Understanding Calcium-Dependent Conformational Changes in S100A1 Protein: A Combination of Molecular Dynamics and Gene Expression Study in Skeletal Muscle

 ,
,  ,
,  ,
, 
 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. System Building
2.2. Simulation Details
2.3. Trajectory Analysis
2.4. CTX-Induced Muscle Regeneration
2.5. C2C12 Cell Culture
2.6. RNA Isolation, cDNA Synthesis, and Real-Time RT-PCR
2.7. Western Blot
2.8. Immunocytochemistry
2.9. Immunohistochemistry
3. Results
3.1. Stability Analysis of Each System
3.2. Effect of Ca2+ on the S100A1 Protein
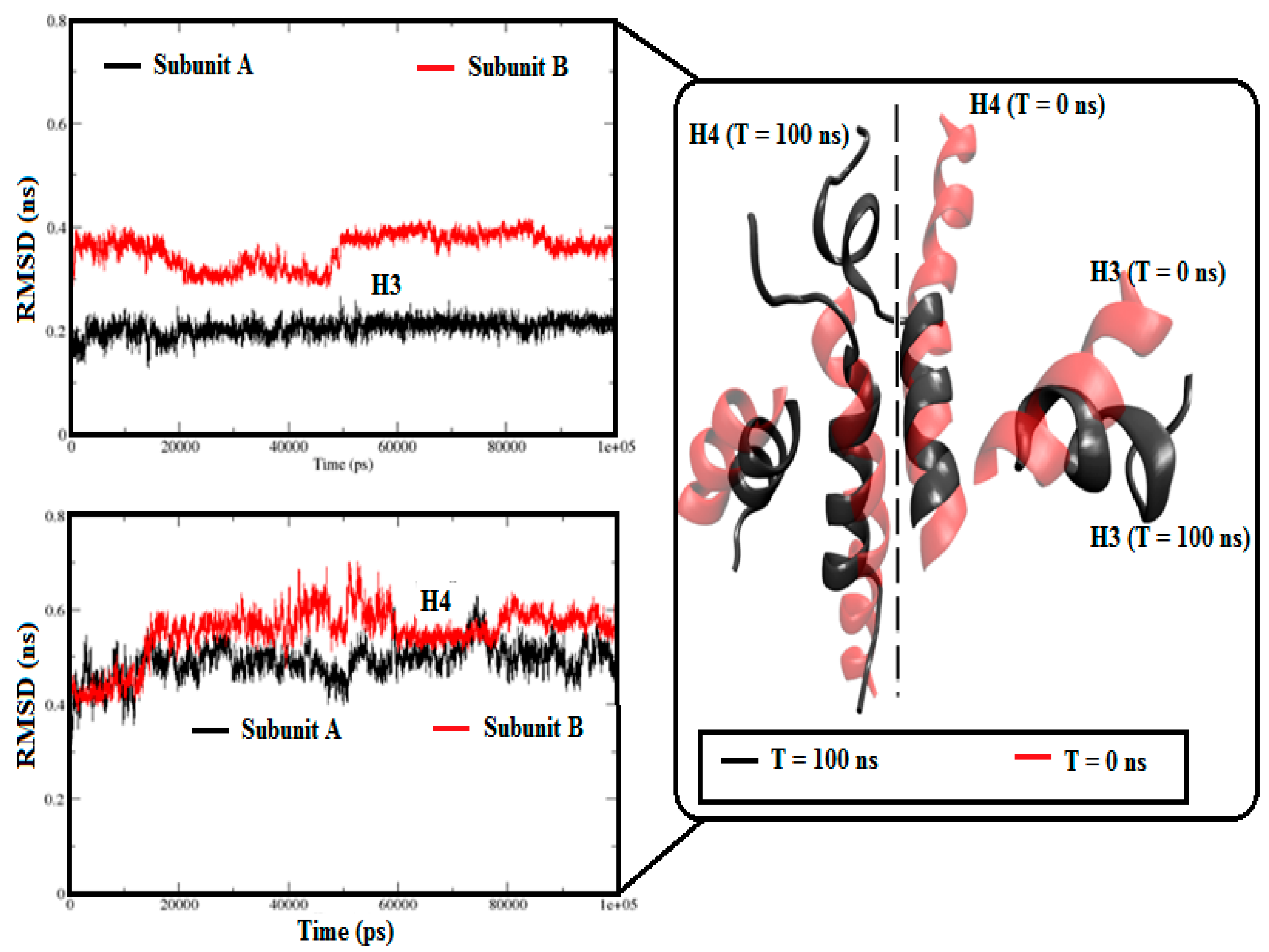
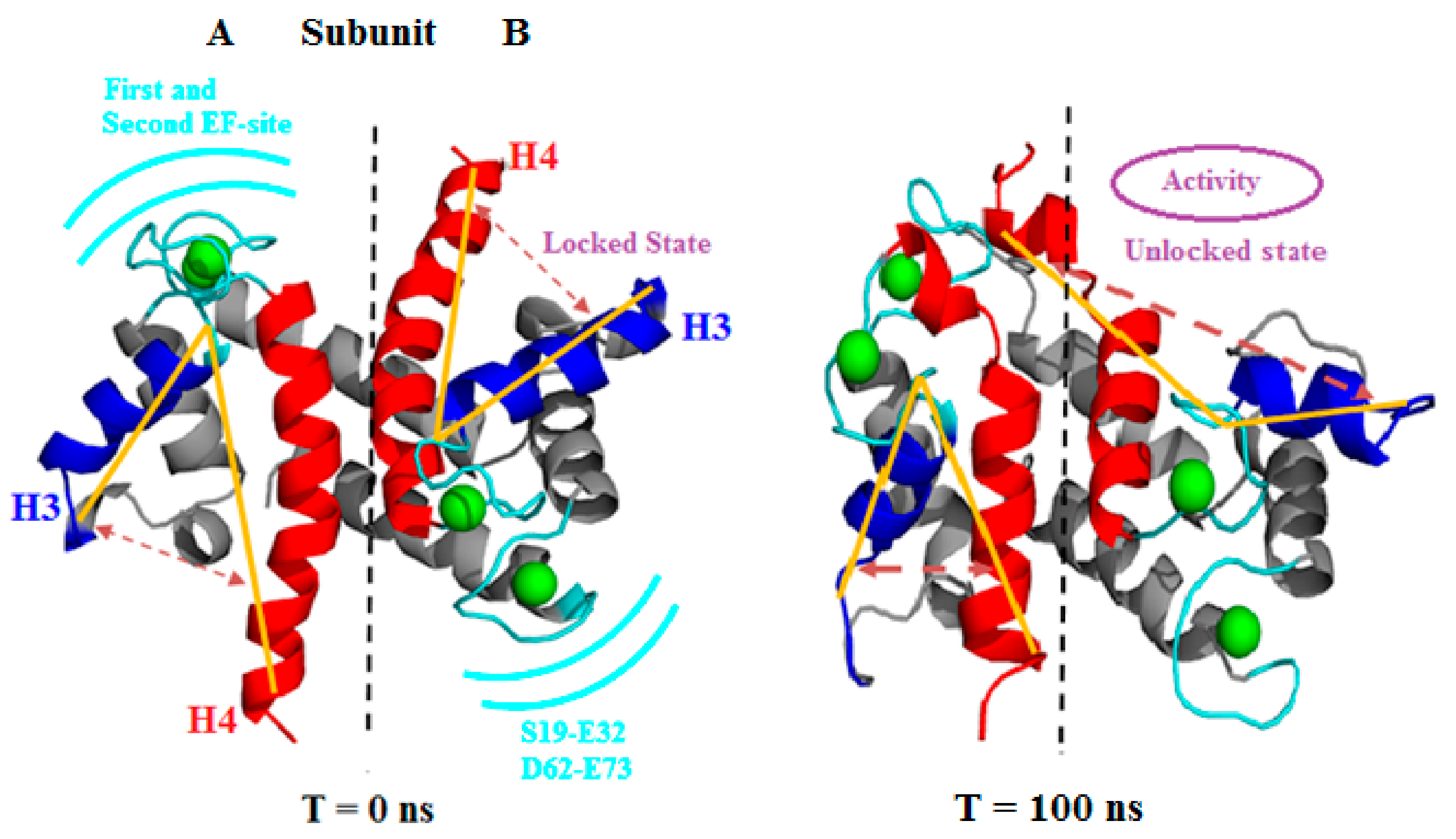
3.2.1. Angle Estimations between Helix-Loop-Helix Domains
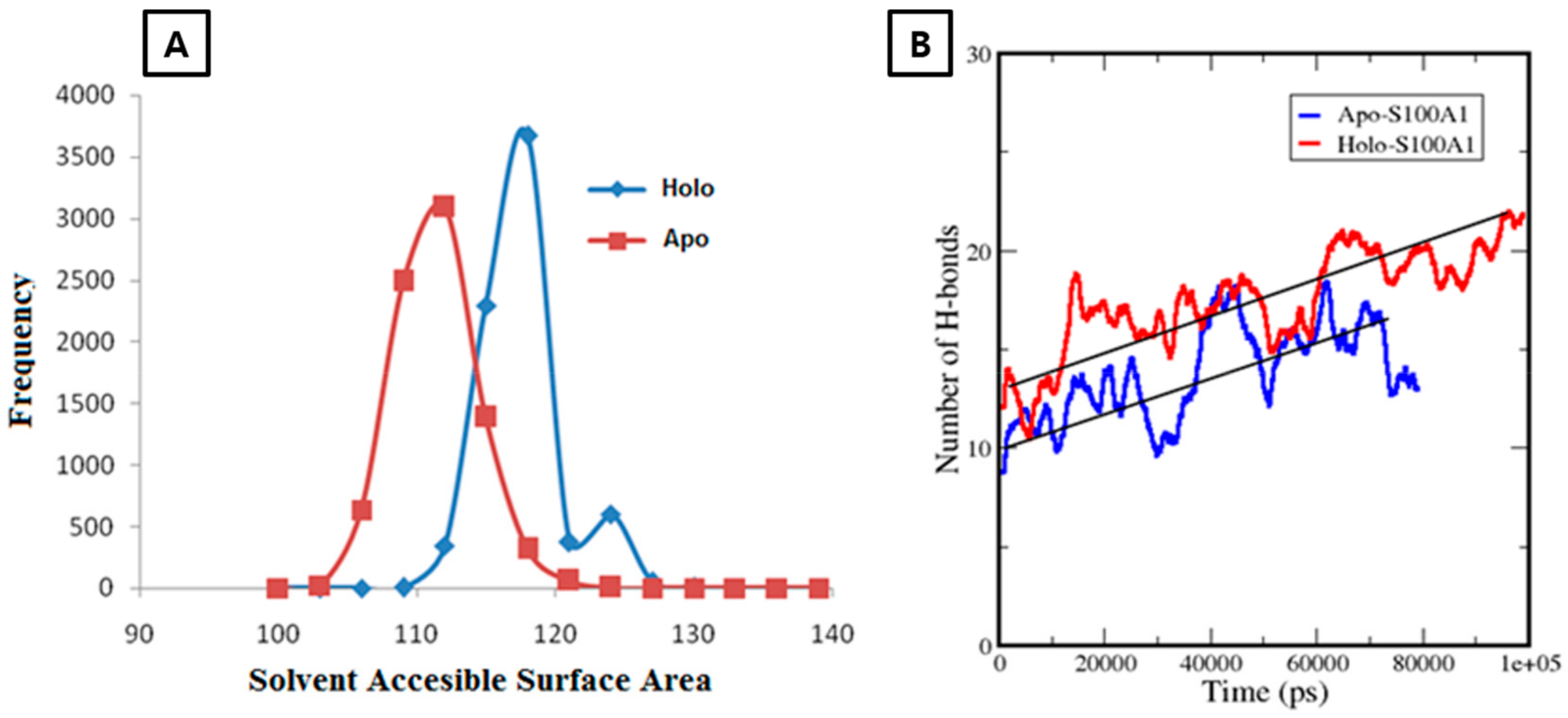
3.2.2. Calculation of a Solvent Accessible Surface Area and Hydrogen Bonds for the Apo-State and Holo-State of the Protein
3.2.3. Temporal Distributions for Apo-State and Holo-State of the Protein
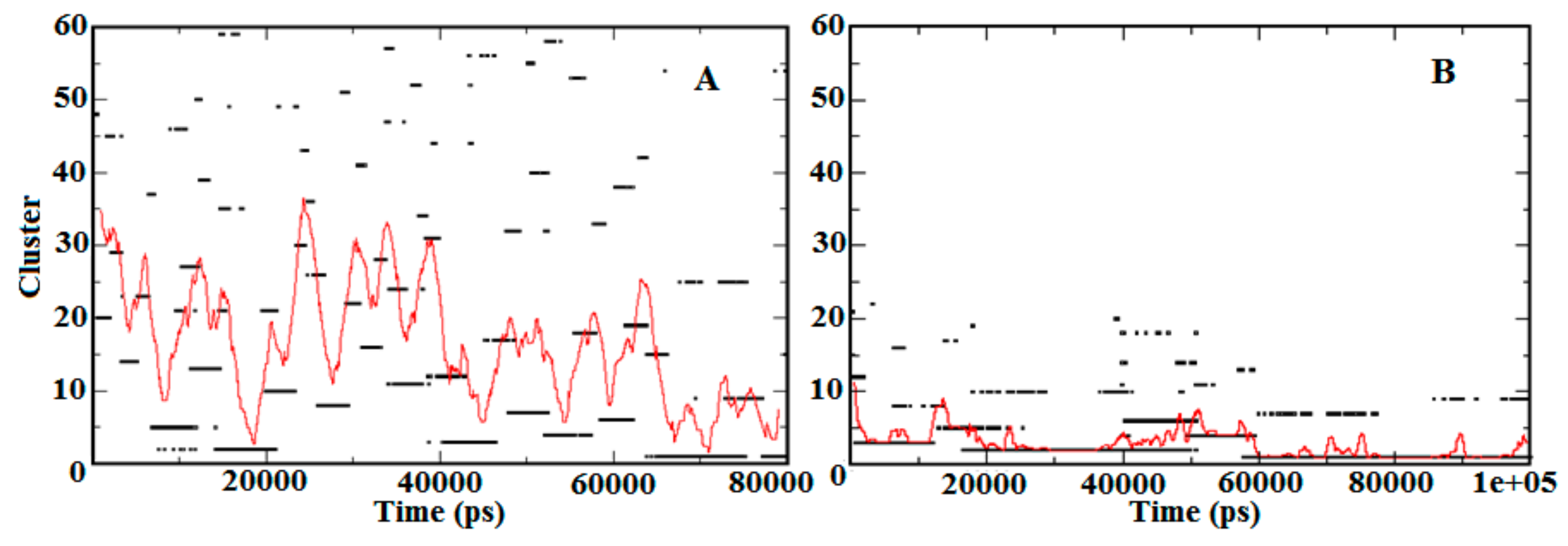
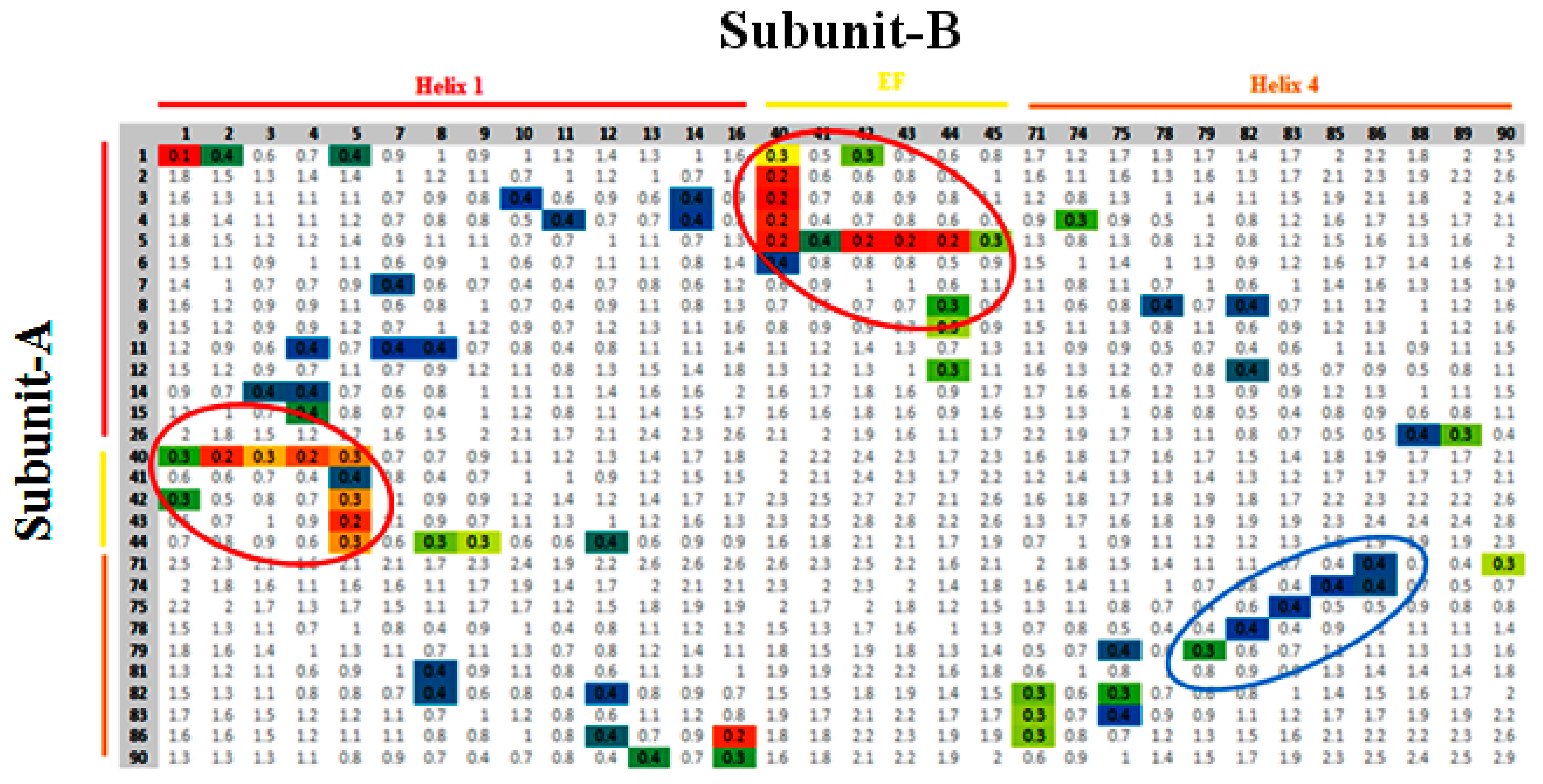
3.3. Contact Map Analysis between Subunits along the Trajectory
3.4. Monitoring of Ca2+ Binding Residues
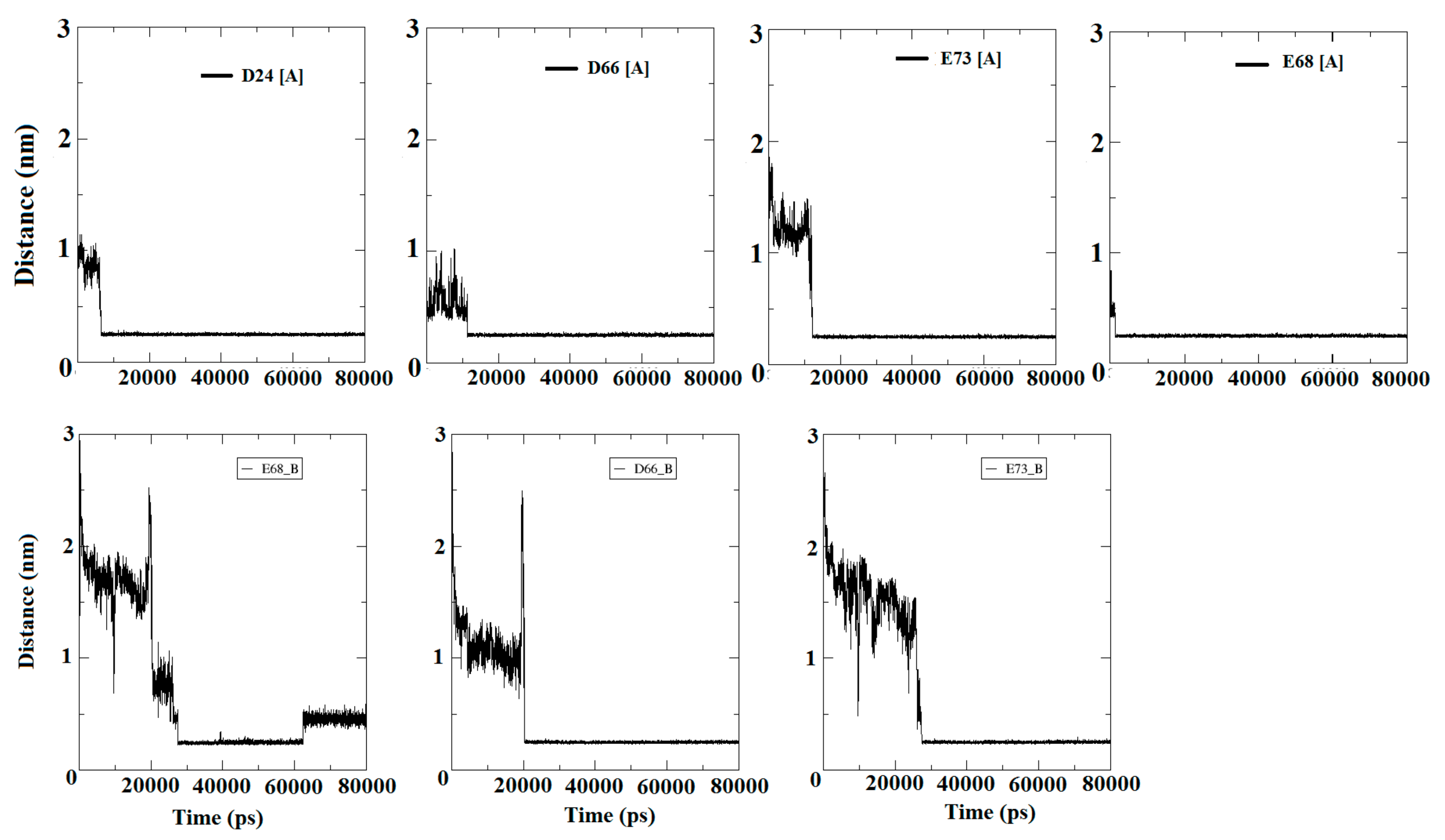
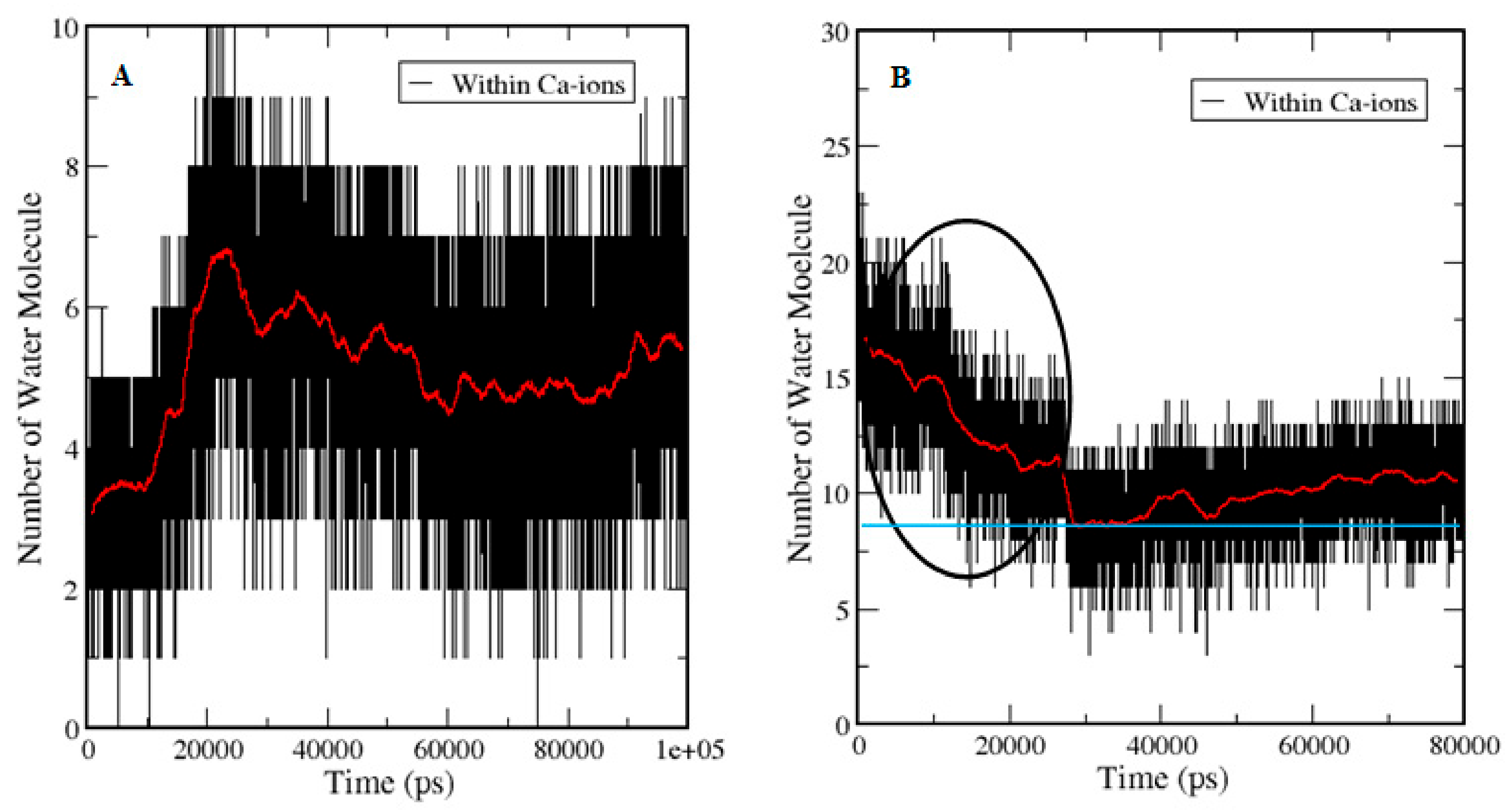
3.5. Entry of Free Ca2+ into the Bulk of the Apo-State Protein
3.5.1. Encounter of Ca2+
3.5.2. Ion Hydration and Dynamics of Water Molecules
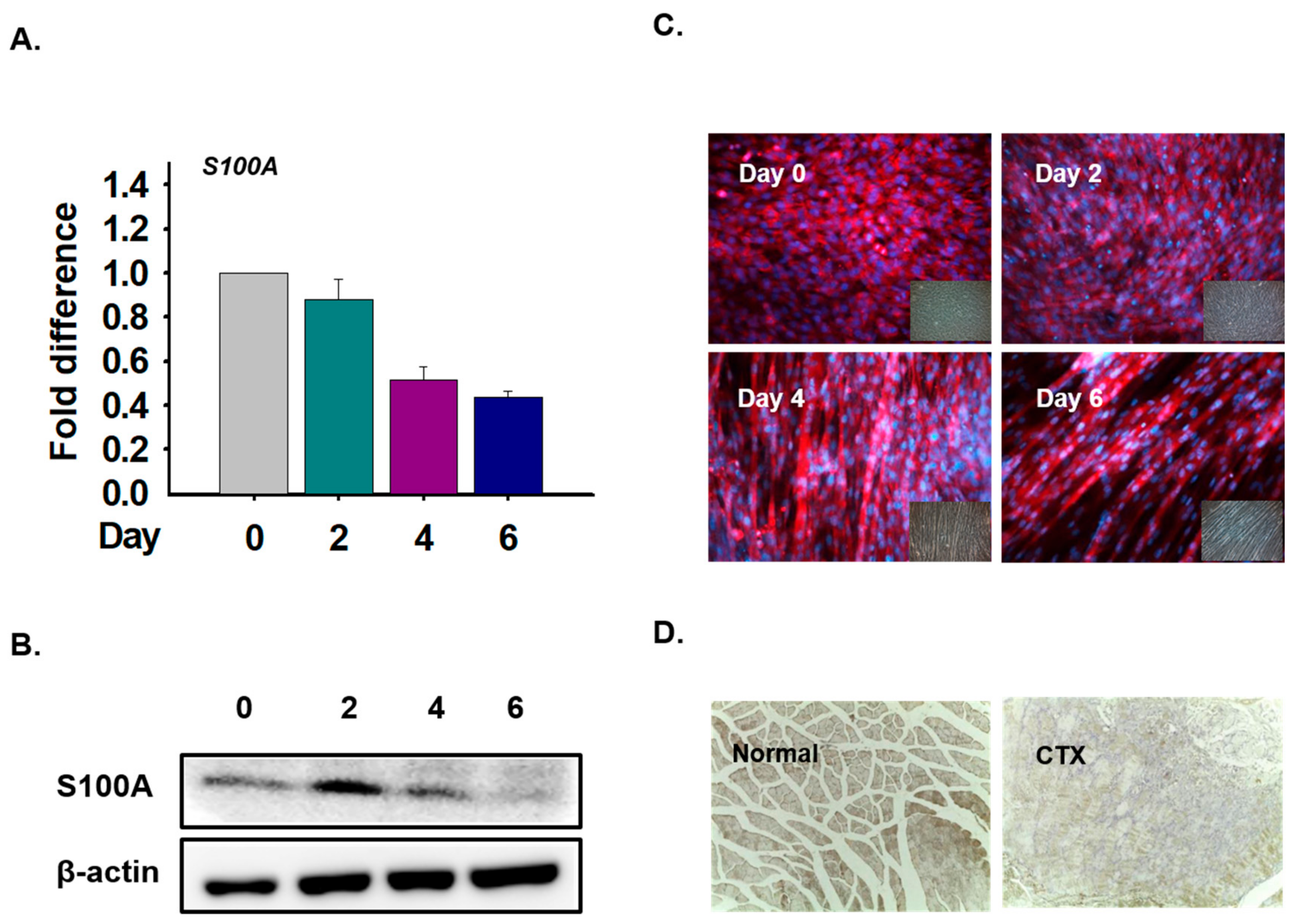
3.6. S100A Expression in Myoblast Differentiation and Muscle Regeneration
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Donato, R.; Cannon, B.R.; Sorci, G.; Riuzzi, F.; Hsu, K.; Weber, D.J.; Geczy, C.L. Functions of S100 proteins. Curr. Mol. Med. 2013, 13, 24–57. [Google Scholar] [CrossRef] [PubMed]
- Völkers, M.; Rohde, D.; Goodman, C.; Most, P. S100A1: A Regulator of Striated Muscle Sarcoplasmic Reticulum Ca<sup> 2. Bio. Med. Res. Int. 2010, 2010. [Google Scholar]
- Kiewitz, R.; Lyons, G.E.; Schäfer, B.W.; Heizmann, C.W. Transcriptional regulation of S100A1 and expression during mouse heart development. BBA-Mol. Cell Res. 2000, 1498, 207–219. [Google Scholar] [CrossRef][Green Version]
- Kraus, C.; Rohde, D.; Weidenhammer, C.; Qiu, G.; Pleger, S.T.; Voelkers, M.; Boerries, M.; Remppis, A.; Katus, H.A.; Most, P. S100A1 in cardiovascular health and disease: Closing the gap between basic science and clinical therapy. J. Mol. Cell. Cardiol. 2009, 47, 445–455. [Google Scholar] [CrossRef] [PubMed]
- Remppis, A.; Greten, T.; Schäfer, B.W.; Hunziker, P.; Erne, P.; Katus, H.A.; Heizmann, C.W. Altered expression of the Ca2+-binding protein S100A1 in human cardiomyopathy. BBA-Mol. Cell Res. 1996, 1313, 253–257. [Google Scholar] [CrossRef]
- Brinks, H.; Rohde, D.; Voelkers, M.; Qiu, G.; Pleger, S.T.; Herzog, N.; Rabinowitz, J.; Ruhparwar, A.; Silvestry, S.; Lerchenmüller, C. S100A1 genetically targeted therapy reverses dysfunction of human failing cardiomyocytes. J. Am. Coll. Cardiol. 2011, 58, 966–973. [Google Scholar] [CrossRef]
- Leclerc, E.; Heizmann, C.W. The importance of Ca2+/Zn2+ signaling S100 proteins and RAGE in translational medicine. Front. Biosci. (Schol Ed.) 2011, 3, 1232–1262. [Google Scholar]
- Carafoli, E. Calcium signaling: A tale for all seasons. Proc. Natl. Acad. Sci. USA 2002, 99, 1115–1122. [Google Scholar] [CrossRef]
- Grabarek, Z. Structural basis for diversity of the EF-hand calcium-binding proteins. J. Mol. Biol. 2006, 359, 509–525. [Google Scholar] [CrossRef]
- Ababou, A.; Zaleska, M.; Pfuhl, M. On the Ca2+ binding and conformational change in EF-hand domains: Experimental evidence of Ca2+-saturated intermediates of N-domain of calmodulin. BBA-Proteins Proteomics 2017, 1865, 640–651. [Google Scholar] [CrossRef]
- Wright, N.T.; Varney, K.M.; Ellis, K.C.; Markowitz, J.; Gitti, R.K.; Zimmer, D.B.; Weber, D.J. The three-dimensional solution structure of Ca2+-bound S100A1 as determined by NMR spectroscopy. J. Mol. Biol. 2005, 353, 410–426. [Google Scholar] [CrossRef] [PubMed]
- Romano, D.R.; Pharris, M.C.; Patel, N.M.; Kinzer-Ursem, T.L. Competitive tuning: Competition’s role in setting the frequency-dependence of Ca2+-dependent proteins. PLoS Comput. Biol. 2017, 13, e1005820. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.T.; Cannon, B.R.; Wilder, P.T.; Morgan, M.T.; Varney, K.M.; Zimmer, D.B.; Weber, D.J. Solution structure of S100A1 bound to the CapZ peptide (TRTK12). J. Mol. Biol. 2009, 386, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Finn, B.E.; Drakenberg, T.; Forsén, S. The structure of apo-calmodulin: A 1H NMR examination of the carboxy-terminal domain. FEBS Lett. 1993, 336, 368–374. [Google Scholar] [CrossRef]
- Finn, B.E.; Evenäs, J.; Drakenberg, T.; Waltho, J.P.; Thulin, E.; Forsén, S. Calcium-induced structural changes and domain autonomy in calmodulin. Nat. Struct. Biol. 1995, 2, 777. [Google Scholar] [CrossRef]
- Tan, R.-Y.; Mabuchi, Y.; Grabarek, Z. Blocking the Ca-induced conformational transitions in calmodulin with disulfide bonds. J. Biol. Chem. 1996, 271, 7479–7483. [Google Scholar] [CrossRef]
- Moorthy, A.K.; Murthy, M. Conformation and structural transitions in the EF-hands of calmodulin. J. Biomol. Struct. Dyn. 2001, 19, 47–57. [Google Scholar] [CrossRef]
- González-Andrade, M.; Rodríguez-Sotres, R.; Madariaga-Mazón, A.; Rivera-Chávez, J.; Mata, R.; Sosa-Peinado, A.; Pozo-Yauner, L.D.; Arias-Olguin, I.I. Insights into molecular interactions between CaM and its inhibitors from molecular dynamics simulations and experimental data. J. Biomol. Struct. Dyn. 2016, 34, 78–91. [Google Scholar] [CrossRef]
- Scott, C.E.; Kekenes-Huskey, P.M. Molecular basis of S100A1 activation at saturating and subsaturating calcium concentrations. Biophys. J. 2016, 110, 1052–1063. [Google Scholar] [CrossRef][Green Version]
- Berchtold, M.W.; Brinkmeier, H.; Muntener, M. Calcium ion in skeletal muscle: Its crucial role for muscle function, plasticity, and disease. Physiol. Rev. 2000, 80, 1215–1265. [Google Scholar] [CrossRef]
- Lee, E.J.; Nam, J.H.; Choi, I. Fibromodulin modulates myoblast differentiation by controlling calcium channel. Biochem. Biophys. Res. Commun. 2018, 503, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kamli, M.R.; Pokharel, S.; Malik, A.; Tareq, K.M.; Roouf Bhat, A.; Park, H.B.; Lee, Y.S.; Kim, S.; Yang, B.; et al. Expressed sequence tags for bovine muscle satellite cells, myotube formed-cells and adipocyte-like cells. PLoS ONE 2013, 8, e79780. [Google Scholar] [CrossRef]
- Bentzinger, C.; von Maltzahn, J.; Rudnicki, M.A. Extrinsic regulation of satellite cell specification. Stem Cell Res. Ther. 2010, 1, 27. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.S.; Dellavalle, A.; Diaz-Manera, J.; Messina, G.; Cossu, G. Repairing skeletal muscle: Regenerative potential of skeletal muscle stem cells. J. Clin. Invest. 2010, 120, 11–19. [Google Scholar] [CrossRef]
- Živković, M.L.; Zaręba-Kozioł, M.; Zhukova, L.; Poznański, J.; Zhukov, I.; Wysłouch-Cieszyńska, A. Post-translational S-nitrosylation is an endogenous factor fine tuning the properties of human S100A1 protein. J. Biol. Chem. 2012, 287, 40457–40470. [Google Scholar] [CrossRef]
- Nowakowski, M.; Ruszczyńska-Bartnik, K.; Budzińska, M.; Jaremko, Ł.; Jaremko, M.; Zdanowski, K.; Bierzyński, A.; Ejchart, A. Impact of calcium binding and thionylation of S100A1 protein on its nuclear magnetic resonance-derived structure and backbone dynamics. Biochemistry 2013, 52, 1149–1159. [Google Scholar] [CrossRef]
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J. GROMACS: Fast, flexible, and free. J. Comput. Chem. 2005, 26, 1701–1718. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A biomolecular force field based on the free enthalpy of hydration and solvation: The GROMOS force-field parameter sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Project, E.; Nachliel, E.; Gutman, M. Parameterization of Ca+ 2–protein interactions for molecular dynamics simulations. J. Comput. Chem. 2008, 29, 1163–1169. [Google Scholar] [CrossRef]
- Yadav, B.S.; Chaturvedi, N.; Yadav, P.K.; Marina, N.; Ganash, M.; Barreto, G.E.; Ashraf, G.M.; Ahmad, K.; Baig, M.H. Protein modeling, molecular network and molecular dynamics study of newly sequenced interleukin-18 (IL-18) gene in Mus musculus. J. Cell Physiol. 2019, 234, 14285–14295. [Google Scholar] [CrossRef]
- Ahmad, K.; Balaramnavar, V.M.; Chaturvedi, N.; Khan, S.; Haque, S.; Lee, Y.H.; Choi, I. Targeting Caspase 8: Using Structural and Ligand-Based Approaches to Identify Potential Leads for the Treatment of Multi-Neurodegenerative Diseases. Molecules 2019, 24, 1827. [Google Scholar] [CrossRef] [PubMed]
- Toukan, K.; Rahman, A. Molecular-dynamics study of atomic motions in water. Phys. Rev. B 1985, 31, 2643. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graphics 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Lee, E.J.; Jan, A.T.; Baig, M.H.; Ashraf, J.M.; Nahm, S.-S.; Kim, Y.-W.; Park, S.-Y.; Choi, I. Fibromodulin: A master regulator of myostatin controlling progression of satellite cells through a myogenic program. FASEB J. 2016, 30, 2708–2719. [Google Scholar] [CrossRef]
- Amber-Vitos, O.; Chaturvedi, N.; Nachliel, E.; Gutman, M.; Tsfadia, Y. The effect of regulating molecules on the structure of the PPAR-RXR complex. BBA-Mol. Cell Biol. Lipids 2016, 1861, 1852–1863. [Google Scholar] [CrossRef]
- Friedman, R.; Nachliel, E.; Gutman, M. Molecular dynamics of a protein surface: Ion-residues interactions. Biophys. J. 2005, 89, 768–781. [Google Scholar] [CrossRef]
- Gdalya, H.; Nachliel, E.; Gutman, M.; Einav, Y.; Tsfadia, Y. The translocation of Na+ ion inside human thrombin accounts for the activation of the enzyme. Isr. J. Chem. 2017, 57, 413–423. [Google Scholar] [CrossRef]
- Wojtas, M.; Hołubowicz, R.; Poznar, M.; Maciejewska, M.; Ożyhar, A.; Dobryszycki, P. Calcium ion binding properties and the effect of phosphorylation on the intrinsically disordered starmaker protein. Biochemistry 2015, 54, 6525–6534. [Google Scholar] [CrossRef]
- Di Tommaso, D.; Ruiz-Agudo, E.; de Leeuw, N.H.; Putnis, A.; Putnis, C.V. Modelling the effects of salt solutions on the hydration of calcium ions. Phys. Chem. Chem. Phys. 2014, 16, 7772–7785. [Google Scholar] [CrossRef]
- Steiner, J.; Bogerts, B.; Schroeter, M.L.; Bernstein, H.-G. S100B protein in neurodegenerative disorders. Clin. Chem. Lab. Med. 2011, 49, 409–424. [Google Scholar] [CrossRef]
- Cmoch, A.; Groves, P.; Palczewska, M.; Pikula, S. S100A proteins in propagation of a calcium signal in norm and pathology. Postepy Biochem. 2012, 58, 429–436. [Google Scholar] [PubMed]
- Chaturvedi, N.; Pandey, P.N. Phylogenetic analysis of gammaproteobacterial arsenate reductase proteins specific to Enterobacteriaceae family, signifying arsenic toxicity. Interdisciplin. Sci.: Comput. Life Sci. 2014, 6, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.; Kajsik, M.; Forsythe, S.; Pandey, P.N. Protein sequences insight into heavy metal tolerance in Cronobacter sakazakii BAA-894 encoded by plasmid pESA3. Arch. Microbial. 2015, 197, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Rustandi, R.R.; Baldisseri, D.M.; Inman, K.G.; Nizner, P.; Hamilton, S.M.; Landar, A.; Landar, A.; Zimmer, D.B.; Weber, D.J. Three-dimensional solution structure of the calcium-signaling protein apo-S100A1 as determined by NMR. Biochemistry 2002, 41, 788–796. [Google Scholar] [CrossRef] [PubMed]
- Wright, N.T.; Prosser, B.L.; Varney, K.M.; Zimmer, D.B.; Schneider, M.F.; Weber, D.J. S100A1 and calmodulin compete for the same binding site on ryanodine receptor. J. Biol. Chem. 2008, 283, 26676–26683. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, N.; Yadav, B.S.; Pandey, P.N.; Tripathi, V. The effect of β-glucan and its potential analog on the structure of Dectin-1 receptor. J. Mol. Graphics Modell. 2017, 74, 315–325. [Google Scholar] [CrossRef]
- Kazakov, A.S.; Sokolov, A.S.; Vologzhannikova, A.A.; Permyakova, M.E.; Khorn, P.A.; Ismailov, R.G.; Denessiouk, K.A.; Denesyuk, A.I.; Rastrygina, V.A.; Baksheeva, V.E. Interleukin-11 binds specific EF-hand proteins via their conserved structural motifs. J. Biomol. Struct. Dyn. 2017, 35, 78–91. [Google Scholar] [CrossRef]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nature Rev. Mol. Cell Biol. 2000, 1, 11. [Google Scholar] [CrossRef]
- Shannon, T.R.; Wang, F.; Puglisi, J.; Weber, C.; Bers, D.M. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys. J. 2004, 87, 3351–3371. [Google Scholar] [CrossRef]
- Su, A.I.; Wiltshire, T.; Batalov, S.; Lapp, H.; Ching, K.A.; Block, D.; Zhang, J.; Soden, R.; Hayakawa, M.; Kreiman, G. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc. Natl. Acad. Sci. USA 2004, 101, 6062–6067. [Google Scholar] [CrossRef]
- Pleger, S.T.; Most, P.; Boucher, M.; Soltys, S.; Chuprun, J.K.; Pleger, W.; Gao, E.; Dasgupta, A.; Rengo, G.; Remppis, A. CLINICAL PERSPECTIVE. Circulation 2007, 115, 2506–2515. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Wright, N.T.; Hernandez-Ochoa, E.O.; Varney, K.M.; Liu, Y.; Olojo, R.O.; Zimmer, D.B.; Weber, D.J.; Schneider, M.F. S100A1 binds to the calmodulin-binding site of ryanodine receptor and modulates skeletal muscle excitation-contraction coupling. J. Biol. Chem. 2008, 283, 5046–5057. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, N.; Prosser, B.L.; Ghassemi, F.; Xu, L.; Pasek, D.A.; Eu, J.P.; Hernández-Ochoa, E.O.; Cannon, B.R.; Wilder, P.T.; Lovering, R.M. Modulation of sarcoplasmic reticulum Ca2+ release in skeletal muscle expressing ryanodine receptor impaired in regulation by calmodulin and S100A1. Am. J. Physiol.-Cell Physiol. 2011, 300, C998–C1012. [Google Scholar] [CrossRef]
- Numa, S.; Tanabe, T.; Takeshima, H.; Mikami, A.; Niidome, T.; Nishimura, S.; Adams, B.; Beam, K. Molecular insights into excitation-contraction coupling. In Proceedings of Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1990; Volume 55, pp. 1–7. [Google Scholar] [CrossRef]
- Mori, Y.; Mori, D.; Chung, U.I.; Tanaka, S.; Heierhorst, J.; Buchou, T.; Baudier, J.; Kawaguchi, H.; Saito, T. S100A1 and S100B are dispensable for endochondral ossification during skeletal development. Biomed. Res. 2014, 35, 243–250. [Google Scholar] [CrossRef] [PubMed]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Serial No | System | PDB id | Number of Ca2+ in Solvent Phase | Duration of Simulations (ns) | |
|---|---|---|---|---|---|
| Simulation-1 | Simulation-2 | ||||
| 1. | Apo-S100A1 | 2LLU | nil | 80 | 0 |
| 2. | Holo-S100A1 | 2LP3 | 4 | 100 | 80 |
| 3. | Entry of free Ca2+ | 2LLU | 4 | 80 | 80 |
| System | Subunit A | Subunit B | ||||
|---|---|---|---|---|---|---|
| 10 ns | 40 ns | 80 ns/100 ns | 10 ns | 40 ns | 80 ns/100 ns | |
| Apo-S100A1 | 64.34 +/− 5.82 | 68.71 +/− 5.18 | 69.82 +/− 4.72 | 46.42 +/− 6.86 | 54.89 +/− 13.34 | 66.94 +/− 17.02 |
| Holo-S100A1 | 65.65 +/−5.12 | 72.19 +/− 7.59 | 79.94 +/− 9.97 | 66.40 +/− 1.64 | 79.66 +/− 6.58 | 106.13 +/− 11.99 |
| System | Time | ||
|---|---|---|---|
| Entry of Ca2+-S100A1 | 10 ns | 40 ns | 80 ns |
| Subunit A | 66.75 +/− 8.23 | 77.50 +/− 5.97 | 80.31 +/− 5.84 |
| Subunit B | 47.34 +/− 5.96 | 42.30 +/− 5.93 | 62.01 +/− 8.76 |
| Residues | Ca2+ Encounter on Subunit A (Time in ps) | TRT (ps) | Ca2+ Encounter on Subunit B (Time in ps) | TRT (ps) | ||
|---|---|---|---|---|---|---|
| FET | SET | FET | SET | |||
| D24 | 7948 | 0 | 72,057 | 9411 | 0 | 33,566 |
| E32 | 0 | 0 | 0 | 0 | 0 | 0 |
| K27 | 58,594 | 77,385 | 22,856 | 0 | 0 | 0 |
| D62 | 0 | 0 | 0 | 588 | 0 | 19,118 |
| D66 | 11,028 | 0 | 68,975 | 20,135 | 59,865 | |
| E68 | 280 | 0 | 79,720 | 22,750 | 0 | 40,004 |
| E73 | 588 | 0 | 79,412 | 28,882 | 0 | 51,118 |
| S29 | 3202 | 11,046 | 5394 | 21,156 | 25,750 | 1548 |
| N64 | 10,046 | 0 | 1980 | 0 | 0 | 0 |
| S19 | 0 | 0 | 0 | 0 | 0 | 0 |
| E63 | 11,699 | 41,928 | 15,686 | 1078 | 0 | 18,138 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chaturvedi, N.; Ahmad, K.; Yadav, B.S.; Lee, E.J.; Sonkar, S.C.; Marina, N.; Choi, I. Understanding Calcium-Dependent Conformational Changes in S100A1 Protein: A Combination of Molecular Dynamics and Gene Expression Study in Skeletal Muscle. Cells 2020, 9, 181. https://doi.org/10.3390/cells9010181
Chaturvedi N, Ahmad K, Yadav BS, Lee EJ, Sonkar SC, Marina N, Choi I. Understanding Calcium-Dependent Conformational Changes in S100A1 Protein: A Combination of Molecular Dynamics and Gene Expression Study in Skeletal Muscle. Cells. 2020; 9(1):181. https://doi.org/10.3390/cells9010181
Chicago/Turabian StyleChaturvedi, Navaneet, Khurshid Ahmad, Brijesh Singh Yadav, Eun Ju Lee, Subash Chandra Sonkar, Ninoslav Marina, and Inho Choi. 2020. "Understanding Calcium-Dependent Conformational Changes in S100A1 Protein: A Combination of Molecular Dynamics and Gene Expression Study in Skeletal Muscle" Cells 9, no. 1: 181. https://doi.org/10.3390/cells9010181
APA StyleChaturvedi, N., Ahmad, K., Yadav, B. S., Lee, E. J., Sonkar, S. C., Marina, N., & Choi, I. (2020). Understanding Calcium-Dependent Conformational Changes in S100A1 Protein: A Combination of Molecular Dynamics and Gene Expression Study in Skeletal Muscle. Cells, 9(1), 181. https://doi.org/10.3390/cells9010181