Hahn, R.E.; Fraser, I.D.; Bishop, S.M.; Acott, T.S.; Brennan, R.G.; Scott, J.D. Interaction of the regulatory subunit (RII) of cAMP-dependent protein kinase with RII-anchoring proteins occurs through an amphipathic helix binding motif. J. Biol. Chem. 1991, 266, 14188–14192. [Google Scholar]
- Gold, M.G.; Lygren, B.; Dokurno, P.; Hoshi, N.; McConnachie, G.; Tasken, K.; Carlson, C.R.; Scott, J.D.; Barford, D. Molecular basis of AKAP specificity for PKA regulatory subunits. Mol. Cell 2006, 24, 383–395. [Google Scholar] [CrossRef]
- Kinderman, F.S.; Kim, C.; von Daake, S.; Ma, Y.; Pham, B.Q.; Spraggon, G.; Xuong, N.H.; Jennings, P.A.; Taylor, S.S. A dynamic mechanism for AKAP binding to RII isoforms of cAMP-dependent protein kinase. Mol. Cell 2006, 24, 397–408. [Google Scholar] [CrossRef]
- Ruppelt, A.; Mosenden, R.; Gronholm, M.; Aandahl, E.M.; Tobin, D.; Carlson, C.R.; Abrahamsen, H.; Herberg, F.W.; Carpen, O.; Tasken, K. Inhibition of T cell activation by cyclic adenosine 5’-monophosphate requires lipid raft targeting of protein kinase A type I by the A-kinase anchoring protein ezrin. J. Immunol. 2007, 179, 5159–5168. [Google Scholar] [CrossRef]
- Pidoux, G.; Witczak, O.; Jarnaess, E.; Myrvold, L.; Urlaub, H.; Stokka, A.J.; Kuntziger, T.; Tasken, K. Optic atrophy 1 is an A-kinase anchoring protein on lipid droplets that mediates adrenergic control of lipolysis. EMBO J. 2011, 30, 4371–4386. [Google Scholar] [CrossRef]
- Means, C.K.; Lygren, B.; Langeberg, L.K.; Jain, A.; Dixon, R.E.; Vega, A.L.; Gold, M.G.; Petrosyan, S.; Taylor, S.S.; Murphy, A.N.; et al. An entirely specific type I A-kinase anchoring protein that can sequester two molecules of protein kinase A at mitochondria. Proc. Natl. Acad. Sci. USA 2011, 108, E1227–E1235. [Google Scholar] [CrossRef]
- Diviani, D.; Maric, D.; Perez Lopez, I.; Cavin, S.; Del Vescovo, C.D. A-kinase anchoring proteins: Molecular regulators of the cardiac stress response. Biochim. Biophys. Acta 2013, 1833, 901–908. [Google Scholar] [CrossRef]
- Kritzer, M.D.; Li, J.; Passariello, C.L.; Gayanilo, M.; Thakur, H.; Dayan, J.; Dodge-Kafka, K.; Kapiloff, M.S. The scaffold protein muscle A-kinase anchoring protein beta orchestrates cardiac myocyte hypertrophic signaling required for the development of heart failure. Circ. Heart Fail. 2014, 7, 663–672. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.D.; Santana, L.F. A-kinase anchoring proteins: Getting to the heart of the matter. Circulation 2010, 121, 1264–1271. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Osman, H.; Delaunay, M.; Kaiser, S. The role of A-kinase anchoring proteins in cardiac oxidative stress. Biochem. Soc. Trans. 2019, 47, 1341–1353. [Google Scholar] [CrossRef] [PubMed]
- Dodge-Kafka, K.; Gildart, M.; Tokarski, K.; Kapiloff, M.S. mAKAPbeta signalosomes—A nodal regulator of gene transcription associated with pathological cardiac remodeling. Cell. Signal. 2019, 63, 109357. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Boccella, N.; Paolillo, R.; Cattaneo, F.; Trimarco, V.; Franzone, A.; D’Apice, S.; Giugliano, G.; Rinaldi, L.; Borzacchiello, D.; et al. Loss of Akap1 Exacerbates Pressure Overload-Induced Cardiac Hypertrophy and Heart Failure. Front. Physiol. 2018, 9, 558. [Google Scholar] [CrossRef]
- Diviani, D.; Soderling, J.; Scott, J.D. AKAP-Lbc anchors protein kinase A and nucleates Galpha 12-selective Rho-mediated stress fiber formation. J. Biol. Chem. 2001, 276, 44247–44257. [Google Scholar] [CrossRef]
- Cavin, S.; Maric, D.; Diviani, D. A-kinase anchoring protein-Lbc promotes pro-fibrotic signaling in cardiac fibroblasts. Biochim. Biophys. Acta 2014, 1843, 335–345. [Google Scholar] [CrossRef]
- Carnegie, G.K.; Smith, F.D.; McConnachie, G.; Langeberg, L.K.; Scott, J.D. AKAP-Lbc nucleates a protein kinase D activation scaffold. Mol. Cell 2004, 15, 889–899. [Google Scholar] [CrossRef]
- Carnegie, G.K.; Soughayer, J.; Smith, F.D.; Pedroja, B.S.; Zhang, F.; Diviani, D.; Bristow, M.R.; Kunkel, M.T.; Newton, A.C.; Langeberg, L.K.; et al. AKAP-Lbc mobilizes a cardiac hypertrophy signaling pathway. Mol. Cell 2008, 32, 169–179. [Google Scholar] [CrossRef]
- Taglieri, D.M.; Johnson, K.R.; Burmeister, B.T.; Monasky, M.M.; Spindler, M.J.; DeSantiago, J.; Banach, K.; Conklin, B.R.; Carnegie, G.K. The C-terminus of the long AKAP13 isoform (AKAP-Lbc) is critical for development of compensatory cardiac hypertrophy. J. Mol. Cell. Cardiol. 2014, 66, 27–40. [Google Scholar] [CrossRef]
- Perez Lopez, I.; Cariolato, L.; Maric, D.; Gillet, L.; Abriel, H.; Diviani, D. A-kinase anchoring protein Lbc coordinates a p38 activating signaling complex controlling compensatory cardiac hypertrophy. Mol. Cell. Biol. 2013, 33, 2903–2917. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Burmeister, B.T.; Johnson, K.R.; Baillie, G.S.; Karginov, A.V.; Skidgel, R.A.; O’Bryan, J.P.; Carnegie, G.K. UCR1C is a novel activator of phosphodiesterase 4 (PDE4) long isoforms and attenuates cardiomyocyte hypertrophy. Cell. Signal. 2015, 27, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Burmeister, B.T.; Taglieri, D.M.; Wang, L.; Carnegie, G.K. Src homology 2 domain-containing phosphatase 2 (Shp2) is a component of the A-kinase-anchoring protein (AKAP)-Lbc complex and is inhibited by protein kinase A (PKA) under pathological hypertrophic conditions in the heart. J. Biol. Chem. 2012, 287, 40535–40546. [Google Scholar] [CrossRef] [PubMed]
- Caso, S.; Maric, D.; Arambasic, M.; Cotecchia, S.; Diviani, D. AKAP-Lbc mediates protection against doxorubicin-induced cardiomyocyte toxicity. Biochim. Biophys. Acta 2017, 1864, 2336–2346. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Raimondi, F.; Del Vescovo, C.D.; Dreyer, E.; Reggi, E.; Osman, H.; Ruggieri, L.; Gonano, C.; Cavin, S.; Box, C.L.; et al. Small-Molecule Protein-Protein Interaction Inhibitor of Oncogenic Rho Signaling. Cell Chem. Biol. 2016, 23, 1135–1146. [Google Scholar] [CrossRef] [PubMed]
- Diviani, D.; Abuin, L.; Cotecchia, S.; Pansier, L. Anchoring of both PKA and 14-3-3 inhibits the Rho-GEF activity of the AKAP-Lbc signaling complex. EMBO J. 2004, 23, 2811–2820. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Smith, F.D.; Stark, C.; Wells, C.D.; Fawcett, J.P.; Kulkarni, S.; Metalnikov, P.; O’Donnell, P.; Taylor, P.; Taylor, L.; et al. Proteomic, functional, and domain-based analysis of in vivo 14-3-3 binding proteins involved in cytoskeletal regulation and cellular organization. Curr. Biol. 2004, 14, 1436–1450. [Google Scholar] [CrossRef]
- Cariolato, L.; Cavin, S.; Diviani, D. A-Kinase Anchoring Protein (AKAP)-Lbc Anchors a PKN-based Signaling Complex Involved in alpha1-Adrenergic Receptor-induced p38 Activation. J. Biol. Chem. 2011, 286, 7925–7937. [Google Scholar] [CrossRef]
- Ibarrola, J.; Sadaba, R.; Martinez-Martinez, E.; Garcia-Pena, A.; Arrieta, V.; Alvarez, V.; Fernandez-Celis, A.; Gainza, A.; Cachofeiro, V.; Santamaria, E.; et al. Aldosterone Impairs Mitochondrial Function in Human Cardiac Fibroblasts via A-Kinase Anchor Protein 12. Sci. Rep. 2018, 8, 6801. [Google Scholar] [CrossRef]
- Radeva, M.Y.; Kugelmann, D.; Spindler, V.; Waschke, J. PKA compartmentalization via AKAP220 and AKAP12 contributes to endothelial barrier regulation. PLoS ONE 2014, 9, e106733. [Google Scholar] [CrossRef]
- Coats, S.R.; Covington, J.W.; Su, M.; Pabon-Pena, L.M.; Eren, M.; Hao, Q.; Vaughan, D.E. SSeCKS gene expression in vascular smooth muscle cells: Regulation by angiotensin II and a potential role in the regulation of PAI-1 gene expression. J. Mol. Cell. Cardiol. 2000, 32, 2207–2219. [Google Scholar] [CrossRef] [PubMed]
- Guillory, A.N.; Yin, X.; Wijaya, C.S.; Diaz Diaz, A.C.; Rababa’h, A.; Singh, S.; Atrooz, F.; Sadayappan, S.; McConnell, B.K. Enhanced cardiac function in Gravin mutant mice involves alterations in the beta-adrenergic receptor signaling cascade. PLoS ONE 2013, 8, e74784. [Google Scholar] [CrossRef] [PubMed]
- Canton, D.A.; Keene, C.D.; Swinney, K.; Langeberg, L.K.; Nguyen, V.; Pelletier, L.; Pawson, T.; Wordeman, L.; Stella, N.; Scott, J.D. Gravin is a transitory effector of polo-like kinase 1 during cell division. Mol. Cell 2012, 48, 547–559. [Google Scholar] [CrossRef]
- Reggi, E.; Diviani, D. The role of A-kinase anchoring proteins in cancer development. Cell. Signal. 2017, 40, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Havekes, R.; Canton, D.A.; Park, A.J.; Huang, T.; Nie, T.; Day, J.P.; Guercio, L.A.; Grimes, Q.; Luczak, V.; Gelman, I.H.; et al. Gravin orchestrates protein kinase A and beta2-adrenergic receptor signaling critical for synaptic plasticity and memory. J. Neurosci. 2012, 32, 18137–18149. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Gelman, I.H. Calmodulin and cyclin D anchoring sites on the Src-suppressed C kinase substrate, SSeCKS. Biochem. Biophys. Res. Commun. 2002, 290, 1368–1375. [Google Scholar] [CrossRef] [PubMed]
- Nauert, J.B.; Klauck, T.M.; Langeberg, L.K.; Scott, J.D. Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr. Biol. 1997, 7, 52–62. [Google Scholar] [CrossRef]
- Tao, J.; Wang, H.Y.; Malbon, C.C. Protein kinase A regulates AKAP250 (gravin) scaffold binding to the beta2-adrenergic receptor. EMBO J. 2003, 22, 6419–6429. [Google Scholar] [CrossRef]
- Li, Y.; Yu, Q.H.; Chu, Y.; Wu, W.M.; Song, J.X.; Zhu, X.B.; Wang, Q. Blockage of AKAP12 accelerates angiotensin II (Ang II)-induced cardiac injury in mice by regulating the transforming growth factor beta1 (TGF-beta1) pathway. Biochem. Biophys. Res. Commun. 2018, 499, 128–135. [Google Scholar] [CrossRef]
- Buonafine, M.; Bonnard, B.; Jaisser, F. Mineralocorticoid Receptor and Cardiovascular Disease. Am. J. Hypertens. 2018, 31, 1165–1174. [Google Scholar] [CrossRef]
- Lee, H.S.; Choi, J.; Son, T.; Wee, H.-J.; Bae, S.-J.; Seo, J.H.; Park, J.H.; Ryu, S.H.; Lee, D.; Jang, M.K.; et al. Altered AKAP12 expression in portal fibroblasts and liver sinusoids mediates transition from hepatic fibrogenesis to fibrosis resolution. Exp. Mol. Med. 2018, 50, 48. [Google Scholar] [CrossRef] [PubMed]
- Porter, K.E.; Turner, N.A. Cardiac fibroblasts: At the heart of myocardial remodeling. Pharmacol. Ther. 2009, 123, 255–278. [Google Scholar] [CrossRef] [PubMed]
- Ubil, E.; Duan, J.; Pillai, I.C.; Rosa-Garrido, M.; Wu, Y.; Bargiacchi, F.; Lu, Y.; Stanbouly, S.; Huang, J.; Rojas, M.; et al. Mesenchymal-endothelial transition contributes to cardiac neovascularization. Nature 2014, 514, 585–590. [Google Scholar] [CrossRef] [PubMed]
- Schrade, K.; Troger, J.; Eldahshan, A.; Zuhlke, K.; Abdul Azeez, K.R.; Elkins, J.M.; Neuenschwander, M.; Oder, A.; Elkewedi, M.; Jaksch, S.; et al. An AKAP-Lbc-RhoA interaction inhibitor promotes the translocation of aquaporin-2 to the plasma membrane of renal collecting duct principal cells. PLoS ONE 2018, 13, e0191423. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delaunay, M.; Osman, H.; Kaiser, S.; Diviani, D. The Role of Cyclic AMP Signaling in Cardiac Fibrosis. Cells 2020, 9, 69. https://doi.org/10.3390/cells9010069
Delaunay M, Osman H, Kaiser S, Diviani D. The Role of Cyclic AMP Signaling in Cardiac Fibrosis. Cells. 2020; 9(1):69. https://doi.org/10.3390/cells9010069
Chicago/Turabian StyleDelaunay, Marion, Halima Osman, Simon Kaiser, and Dario Diviani. 2020. "The Role of Cyclic AMP Signaling in Cardiac Fibrosis" Cells 9, no. 1: 69. https://doi.org/10.3390/cells9010069
APA StyleDelaunay, M., Osman, H., Kaiser, S., & Diviani, D. (2020). The Role of Cyclic AMP Signaling in Cardiac Fibrosis. Cells, 9(1), 69. https://doi.org/10.3390/cells9010069