Mitochondrial Mechanisms of Neuromuscular Junction Degeneration with Aging



Abstract
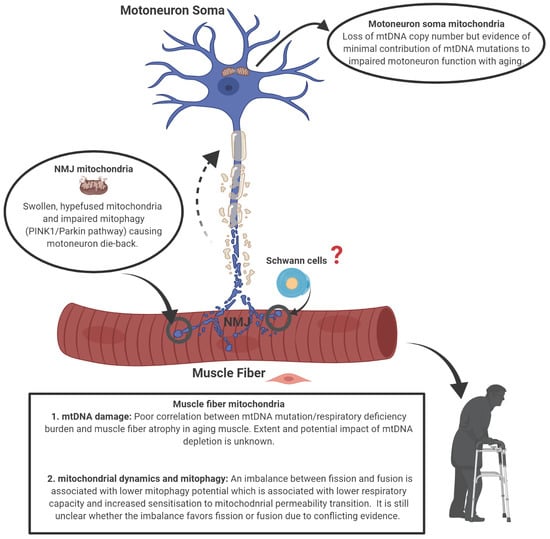
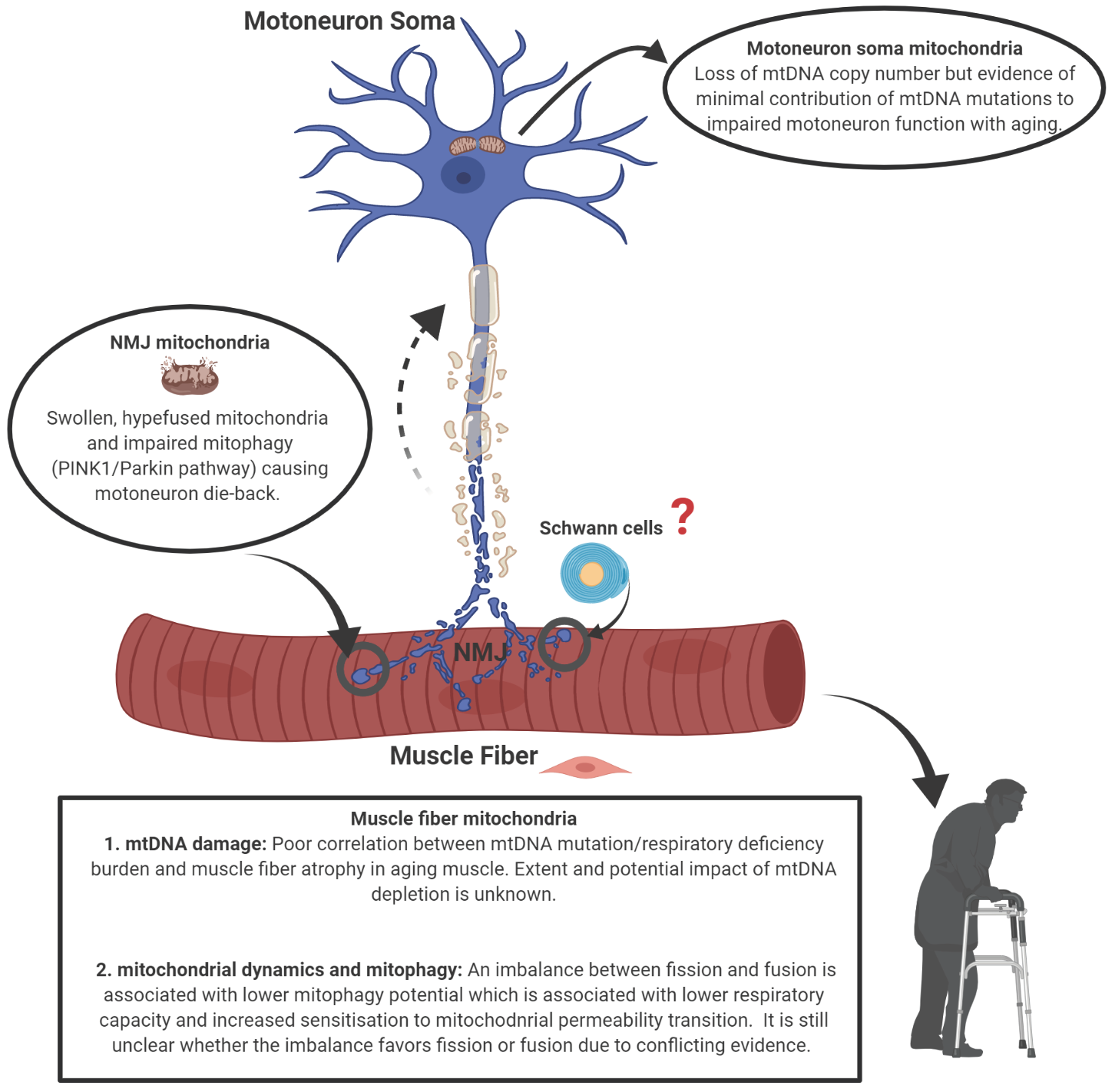
1. Introduction
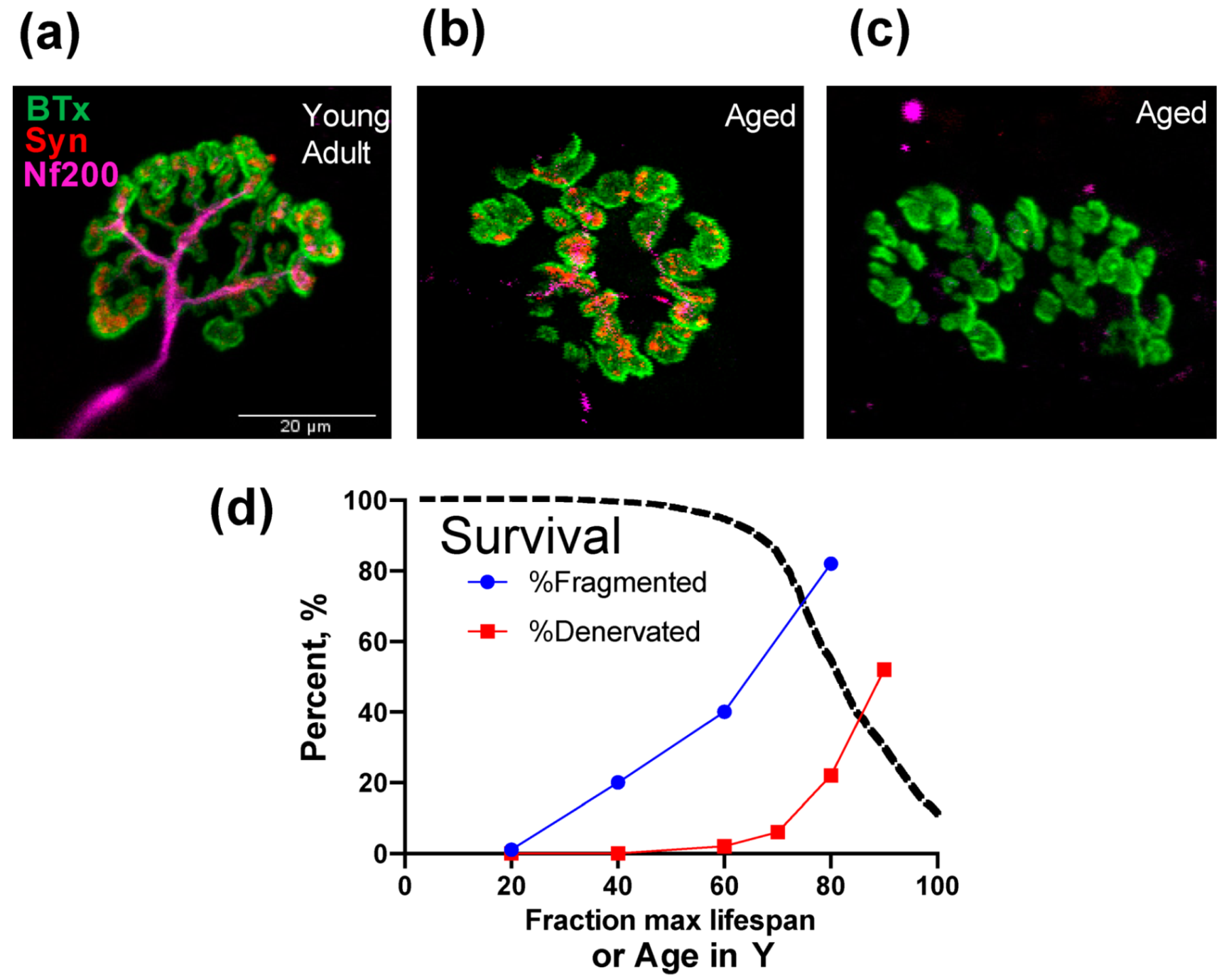
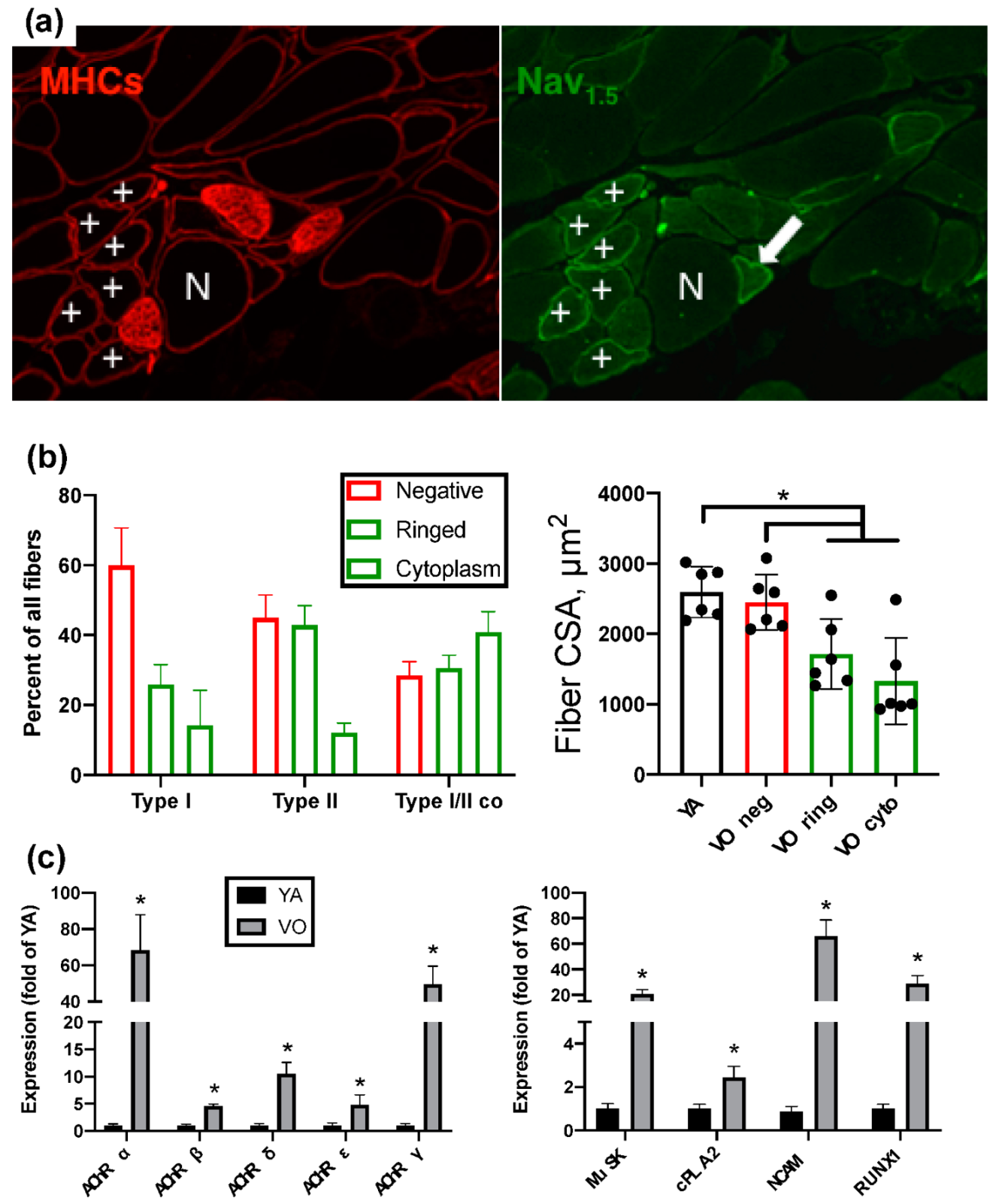
2. Neuromuscular Junction, Denervation-Reinnervation and Atrophy of Aging Muscle
3. Mitochondrial Alterations with Aging
3.1. Mitochondrial Morphology
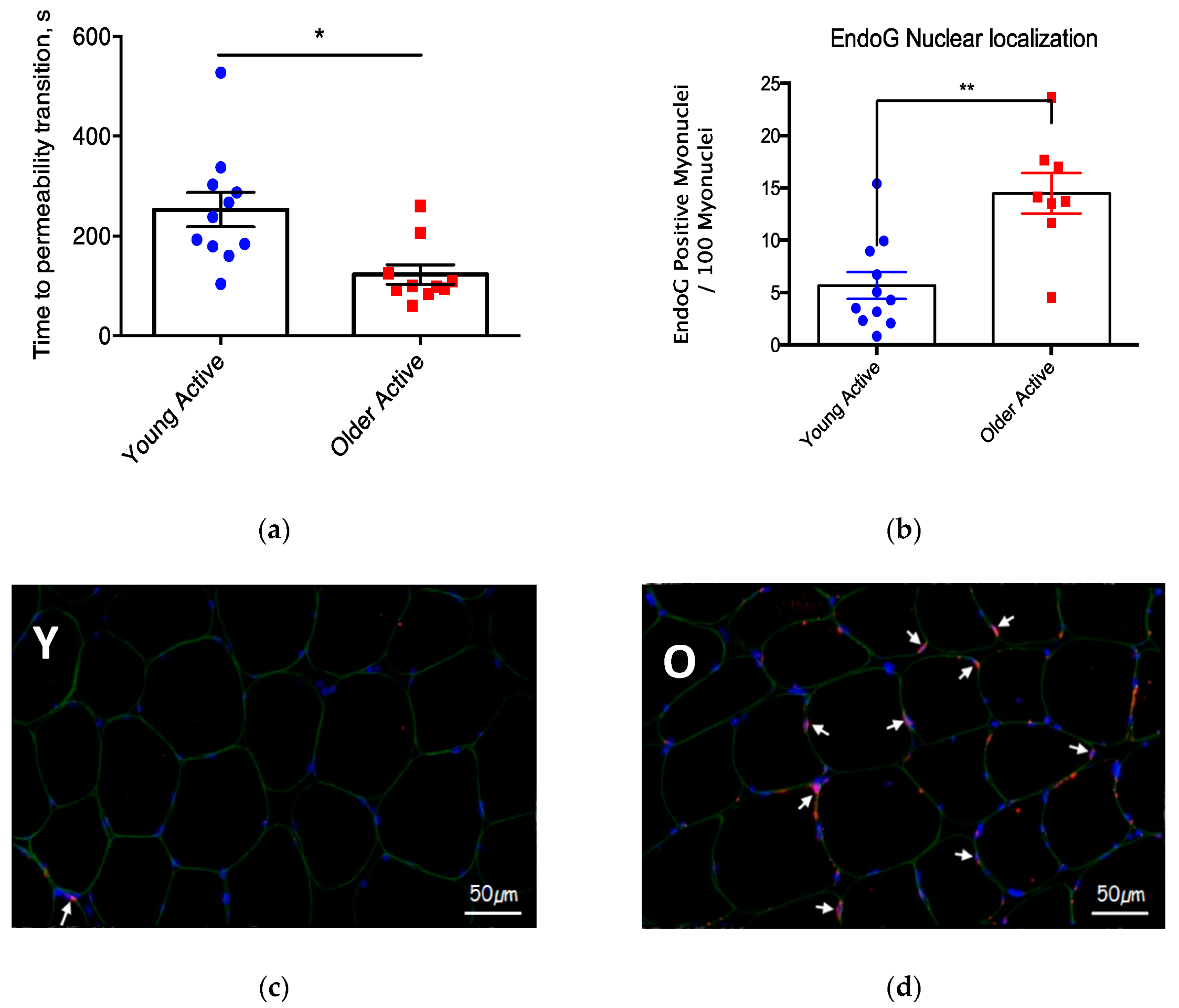
3.2. Mitochondrial Function
4. Mechanisms of Mitochondrial Dysfunction with Aging
4.1. mtDNA Alterations
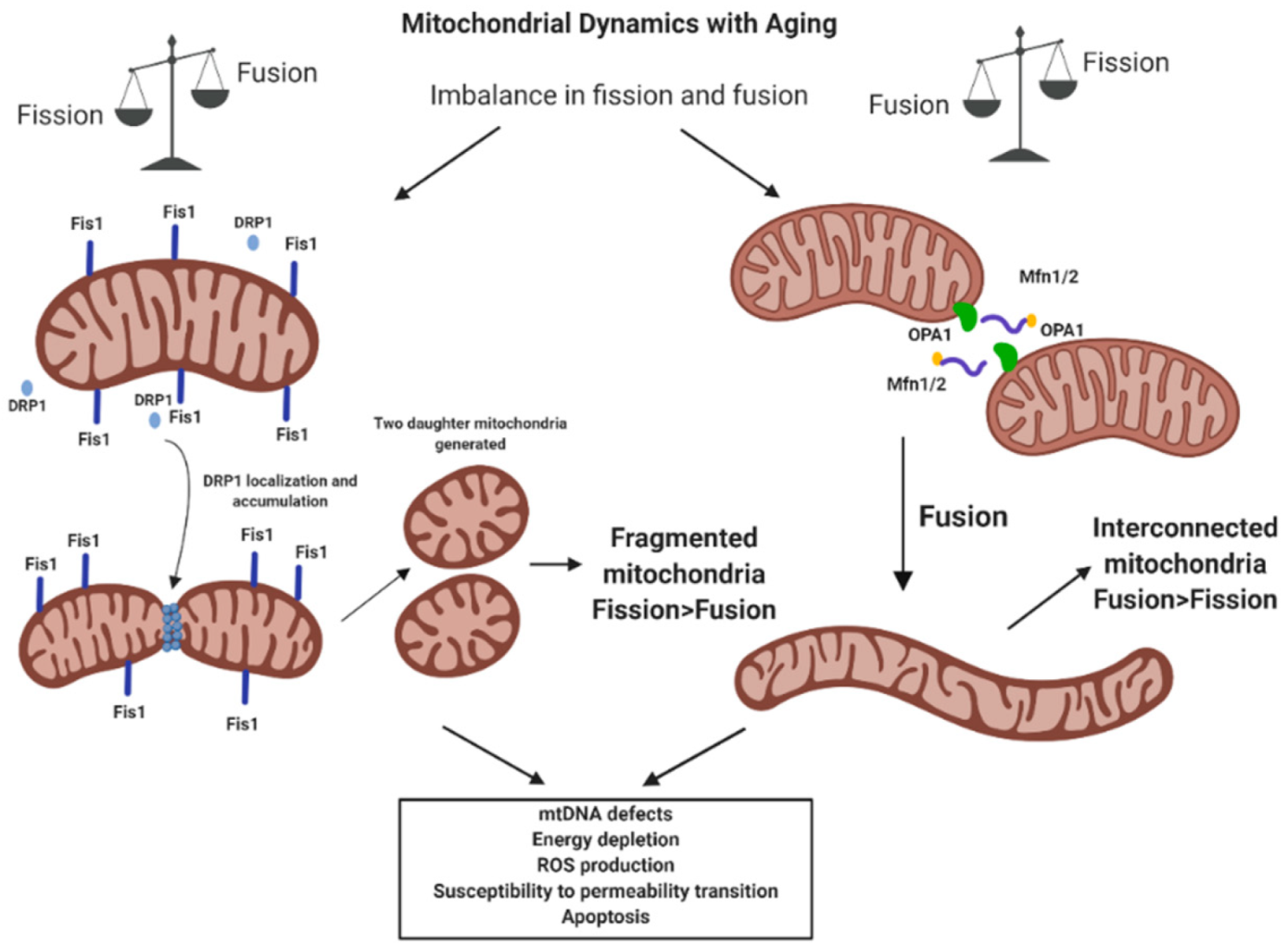
4.2. Mitochondrial Dynamics and Mitostasis
4.3. The Special Problem of Mitostasis in Motoneurons
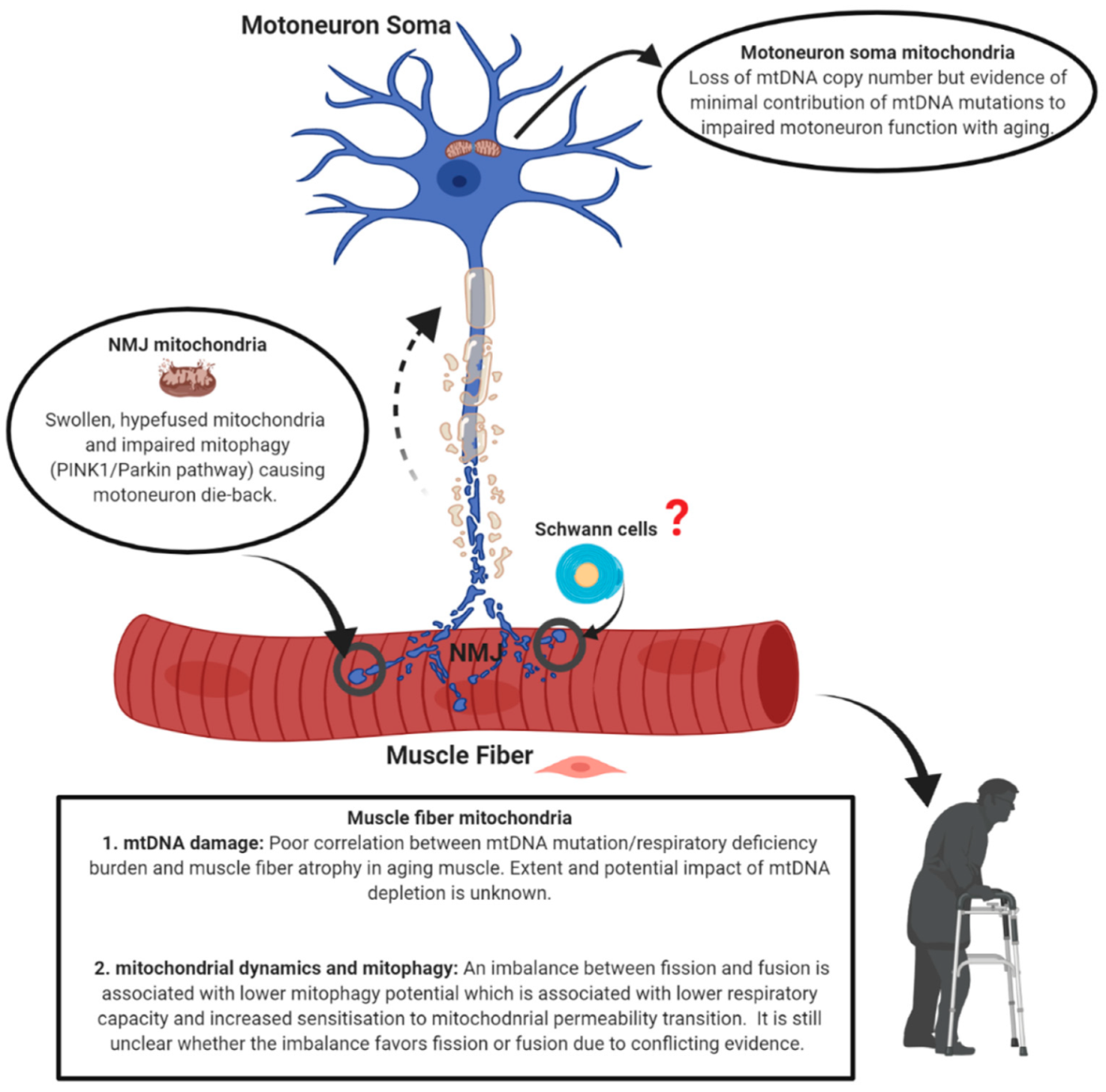
5. Conclusions
Funding
Conflicts of Interest
References
- Angulo, J.; El Assar, M.; Rodriguez-Manas, L. Frailty and sarcopenia as the basis for the phenotypic manifestation of chronic diseases in older adults. Mol. Asp. Med. 2016, 50, 1–32. [Google Scholar] [CrossRef]
- Beaudart, C.; Zaaria, M.; Pasleau, F.; Reginster, J.Y.; Bruyere, O. Health Outcomes of Sarcopenia: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0169548. [Google Scholar] [CrossRef] [PubMed]
- Larsson, L.; Degens, H.; Li, M.; Salviati, L.; Lee, Y.I.; Thompson, W.; Kirkland, J.L.; Sandri, M. Sarcopenia: Aging-Related Loss of Muscle Mass and Function. Physiol. Rev. 2019, 99, 427–511. [Google Scholar] [CrossRef] [PubMed]
- Piasecki, M.; Ireland, A.; Jones, D.A.; McPhee, J.S. Age-dependent motor unit remodelling in human limb muscles. Biogerontology 2016, 17, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Fuglevand, A.J.; Segal, S.S. Simulation of motor unit recruitment and microvascular unit perfusion: Spatial considerations. J. Appl. Physiol. 1997, 83, 1223–1234. [Google Scholar] [CrossRef]
- Imboden, M.T.; Swartz, A.M.; Finch, H.W.; Harber, M.P.; Kaminsky, L.A. Reference standards for lean mass measures using GE dual energy x-ray absorptiometry in Caucasian adults. PLoS ONE 2017, 12, e0176161. [Google Scholar] [CrossRef]
- Rowan, S.L.; Rygiel, K.; Purves-Smith, F.M.; Solbak, N.M.; Turnbull, D.M.; Hepple, R.T. Denervation causes fiber atrophy and Myosin heavy chain co-expression in senescent skeletal muscle. PLoS ONE 2012, 7, e29082. [Google Scholar] [CrossRef]
- Rowan, S.L.; Purves-Smith, F.M.; Solbak, N.M.; Hepple, R.T. Accumulation of severely atrophic myofibers marks the acceleration of sarcopenia in slow and fast twitch muscles. Exp. Gerontol. 2011, 46, 660–669. [Google Scholar] [CrossRef]
- Hepple, R.T.; Hagen, J.L.; Krause, D.J.; Baker, D.J. Skeletal muscle aging in F344BN F1-hybrid rats: II. Improved contractile economy in senescence helps compensate for reduced ATP generating capacity. J. Gerontol. A Biol. Sci. Med. Sci. 2004, 59, 1111–1119. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Picard, M.; Turnbull, D.M. The ageing neuromuscular system and sarcopenia: A mitochondrial perspective. J. Physiol. 2016, 594, 4499–4512. [Google Scholar] [CrossRef]
- Comley, L.H.; Nijssen, J.; Frost-Nylen, J.; Hedlund, E. Cross-disease comparison of amyotrophic lateral sclerosis and spinal muscular atrophy reveals conservation of selective vulnerability but differential neuromuscular junction pathology. J. Comp. Neurol. 2016, 524, 1424–1442. [Google Scholar] [CrossRef] [PubMed]
- Martineau, E.; Di Polo, A.; Vande Velde, C.; Robitaille, R. Dynamic neuromuscular remodeling precedes motor-unit loss in a mouse model of ALS. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Deschenes, M.R.; Roby, M.A.; Eason, M.K.; Harris, M.B. Remodeling of the neuromuscular junction precedes sarcopenia related alterations in myofibers. Exp. Gerontol. 2010, 45, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Spendiff, S.; Vuda, M.; Gouspillou, G.; Aare, S.; Perez, A.; Morais, J.A.; Jagoe, R.T.; Filion, M.E.; Glicksman, R.; Kapchinsky, S.; et al. Denervation drives mitochondrial dysfunction in skeletal muscle of octogenarians. J. Physiol. 2016, 594, 7361–7379. [Google Scholar] [CrossRef]
- Gutmann, E.; Hanzlikova, V. Motor unit in old age. Nature 1966, 209, 921–922. [Google Scholar] [CrossRef]
- Oda, K. Age changes of motor innervation and acetylcholine receptor distribution on human skeletal muscle fibres. J. Neurol. Sci. 1984, 66, 327–338. [Google Scholar] [CrossRef]
- Arizono, N.; Koreto, O.; Iwai, Y.; Hidaka, T.; Takeoka, O. Morphometric analysis of human neuromuscular junction in different ages. Acta Pathol. Jpn. 1984, 34, 1243–1249. [Google Scholar] [CrossRef]
- Wokke, J.H.; Jennekens, F.G.; van den Oord, C.J.; Veldman, H.; Smit, L.M.; Leppink, G.J. Morphological changes in the human end plate with age. J. Neurol. Sci. 1990, 95, 291–310. [Google Scholar] [CrossRef]
- Valdez, G.; Tapia, J.C.; Kang, H.; Clemenson, G.D., Jr.; Gage, F.H.; Lichtman, J.W.; Sanes, J.R. Attenuation of age-related changes in mouse neuromuscular synapses by caloric restriction and exercise. Proc. Natl. Acad. Sci. USA 2010, 107, 14863–14868. [Google Scholar] [CrossRef]
- Willadt, S.; Nash, M.; Slater, C.R. Age-related fragmentation of the motor endplate is not associated with impaired neuromuscular transmission in the mouse diaphragm. Sci. Rep. 2016, 6, 24849. [Google Scholar] [CrossRef]
- Urbanchek, M.G.; Picken, E.B.; Kalliainen, L.K.; Kuzon, W.M., Jr. Specific force deficit in skeletal muscles of old rats is partially explained by the existence of denervated muscle fibers. J. Gerontol. A Biol. Sci. Med. Sci. 2001, 56, B191–B197. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.M.; Zheng, Z.; Messi, M.L.; Delbono, O. Extension and magnitude of denervation in skeletal muscle from ageing mice. J. Physiol. Online 2005, 565, 757–764. [Google Scholar] [CrossRef] [PubMed]
- Sonjak, V.; Jacob, K.; Morais, J.A.; Rivera-Zengotita, M.; Spendiff, S.; Spake, C.; Taivassalo, T.; Chevalier, S.; Hepple, R.T. Fidelity of muscle fibre reinnervation modulates ageing muscle impact in elderly women. J. Physiol. 2019, 597, 5009–5023. [Google Scholar] [CrossRef]
- Aare, S.; Spendiff, S.; Vuda, M.; Elkrief, D.; Perez, A.; Wu, Q.; Mayaki, D.; Hussain, S.N.; Hettwer, S.; Hepple, R.T. Failed reinnervation in aging skeletal muscle. Skelet. Muscle 2016, 6, 29. [Google Scholar] [CrossRef] [PubMed]
- Piasecki, M.; Ireland, A.; Piasecki, J.; Stashuk, D.W.; Swiecicka, A.; Rutter, M.K.; Jones, D.A.; McPhee, J.S. Failure to expand the motor unit size to compensate for declining motor unit numbers distinguishes sarcopenic from non-sarcopenic older men. J. Physiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mosole, S.; Carraro, U.; Kern, H.; Loefler, S.; Fruhmann, H.; Vogelauer, M.; Burggraf, S.; Mayr, W.; Krenn, M.; Paternostro-Sluga, T.; et al. Long-term high-level exercise promotes muscle reinnervation with age. J. Neuropathol. Exp. Neurol. 2014, 73, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Alway, S.E.; Mohamed, J.S.; Myers, M.J. Mitochondria Initiate and Regulate Sarcopenia. Exerc. Sport Sci. Rev. 2017, 45, 58–69. [Google Scholar] [CrossRef]
- Grimm, A.; Eckert, A. Brain aging and neurodegeneration: From a mitochondrial point of view. J. Neurochem. 2017, 143, 418–431. [Google Scholar] [CrossRef]
- Leduc-Gaudet, J.P.; Picard, M.; St-Jean Pelletier, F.; Sgarioto, N.; Auger, M.J.; Vallee, J.; Robitaille, R.; St-Pierre, D.H.; Gouspillou, G. Mitochondrial morphology is altered in atrophied skeletal muscle of aged mice. Oncotarget 2015, 6, 17923–17937. [Google Scholar] [CrossRef]
- Chabi, B.; Ljubicic, V.; Menzies, K.J.; Huang, J.H.; Saleem, A.; Hood, D.A. Mitochondrial function and apoptotic susceptibility in aging skeletal muscle. Aging Cell 2008, 7, 2–12. [Google Scholar] [CrossRef]
- Picard, M.; Ritchie, D.; Wright, K.J.; Romestaing, C.; Thomas, M.M.; Rowan, S.L.; Taivassalo, T.; Hepple, R.T. Mitochondrial functional impairment with aging is exaggerated in isolated mitochondria compared to permeabilized myofibers. Aging Cell 2010, 9, 1032–1046. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Sgarioto, N.; Kapchinsky, S.; Purves-Smith, F.; Norris, B.; Pion, C.H.; Barbat-Artigas, S.; Lemieux, F.; Taivassalo, T.; Morais, J.A.; et al. Increased sensitivity to mitochondrial permeability transition and myonuclear translocation of endonuclease G in atrophied muscle of physically active older humans. FASEB J. 2014, 28, 1621–1633. [Google Scholar] [CrossRef] [PubMed]
- Sonjak, V.; Jacob, K.J.; Spendiff, S.; Vuda, M.; Perez, A.; Miguez, K.; Minozzo, F.C.; Spake, C.; Morais, J.A.; Hepple, R.T. Reduced Mitochondrial Content, Elevated Reactive Oxygen Species, and Modulation by Denervation in Skeletal Muscle of Prefrail or Frail Elderly Women. J. Gerontol. A Biol. Sci. Med. Sci. 2019, 74, 1887–1895. [Google Scholar] [CrossRef]
- Bakeeva, L.E.; Chentsov, Y.S.; Skulachev, V.P. Mitochondrial framework (reticulum mitochondriale) in rat diaphragm muscle. Biochim. Biophys. Acta 1978, 501, 349–369. [Google Scholar] [CrossRef]
- Ogata, T.; Yamasaki, Y. Ultra-high-resolution scanning electron microscopy of mitochondria and sarcoplasmic reticulum arrangement in human red, white, and intermediate muscle fibers. Anat. Rec. 1997, 248, 214–223. [Google Scholar] [CrossRef]
- Huertas, J.R.; Ruiz-Ojeda, F.J.; Plaza-Diaz, J.; Nordsborg, N.B.; Martin-Albo, J.; Rueda-Robles, A.; Casuso, R.A. Human muscular mitochondrial fusion in athletes during exercise. FASEB J. 2019. [Google Scholar] [CrossRef]
- Kirkwood, S.P.; Munn, E.A.; Brooks, G.A. Mitochondrial reticulum in limb skeletal muscle. Am. J. Physiol. 1986, 251, C395–C402. [Google Scholar] [CrossRef]
- Bleck, C.K.E.; Kim, Y.; Willingham, T.B.; Glancy, B. Subcellular connectomic analyses of energy networks in striated muscle. Nat. Commun. 2018, 9, 5111. [Google Scholar] [CrossRef]
- Vincent, A.E.; White, K.; Davey, T.; Philips, J.; Ogden, R.T.; Lawless, C.; Warren, C.; Hall, M.G.; Ng, Y.S.; Falkous, G.; et al. Quantitative 3D Mapping of the Human Skeletal Muscle Mitochondrial Network. Cell Rep. 2019, 27, 321. [Google Scholar] [CrossRef]
- Glancy, B.; Hartnell, L.M.; Malide, D.; Yu, Z.X.; Combs, C.A.; Connelly, P.S.; Subramaniam, S.; Balaban, R.S. Mitochondrial reticulum for cellular energy distribution in muscle. Nature 2015, 523, 617–620. [Google Scholar] [CrossRef]
- Glancy, B.; Hartnell, L.M.; Combs, C.A.; Femnou, A.; Sun, J.; Murphy, E.; Subramaniam, S.; Balaban, R.S. Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep. 2017, 19, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.H.; Joseph, A.M.; Ljubicic, V.; Iqbal, S.; Hood, D.A. Effect of age on the processing and import of matrix-destined mitochondrial proteins in skeletal muscle. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 138–146. [Google Scholar] [CrossRef] [PubMed]
- Poggi, P.; Marchetti, C.; Scelsi, R. Automatic morphometric analysis of skeletal muscle fibers in the aging man. Anat. Rec. 1987, 217, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T. Fiber typing in aging muscle. Exerc. Sport Sci. Rev. 2014, 42, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, S.; Ostojic, O.; Singh, K.; Joseph, A.M.; Hood, D.A. Expression of mitochondrial fission and fusion regulatory proteins in skeletal muscle during chronic use and disuse. Muscle Nerve 2013, 48, 963–970. [Google Scholar] [CrossRef] [PubMed]
- Romanello, V.; Guadagnin, E.; Gomes, L.; Roder, I.; Sandri, C.; Petersen, Y.; Milan, G.; Masiero, E.; Del Piccolo, P.; Foretz, M.; et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J. 2010, 29, 1774–1785. [Google Scholar] [CrossRef]
- Garcia, M.L.; Fernandez, A.; Solas, M.T. Mitochondria, motor neurons and aging. J. Neurol. Sci. 2013, 330, 18–26. [Google Scholar] [CrossRef]
- Trounce, I.; Byrne, E.; Marzuki, S. Decline in skeletal muscle mitochondrial respiratory chain function: Possible factor in ageing. Lancet 1989, 1, 637–639. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Ritchie, D.; Wright, K.J.; Thomas, M.M.; Romestaing, C.; Hepple, R.T. Mitochondrial structure and function are disrupted by standard isolation methods. PLoS ONE 2011, 6, e18317. [Google Scholar] [CrossRef]
- Saks, V.A.; Veksler, V.I.; Kuznetsov, A.V.; Kay, L.; Sikk, P.; Tiivel, T.; Tranqui, L.; Olivares, J.; Winkler, K.; Wiedemann, F.; et al. Permeabilized cell and skinned fiber techniques in studies of mitochondrial function in vivo. Mol. Cell. Biochem. 1998, 184, 81–100. [Google Scholar] [CrossRef]
- Picard, M.; Taivassalo, T.; Gouspillou, G.; Hepple, R.T. Mitochondria: Isolation, Structure and Function. J. Physiol. 2011, 589, 4413–4421. [Google Scholar] [CrossRef] [PubMed]
- Hepple, R.T. Impact of aging on mitochondrial function in cardiac and skeletal muscle. Free Radic. Biol. Med. 2016, 98, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Gonzalez de Aguilar, J.L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef]
- Bevilacqua, L.; Ramsey, J.J.; Hagopian, K.; Weindruch, R.; Harper, M.E. Long-term caloric restriction increases UCP3 content but decreases proton leak and reactive oxygen species production in rat skeletal muscle mitochondria. AJP Endocrinol. Metab. 2005, 289, E429–E438. [Google Scholar] [CrossRef] [PubMed]
- Echtay, K.S. Mitochondrial uncoupling proteins—What is their physiological role? Free Radic. Biol. Med. 2007, 43, 1351–1371. [Google Scholar] [CrossRef] [PubMed]
- Chung, T.; Park, J.S.; Kim, S.; Montes, N.; Walston, J.; Hoke, A. Evidence for dying-back axonal degeneration in age-associated skeletal muscle decline. Muscle Nerve 2017, 55, 894–901. [Google Scholar] [CrossRef]
- Audouard, E.; Van Hees, L.; Suain, V.; Yilmaz, Z.; Poncelet, L.; Leroy, K.; Brion, J.P. Motor deficit in a tauopathy model is induced by disturbances of axonal transport leading to dying-back degeneration and denervation of neuromuscular junctions. Am. J. Pathol. 2015, 185, 2685–2697. [Google Scholar] [CrossRef]
- Yin, Z.; Valkenburg, F.; Hornix, B.E.; Mantingh-Otter, I.; Zhou, X.; Mari, M.; Reggiori, F.; Van Dam, D.; Eggen, B.J.L.; De Deyn, P.P.; et al. Progressive Motor Deficit is Mediated by the Denervation of Neuromuscular Junctions and Axonal Degeneration in Transgenic Mice Expressing Mutant (P301S) Tau Protein. J. Alzheimer Dis. JAD 2017, 60, S41–S57. [Google Scholar] [CrossRef]
- So, E.; Mitchell, J.C.; Memmi, C.; Chennell, G.; Vizcay-Barrena, G.; Allison, L.; Shaw, C.E.; Vance, C. Mitochondrial abnormalities and disruption of the neuromuscular junction precede the clinical phenotype and motor neuron loss in hFUSWT transgenic mice. Hum. Mol. Genet. 2018, 27, 463–474. [Google Scholar] [CrossRef]
- Rogers, R.S.; Tungtur, S.; Tanaka, T.; Nadeau, L.L.; Badawi, Y.; Wang, H.; Ni, H.M.; Ding, W.X.; Nishimune, H. Impaired Mitophagy Plays a Role in Denervation of Neuromuscular Junctions in ALS Mice. Front. Neurosci. 2017, 11, 473. [Google Scholar] [CrossRef]
- Gustafsson, C.M.; Falkenberg, M.; Larsson, N.G. Maintenance and Expression of Mammalian Mitochondrial DNA. Annu. Rev. Biochem. 2016, 85, 133–160. [Google Scholar] [CrossRef] [PubMed]
- Simonetti, S.; Chen, X.; DiMauro, S.; Schon, E.A. Accumulation of deletions in human mitochondrial DNA during normal aging: Analysis by quantitative PCR. Biochim. Biophys. Acta 1992, 1180, 113–122. [Google Scholar] [CrossRef]
- Melov, S.; Shoffner, J.M.; Kaufman, A.; Wallace, D.C. Marked increase in the number and variety of mitochondrial DNA rearrangements in aging human skeletal muscle. Nucleic Acids Res. 1995, 23, 4122–4126. [Google Scholar] [CrossRef] [PubMed]
- Liu, V.W.; Zhang, C.; Nagley, P. Mutations in mitochondrial DNA accumulate differentially in three different human tissues during ageing. Nucleic Acids Res. 1998, 26, 1268–1275. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef]
- Rossignol, R.; Faustin, B.; Rocher, C.; Malgat, M.; Mazat, J.P.; Letellier, T. Mitochondrial threshold effects. Biochem. J. 2003, 370, 751–762. [Google Scholar] [CrossRef]
- Tuppen, H.A.; Blakely, E.L.; Turnbull, D.M.; Taylor, R.W. Mitochondrial DNA mutations and human disease. Biochim. Biophys. Acta 2010, 1797, 113–128. [Google Scholar] [CrossRef]
- Tzoulis, C.; Tran, G.T.; Coxhead, J.; Bertelsen, B.; Lilleng, P.K.; Balafkan, N.; Payne, B.; Miletic, H.; Chinnery, P.F.; Bindoff, L.A. Molecular pathogenesis of polymerase gamma-related neurodegeneration. Ann. Neurol. 2014, 76, 66–81. [Google Scholar] [CrossRef]
- Konokhova, Y.; Spendiff, S.; Jagoe, R.T.; Aare, S.; Kapchinsky, S.; MacMillan, N.J.; Rozakis, P.; Picard, M.; Aubertin-Leheudre, M.; Pion, C.H.; et al. Failed upregulation of TFAM protein and mitochondrial DNA in oxidatively deficient fibers of chronic obstructive pulmonary disease locomotor muscle. Skelet. Muscle 2016, 6, 10. [Google Scholar] [CrossRef]
- Stewart, J.B.; Chinnery, P.F. The dynamics of mitochondrial DNA heteroplasmy: Implications for human health and disease. Nat. Rev. Genet. 2015, 16, 530–542. [Google Scholar] [CrossRef]
- Lee, C.M.; Lopez, M.E.; Weindruch, R.; Aiken, J.M. Association of age-related mitochondrial abnormalities with skeletal muscle fiber atrophy. Free Radic. Biol. Med. 1998, 25, 964–972. [Google Scholar] [CrossRef]
- Wanagat, J.; Cao, Z.; Pathare, P.; Aiken, J.M. Mitochondrial DNA deletion mutations colocalize with segmental electron transport system abnormalities, muscle fiber atrophy, fiber splitting, and oxidative damage in sarcopenia. FASEB J. 2001, 15, 322–332. [Google Scholar] [CrossRef] [PubMed]
- Cheema, N.; Herbst, A.; McKenzie, D.; Aiken, J.M. Apoptosis and necrosis mediate skeletal muscle fiber loss in age-induced mitochondrial enzymatic abnormalities. Aging Cell 2015. [Google Scholar] [CrossRef] [PubMed]
- Bua, E.; Johnson, J.; Herbst, A.; Delong, B.; McKenzie, D.; Salamat, S.; Aiken, J.M. Mitochondrial DNA-deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 2006, 79, 469–480. [Google Scholar] [CrossRef]
- Jacobs, H.T. Rebuttal to Pak et al.: New data, old chestnuts. Aging Cell 2003, 2, 19–20. [Google Scholar] [CrossRef]
- Taivassalo, T.; Jensen, T.D.; Kennaway, N.; DiMauro, S.; Vissing, J.; Haller, R.G. The spectrum of exercise tolerance in mitochondrial myopathies: A study of 40 patients. Brain 2003, 126, 413–423. [Google Scholar] [CrossRef]
- Trifunovic, A.; Wredenberg, A.; Falkenberg, M.; Spelbrink, J.N.; Rovio, A.T.; Bruder, C.E.; Bohlooly, Y.; Gidlof, S.; Oldfors, A.; Wibom, R.; et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 2004, 429, 417–423. [Google Scholar] [CrossRef]
- Hiona, A.; Sanz, A.; Kujoth, G.C.; Pamplona, R.; Seo, A.Y.; Hofer, T.; Someya, S.; Miyakawa, T.; Nakayama, C.; Samhan-Arias, A.K.; et al. Mitochondrial DNA Mutations Induce Mitochondrial Dysfunction, Apoptosis and Sarcopenia in Skeletal Muscle of Mitochondrial DNA Mutator Mice. PLoS ONE 2010, 5, e11468. [Google Scholar] [CrossRef]
- Picard, M.; Ritchie, D.; Thomas, M.M.; Wright, K.J.; Hepple, R.T. Alterations in intrinsic mitochondrial function with aging are fiber type-specific and do not explain differential atrophy between muscles. Aging Cell 2011, 10, 1047–1055. [Google Scholar] [CrossRef]
- Rygiel, K.A.; Grady, J.P.; Turnbull, D.M. Respiratory chain deficiency in aged spinal motor neurons. Neurobiol. Aging 2014, 35, 2230–2238. [Google Scholar] [CrossRef]
- Chan, D.C. Fusion and fission: Interlinked processes critical for mitochondrial health. Annu. Rev. Genet. 2012, 46, 265–287. [Google Scholar] [CrossRef] [PubMed]
- Twig, G.; Elorza, A.; Molina, A.J.; Mohamed, H.; Wikstrom, J.D.; Walzer, G.; Stiles, L.; Haigh, S.E.; Katz, S.; Las, G.; et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008, 27, 433–446. [Google Scholar] [CrossRef] [PubMed]
- Dorn, G.W., 2nd. Evolving Concepts of Mitochondrial Dynamics. Annu. Rev. Physiol. 2019, 81, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Tezze, C.; Romanello, V.; Desbats, M.A.; Fadini, G.P.; Albiero, M.; Favaro, G.; Ciciliot, S.; Soriano, M.E.; Morbidoni, V.; Cerqua, C.; et al. Age-Associated Loss of OPA1 in Muscle Impacts Muscle Mass, Metabolic Homeostasis, Systemic Inflammation, and Epithelial Senescence. Cell Metab. 2017, 25, 1374–1389. [Google Scholar] [CrossRef]
- Sebastian, D.; Sorianello, E.; Segales, J.; Irazoki, A.; Ruiz-Bonilla, V.; Sala, D.; Planet, E.; Berenguer-Llergo, A.; Munoz, J.P.; Sanchez-Feutrie, M.; et al. Mfn2 deficiency links age-related sarcopenia and impaired autophagy to activation of an adaptive mitophagy pathway. EMBO J. 2016, 35, 1677–1693. [Google Scholar] [CrossRef] [PubMed]
- Verstreken, P.; Ly, C.V.; Venken, K.J.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef]
- Favaro, G.; Romanello, V.; Varanita, T.; Andrea Desbats, M.; Morbidoni, V.; Tezze, C.; Albiero, M.; Canato, M.; Gherardi, G.; De Stefani, D.; et al. DRP1-mediated mitochondrial shape controls calcium homeostasis and muscle mass. Nat. Commun. 2019, 10, 2576. [Google Scholar] [CrossRef]
- Romanello, V.; Scalabrin, M.; Albiero, M.; Blaauw, B.; Scorrano, L.; Sandri, M. Inhibition of the Fission Machinery Mitigates OPA1 Impairment in Adult Skeletal Muscles. Cells 2019, 8, 597. [Google Scholar] [CrossRef]
- Chandhok, G.; Lazarou, M.; Neumann, B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol. Rev. Camb. Philos. Soc. 2017. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef]
- Karunadharma, P.P.; Basisty, N.; Chiao, Y.A.; Dai, D.F.; Drake, R.; Levy, N.; Koh, W.J.; Emond, M.J.; Kruse, S.; Marcinek, D.; et al. Respiratory chain protein turnover rates in mice are highly heterogeneous but strikingly conserved across tissues, ages, and treatments. FASEB J. 2015, 29, 3582–3592. [Google Scholar] [CrossRef] [PubMed]
- Konig, T.; Troder, S.E.; Bakka, K.; Korwitz, A.; Richter-Dennerlein, R.; Lampe, P.A.; Patron, M.; Muhlmeister, M.; Guerrero-Castillo, S.; Brandt, U.; et al. The m-AAA Protease Associated with Neurodegeneration Limits MCU Activity in Mitochondria. Mol. Cell 2016, 64, 148–162. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Carelli, V.; Manfredi, G.; Chan, D.C. Proteolytic cleavage of Opa1 stimulates mitochondrial inner membrane fusion and couples fusion to oxidative phosphorylation. Cell Metab. 2014, 19, 630–641. [Google Scholar] [CrossRef]
- Sprenger, H.G.; Wani, G.; Hesseling, A.; Konig, T.; Patron, M.; MacVicar, T.; Ahola, S.; Wai, T.; Barth, E.; Rugarli, E.I.; et al. Loss of the mitochondrial i-AAA protease YME1L leads to ocular dysfunction and spinal axonopathy. EMBO Mol. Med. 2019, 11. [Google Scholar] [CrossRef] [PubMed]
- Mouton-Liger, F.; Jacoupy, M.; Corvol, J.C.; Corti, O. PINK1/Parkin-Dependent Mitochondrial Surveillance: From Pleiotropy to Parkinson’s Disease. Front. Mol. Neurosci. 2017, 10, 120. [Google Scholar] [CrossRef] [PubMed]
- Drake, J.C.; Laker, R.C.; Wilson, R.J.; Zhang, M.; Yan, Z. Exercise-induced mitophagy in skeletal muscle occurs in the absence of stabilization of Pink1 on mitochondria. Cell Cycle 2019, 18, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.C.W.; Erlich, A.T.; Crilly, M.J.; Hood, D.A. Parkin is required for exercise-induced mitophagy in muscle: Impact of aging. Am. J. Physiol. Endocrinol. Metab. 2018, 315, E404–E415. [Google Scholar] [CrossRef]
- Gouspillou, G.; Godin, R.; Piquereau, J.; Picard, M.; Mofarrahi, M.; Mathew, J.; Purves-Smith, F.M.; Sgarioto, N.; Hepple, R.T.; Burelle, Y.; et al. Protective role of Parkin in skeletal muscle contractile and mitochondrial function. J. Physiol. 2018, 596, 2565–2579. [Google Scholar] [CrossRef]
- Peker, N.; Donipadi, V.; Sharma, M.; McFarlane, C.; Kambadur, R. Loss of Parkin impairs mitochondrial function and leads to muscle atrophy. Am. J. Physiol. Cell Physiol. 2018, 315, C164–C185. [Google Scholar] [CrossRef]
- Kelly, N.A.; Hammond, K.G.; Bickel, C.S.; Windham, S.T.; Tuggle, S.C.; Bamman, M.M. Effects of aging and Parkinson’s disease on motor unit remodeling: Influence of resistance exercise training. J. Appl. Physiol. 2018, 124, 888–898. [Google Scholar] [CrossRef]
- McWilliams, T.G.; Prescott, A.R.; Montava-Garriga, L.; Ball, G.; Singh, F.; Barini, E.; Muqit, M.M.K.; Brooks, S.P.; Ganley, I.G. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab. 2018, 27, 439–449. [Google Scholar] [CrossRef] [PubMed]
- Cornelissen, T.; Vilain, S.; Vints, K.; Gounko, N.; Verstreken, P.; Vandenberghe, W. Deficiency of parkin and PINK1 impairs age-dependent mitophagy in Drosophila. Elife 2018, 7. [Google Scholar] [CrossRef] [PubMed]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, A.; McLelland, G.L.; Fon, E.A.; McBride, H.M. A new pathway for mitochondrial quality control: Mitochondrial-derived vesicles. EMBO J. 2014, 33, 2142–2156. [Google Scholar] [CrossRef]
- Qaisar, R.; Larsson, L. What determines myonuclear domain size? Indian J. Physiol. Pharmacol. 2014, 58, 1–12. [Google Scholar]
- Gutmann, E.; Hanzlikova, V. Basic mechanisms of aging in the neuromuscular system. Mech. Ageing Dev. 1973, 1, 327–349. [Google Scholar] [CrossRef]
- Ashrafi, G.; Schlehe, J.S.; LaVoie, M.J.; Schwarz, T.L. Mitophagy of damaged mitochondria occurs locally in distal neuronal axons and requires PINK1 and Parkin. J. Cell Biol. 2014, 206, 655–670. [Google Scholar] [CrossRef]
- Harbauer, A.B. Mitochondrial health maintenance in axons. Biochem. Soc. Trans. 2017, 45, 1045–1052. [Google Scholar] [CrossRef]
- Amiri, M.; Hollenbeck, P.J. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev. Neurobiol. 2008, 68, 1348–1361. [Google Scholar] [CrossRef]
- Hayakawa, K.; Esposito, E.; Wang, X.; Terasaki, Y.; Liu, Y.; Xing, C.; Ji, X.; Lo, E.H. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 2016, 535, 551–555. [Google Scholar] [CrossRef]
- Duregotti, E.; Negro, S.; Scorzeto, M.; Zornetta, I.; Dickinson, B.C.; Chang, C.J.; Montecucco, C.; Rigoni, M. Mitochondrial alarmins released by degenerating motor axon terminals activate perisynaptic Schwann cells. Proc. Natl. Acad. Sci. USA 2015, 112, E497–E505. [Google Scholar] [CrossRef] [PubMed]
- Negro, S.; Bergamin, E.; Rodella, U.; Duregotti, E.; Scorzeto, M.; Jalink, K.; Montecucco, C.; Rigoni, M. ATP Released by Injured Neurons Activates Schwann Cells. Front. Cell. Neurosci. 2016, 10, 134. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Aspect | Tissue | References |
|---|---|---|
| Mitochondrial structure | Muscle | [29,42,43] |
| Mitochondrial structure | Neuron | [47] |
| Mitochondrial function | Muscle | [14,30,31,32,33,79] |
| Mitochondrial function | Neuron | None |
| Mechanism | Tissue | References |
|---|---|---|
| mtDNA alteration | Muscle | [62,63,64,72,73,74,78] |
| mtDNA alteration | Neuron | [77,80] |
| Mitochondrial dynamics | Muscle | [84,85,87,88] |
| Mitochondrial dynamics | Neuron | [86,89] |
| Mitostasis | Muscle | [97,98,99] |
| Mitostasis | Neuron | [60,94,100,102,103,107,109,110] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anagnostou, M.-E.; Hepple, R.T. Mitochondrial Mechanisms of Neuromuscular Junction Degeneration with Aging. Cells 2020, 9, 197. https://doi.org/10.3390/cells9010197
Anagnostou M-E, Hepple RT. Mitochondrial Mechanisms of Neuromuscular Junction Degeneration with Aging. Cells. 2020; 9(1):197. https://doi.org/10.3390/cells9010197
Chicago/Turabian StyleAnagnostou, Maria-Eleni, and Russell T. Hepple. 2020. "Mitochondrial Mechanisms of Neuromuscular Junction Degeneration with Aging" Cells 9, no. 1: 197. https://doi.org/10.3390/cells9010197
APA StyleAnagnostou, M.-E., & Hepple, R. T. (2020). Mitochondrial Mechanisms of Neuromuscular Junction Degeneration with Aging. Cells, 9(1), 197. https://doi.org/10.3390/cells9010197