Single-Cell Expression Variability Implies Cell Function



Abstract
1. Introduction
2. Materials and Methods
2.1. LCL Cell Culture and scRNA-seq Experiment
2.2. Non-LCL scRNA-seq Data Sets
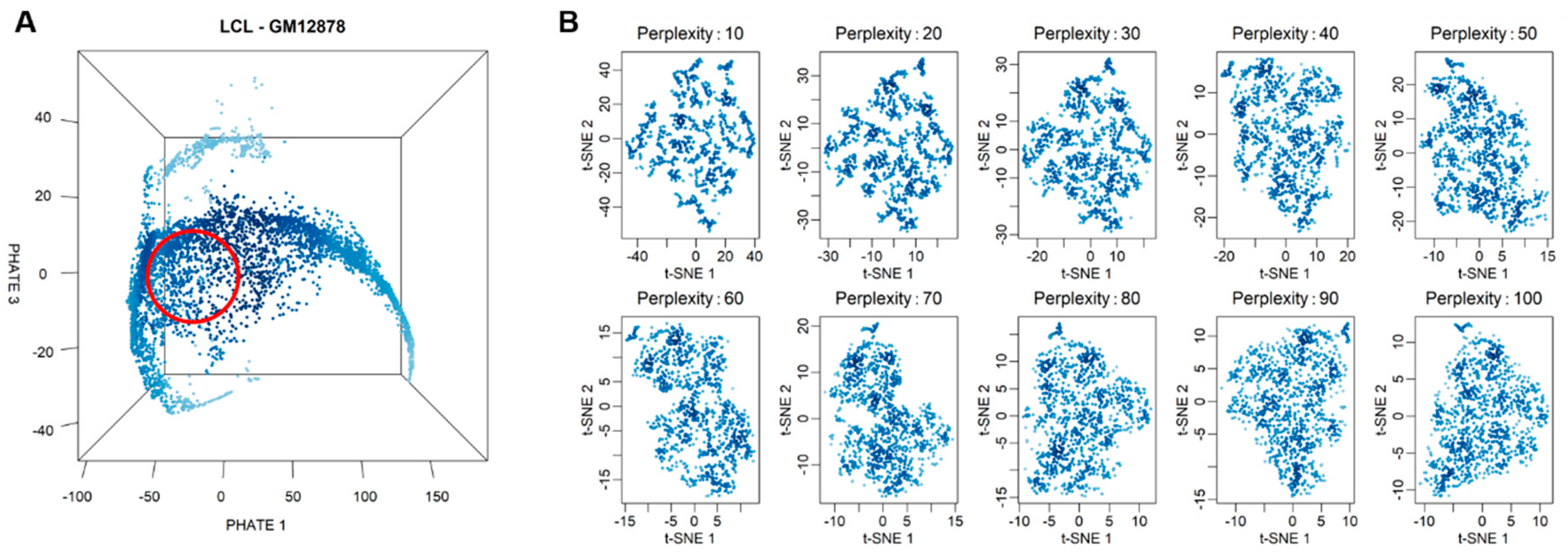
2.3. Selection of Highly Homogeneous Populations of Cells
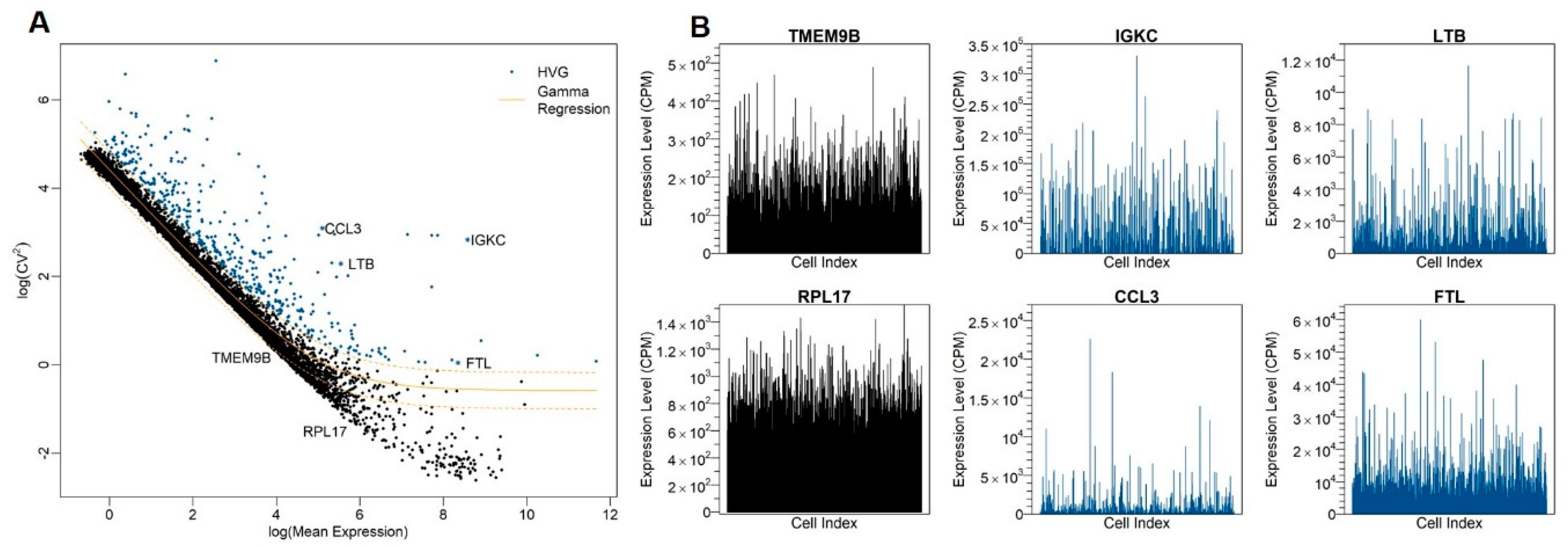
2.4. Identification of HVGs
2.5. Function Enrichment Analyses
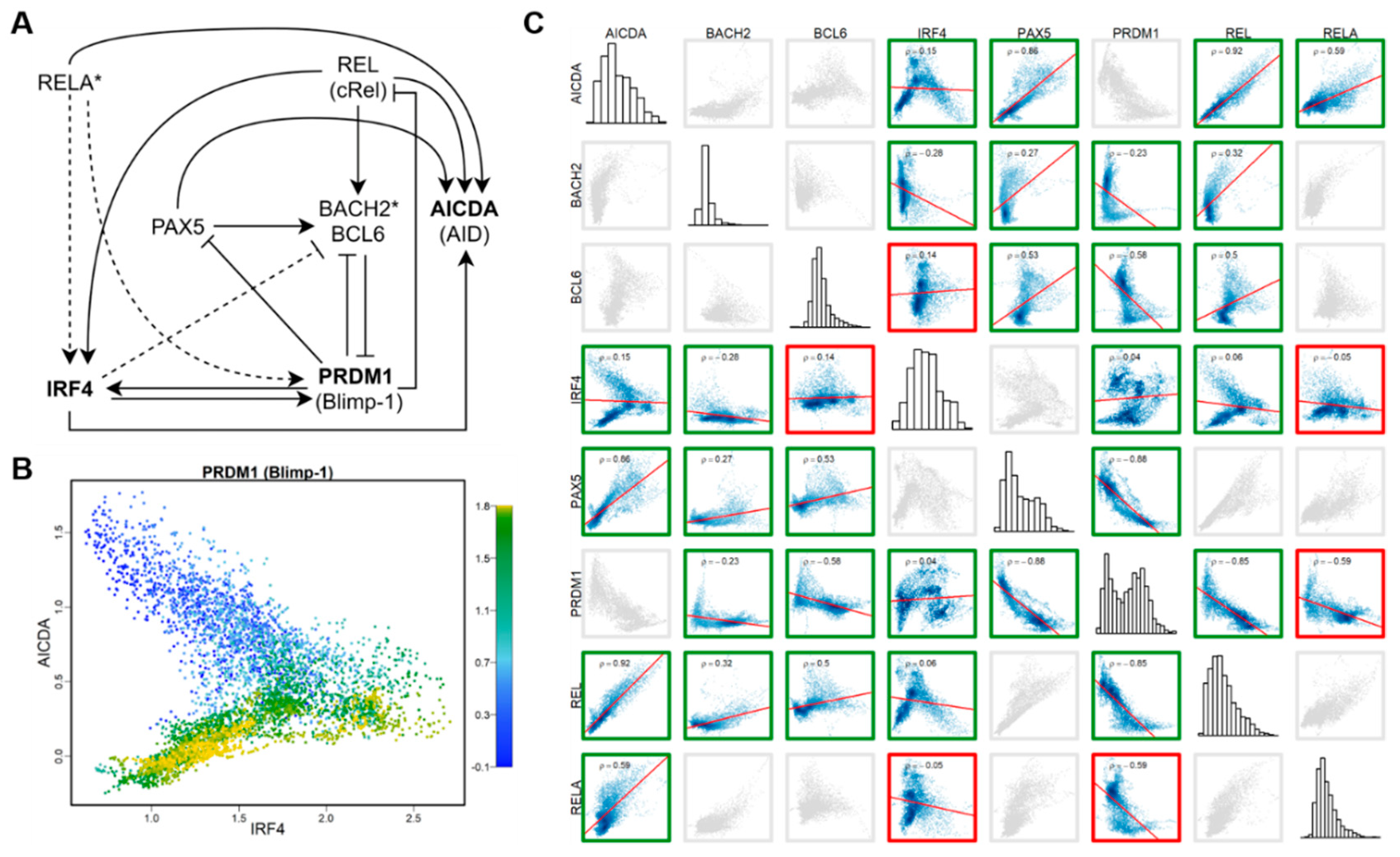
2.6. Analyses of co-Expression Network and Regulatory Regions of HVGs
2.7. Data Availability
- LCLs GM12878 scRNA-seq data in the GEO database with accession number GSE126321: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE126321
- LAEC scRNA-seq data in the GEO database with accession number GSE115982: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE115982
- DF scRNA-seq data in ArrayExpress database with accession number E-MTAB-5988: https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-5988
- Human iPSC scRNA-seq data in ArrayExpress database with accession number E-MTAB-6687: https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6687
- Fibroblasts scRNA-seq data in GEO database with accession number GSM2894834: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM2894834
- Fibroblasts scRNA-seq data in GEO database with accession number GSM2894835: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSM2894835
- Human iPSC scRNA-seq data in ArrayExpress database with accession number E-MTAB-6268: https://www.ebi.ac.uk/arrayexpress/experiments/E-MTAB-6268/
- Computer codes used to analyze data: https://github.com/cailab-tamu/HVG
3. Results
3.1. Single-Cell RNA Sequencing and Selection of Highly Homogenous Cells
3.2. Identification of Highly Variable Genes
3.3. Cell-Type Origin Determines the Function of Highly Variable Genes
3.4. Functions Associated to HVGs are Conserved Across Tissues and Subpopulations of Cells
3.5. HVGs as Part of the Regulatory Network with High Cell-Type Specificity
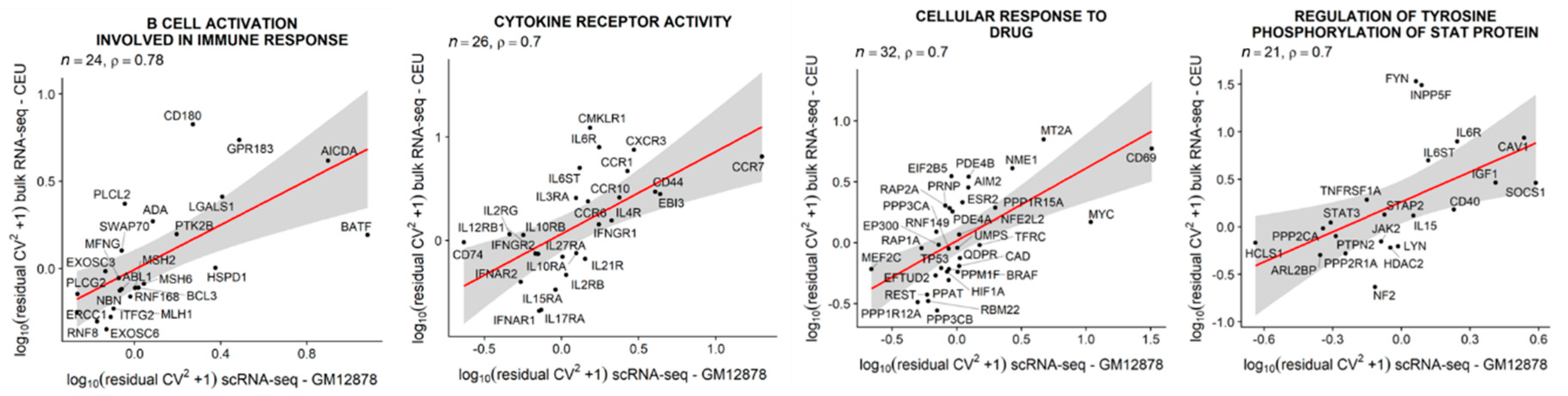
3.6. Single-Cell Expression Variability in LCLs is Positively Correlated with between-Individual Expression Variability
3.7. No Enriched Functions Associated with HVGs Identified in Human Induced Pluripotent Stem Cells (iPSCs)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, X.; Li, T.; Liu, F.; Chen, Y.; Yao, J.; Li, Z.; Huang, Y.; Wang, J. Comparative Analysis of Droplet-Based Ultra-High-Throughput Single-Cell RNA-Seq Systems. Mol. Cell 2019, 73, 130–142 e135. [Google Scholar] [CrossRef] [PubMed]
- Buettner, F.; Natarajan, K.N.; Casale, F.P.; Proserpio, V.; Scialdone, A.; Theis, F.J.; Teichmann, S.A.; Marioni, J.C.; Stegle, O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nat. Biotechnol. 2015, 33, 155–160. [Google Scholar] [CrossRef]
- Trapnell, C. Defining cell types and states with single-cell genomics. Genome Res. 2015, 25, 1491–1498. [Google Scholar] [CrossRef]
- Ko, M.S. Induction mechanism of a single gene molecule: Stochastic or deterministic? Bioessays 1992, 14, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Fiering, S.; Whitelaw, E.; Martin, D.I. To be or not to be active: The stochastic nature of enhancer action. Bioessays 2000, 22, 381–387. [Google Scholar] [CrossRef]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Eldar, A.; Elowitz, M.B. Functional roles for noise in genetic circuits. Nature 2010, 467, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Pelkmans, L. Cell Biology. Using cell-to-cell variability--a new era in molecular biology. Science 2012, 336, 425–426. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Tan, Y.; Cahan, P. Understanding development and stem cells using single cell-based analyses of gene expression. Development 2017, 144, 17–32. [Google Scholar] [CrossRef] [PubMed]
- Wernet, M.F.; Mazzoni, E.O.; Celik, A.; Duncan, D.M.; Duncan, I.; Desplan, C. Stochastic spineless expression creates the retinal mosaic for colour vision. Nature 2006, 440, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.H.; Hemberg, M.; Barahona, M.; Ingber, D.E.; Huang, S. Transcriptome-wide noise controls lineage choice in mammalian progenitor cells. Nature 2008, 453, 544–547. [Google Scholar] [CrossRef] [PubMed]
- Faure, A.J.; Schmiedel, J.M.; Lehner, B. Systematic Analysis of the Determinants of Gene Expression Noise in Embryonic Stem Cells. Cell Syst. 2017, 5, 471–484. [Google Scholar] [CrossRef]
- Martinez-Jimenez, C.P.; Eling, N.; Chen, H.C.; Vallejos, C.A.; Kolodziejczyk, A.A.; Connor, F.; Stojic, L.; Rayner, T.F.; Stubbington, M.J.T.; Teichmann, S.A.; et al. Aging increases cell-to-cell transcriptional variability upon immune stimulation. Science 2017, 355, 1433–1436. [Google Scholar] [CrossRef] [PubMed]
- Wiley, C.D.; Flynn, J.M.; Morrissey, C.; Lebofsky, R.; Shuga, J.; Dong, X.; Unger, M.A.; Vijg, J.; Melov, S.; Campisi, J. Analysis of individual cells identifies cell-to-cell variability following induction of cellular senescence. Aging Cell 2017, 16, 1043–1050. [Google Scholar] [CrossRef]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308 e1236. [Google Scholar] [CrossRef]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andreasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [PubMed]
- Dueck, H.; Eberwine, J.; Kim, J. Variation is function: Are single cell differences functionally important?: Testing the hypothesis that single cell variation is required for aggregate function. Bioessays 2016, 38, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Tay, S.; Hughey, J.J.; Lee, T.K.; Lipniacki, T.; Quake, S.R.; Covert, M.W. Single-cell NF-kappaB dynamics reveal digital activation and analogue information processing. Nature 2010, 466, 267–271. [Google Scholar] [CrossRef]
- Raj, A.; Rifkin, S.A.; Andersen, E.; van Oudenaarden, A. Variability in gene expression underlies incomplete penetrance. Nature 2010, 463, 913–918. [Google Scholar] [CrossRef]
- Mitchell, S.; Roy, K.; Zangle, T.A.; Hoffmann, A. Nongenetic origins of cell-to-cell variability in B lymphocyte proliferation. Proc. Natl. Acad. Sci. USA 2018, 115, E2888–E2897. [Google Scholar] [CrossRef]
- Snijder, B.; Sacher, R.; Ramo, P.; Damm, E.M.; Liberali, P.; Pelkmans, L. Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 2009, 461, 520–523. [Google Scholar] [CrossRef] [PubMed]
- Kernfeld, E.M.; Genga, R.M.J.; Neherin, K.; Magaletta, M.E.; Xu, P.; Maehr, R. A Single-Cell Transcriptomic Atlas of Thymus Organogenesis Resolves Cell Types and Developmental Maturation. Immunity 2018, 48, 1258–1270 e1256. [Google Scholar] [CrossRef] [PubMed]
- Miragaia, R.J.; Zhang, X.; Gomes, T.; Svensson, V.; Ilicic, T.; Henriksson, J.; Kar, G.; Lonnberg, T. Single-cell RNA-sequencing resolves self-antigen expression during mTEC development. Sci. Rep. 2018, 8, 685. [Google Scholar] [CrossRef] [PubMed]
- McDavid, A.; Dennis, L.; Danaher, P.; Finak, G.; Krouse, M.; Wang, A.; Webster, P.; Beechem, J.; Gottardo, R. Modeling bi-modality improves characterization of cell cycle on gene expression in single cells. PLoS Comput. Biol. 2014, 10, e1003696. [Google Scholar] [CrossRef] [PubMed]
- Svensson, V.; Vento-Tormo, R.; Teichmann, S.A. Exponential scaling of single-cell RNA-seq in the past decade. Nat. Protoc. 2018, 13, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Geiler-Samerotte, K.A.; Bauer, C.R.; Li, S.; Ziv, N.; Gresham, D.; Siegal, M.L. The details in the distributions: Why and how to study phenotypic variability. Curr. Opin. Biotechnol. 2013, 24, 752–759. [Google Scholar] [CrossRef]
- Mar, J.C. The rise of the distributions: Why non-normality is important for understanding the transcriptome and beyond. Biophys. Rev. 2019, 11, 89–94. [Google Scholar] [CrossRef]
- Marinov, G.K.; Williams, B.A.; McCue, K.; Schroth, G.P.; Gertz, J.; Myers, R.M.; Wold, B.J. From single-cell to cell-pool transcriptomes: Stochasticity in gene expression and RNA splicing. Genome Res. 2014, 24, 496–510. [Google Scholar] [CrossRef]
- Li, B.; You, L. Predictive power of cell-to-cell variability. Quant. Biol. 2013, 1, 131–139. [Google Scholar] [CrossRef]
- Moon, K.R.; van Dijk, D.; Wang, Z.; Gigante, S.; Burkhardt, D.; Chen, W.; Yim, K.; Elzen, A.V.D.; Hirn, M.J.; Coifman, R.R.; et al. Visualizing structure and transitions in high-dimensional biological data. Nat Biotechnol. 2019, 37, 1482–1492. [Google Scholar] [CrossRef]
- Osorio, D.; Yu, X.; Yu, P.; Serpedin, E.; Cai, J.J. Single-cell RNA sequencing of a European and an African lymphoblastoid cell line. Sci. Data 2019, 6, 112. [Google Scholar] [CrossRef] [PubMed]
- Habiel, D.M.; Espindola, M.S.; Jones, I.C.; Coelho, A.L.; Stripp, B.; Hogaboam, C.M. CCR10+ epithelial cells from idiopathic pulmonary fibrosis lungs drive remodeling. Jci Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Hagai, T.; Chen, X.; Miragaia, R.J.; Rostom, R.; Gomes, T.; Kunowska, N.; Henriksson, J.; Park, J.E.; Proserpio, V.; Donati, G.; et al. Gene expression variability across cells and species shapes innate immunity. Nature 2018, 563, 197–202. [Google Scholar] [CrossRef] [PubMed]
- Friedman, C.E.; Nguyen, Q.; Lukowski, S.W.; Helfer, A.; Chiu, H.S.; Miklas, J.; Levy, S.; Suo, S.; Han, J.-D.J.; Osteil, P. Single-cell transcriptomic analysis of cardiac differentiation from human PSCs reveals HOPX-dependent cardiomyocyte maturation. Cell Stem Cell 2018, 23, 586–598. e588. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.; Hoffman, P.; Smibert, P.; Papalexi, E.; Satija, R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 2018, 36, 411–420. [Google Scholar] [CrossRef]
- Brennecke, P.; Anders, S.; Kim, J.K.; Kolodziejczyk, A.A.; Zhang, X.; Proserpio, V.; Baying, B.; Benes, V.; Teichmann, S.A.; Marioni, J.C.; et al. Accounting for technical noise in single-cell RNA-seq experiments. Nat. Methods 2013, 10, 1093–1095. [Google Scholar] [CrossRef]
- Cai, J.J. scGEAToolbox: A Matlab toolbox for single-cell RNA sequencing data analysis. Bioinformatics 2019. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. Bmc Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Eden, E.; Navon, R.; Steinfeld, I.; Lipson, D.; Yakhini, Z. GOrilla: A tool for discovery and visualization of enriched GO terms in ranked gene lists. Bmc Bioinform. 2009, 10, 48. [Google Scholar] [CrossRef] [PubMed]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- van Dijk, D.; Sharma, R.; Nainys, J.; Yim, K.; Kathail, P.; Carr, A.J.; Burdziak, C.; Moon, K.R.; Chaffer, C.L.; Pattabiraman, D.; et al. Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 2018, 174, 716–729 e727. [Google Scholar] [CrossRef] [PubMed]
- Konganti, K.; Wang, G.; Yang, E.; Cai, J.J. SBEToolbox: A Matlab toolbox for biological network analysis. Evol. Bioinform. Online 2013, 9, 179–182. [Google Scholar] [CrossRef] [PubMed]
- McLean, C.Y.; Bristor, D.; Hiller, M.; Clarke, S.L.; Schaar, B.T.; Lowe, C.B.; Wenger, A.M.; Bejerano, G. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010, 28, 495–501. [Google Scholar] [CrossRef]
- Smedley, D.; Haider, S.; Ballester, B.; Holland, R.; London, D.; Thorisson, G.; Kasprzyk, A. BioMart--biological queries made easy. Bmc Genom. 2009, 10, 22. [Google Scholar] [CrossRef]
- Khan, A.; Fornes, O.; Stigliani, A.; Gheorghe, M.; Castro-Mondragon, J.A.; van der Lee, R.; Bessy, A.; Cheneby, J.; Kulkarni, S.R.; Tan, G.; et al. JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucleic Acids Res. 2018, 46, D1284. [Google Scholar] [CrossRef]
- Kivioja, T.; Vaharautio, A.; Karlsson, K.; Bonke, M.; Enge, M.; Linnarsson, S.; Taipale, J. Counting absolute numbers of molecules using unique molecular identifiers. Nat. Methods 2011, 9, 72–74. [Google Scholar] [CrossRef]
- van der Maaten, L.; Hinton, G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008, 9, 2579–2605. [Google Scholar]
- Becht, E.; McInnes, L.; Healy, J.; Dutertre, C.A.; Kwok, I.W.H.; Ng, L.G.; Ginhoux, F.; Newell, E.W. Dimensionality reduction for visualizing single-cell data using UMAP. Nat. Biotechnol. 2018. [Google Scholar] [CrossRef]
- Chen, H.I.; Jin, Y.; Huang, Y.; Chen, Y. Detection of high variability in gene expression from single-cell RNA-seq profiling. Bmc Genom. 2016, 17 (Suppl. S7), 508. [Google Scholar] [CrossRef] [PubMed]
- Tabib, T.; Morse, C.; Wang, T.; Chen, W.; Lafyatis, R. SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin. J. Invest. Derm. 2018, 138, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Singhal, P.K.; Sassi, S.; Lan, L.; Au, P.; Halvorsen, S.C.; Fukumura, D.; Jain, R.K.; Seed, B. Mouse embryonic fibroblasts exhibit extensive developmental and phenotypic diversity. Proc. Natl. Acad. Sci. USA 2016, 113, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Mantsoki, A.; Devailly, G.; Joshi, A. Gene expression variability in mammalian embryonic stem cells using single cell RNA-seq data. Comput. Biol. Chem. 2016, 63, 52–61. [Google Scholar] [CrossRef]
- Courtois, G.; Smahi, A.; Reichenbach, J.; Doffinger, R.; Cancrini, C.; Bonnet, M.; Puel, A.; Chable-Bessia, C.; Yamaoka, S.; Feinberg, J.; et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J. Clin. Invest. 2003, 112, 1108–1115. [Google Scholar] [CrossRef]
- Lopez-Granados, E.; Keenan, J.E.; Kinney, M.C.; Leo, H.; Jain, N.; Ma, C.A.; Quinones, R.; Gelfand, E.W.; Jain, A. A novel mutation in NFKBIA/IKBA results in a degradation-resistant N-truncated protein and is associated with ectodermal dysplasia with immunodeficiency. Hum. Mutat. 2008, 29, 861–868. [Google Scholar] [CrossRef]
- Nutt, S.L.; Hodgkin, P.D.; Tarlinton, D.M.; Corcoran, L.M. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 2015, 15, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Sciammas, R.; Li, Y.; Warmflash, A.; Song, Y.; Dinner, A.R.; Singh, H. An incoherent regulatory network architecture that orchestrates B cell diversification in response to antigen signaling. Mol. Syst. Biol. 2011, 7, 495. [Google Scholar] [CrossRef]
- Roy, K.; Mitchell, S.; Liu, Y.; Ohta, S.; Lin, Y.S.; Metzig, M.O.; Nutt, S.L.; Hoffmann, A. A Regulatory Circuit Controlling the Dynamics of NFkappaB cRel Transitions B Cells from Proliferation to Plasma Cell Differentiation. Immunity 2019, 50, 616–628 e616. [Google Scholar] [CrossRef]
- Lappalainen, T.; Sammeth, M.; Friedlander, M.R.; AC’t Hoen, P.A.; Monlong, J.; Rivas, M.A.; Gonzalez-Porta, M.; Kurbatova, N.; Griebel, T.; Ferreira, P.G.; et al. Transcriptome and genome sequencing uncovers functional variation in humans. Nature 2013, 501, 506–511. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, Q.H.; Lukowski, S.W.; Chiu, H.S.; Senabouth, A.; Bruxner, T.J.C.; Christ, A.N.; Palpant, N.J.; Powell, J.E. Single-cell RNA-seq of human induced pluripotent stem cells reveals cellular heterogeneity and cell state transitions between subpopulations. Genome Res. 2018, 28, 1053–1066. [Google Scholar] [CrossRef] [PubMed]
- Mandegar, M.A.; Huebsch, N.; Frolov, E.B.; Shin, E.; Truong, A.; Olvera, M.P.; Chan, A.H.; Miyaoka, Y.; Holmes, K.; Spencer, C.I.; et al. CRISPR Interference Efficiently Induces Specific and Reversible Gene Silencing in Human iPSCs. Cell Stem Cell 2016, 18, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Kaern, M.; Elston, T.C.; Blake, W.J.; Collins, J.J. Stochasticity in gene expression: From theories to phenotypes. Nat. Rev. Genet. 2005, 6, 451–464. [Google Scholar] [CrossRef] [PubMed]
- Raser, J.M.; O’Shea, E.K. Noise in gene expression: Origins, consequences, and control. Science 2005, 309, 2010–2013. [Google Scholar] [CrossRef]
- Chalancon, G.; Ravarani, C.N.; Balaji, S.; Martinez-Arias, A.; Aravind, L.; Jothi, R.; Babu, M.M. Interplay between gene expression noise and regulatory network architecture. Trends Genet. 2012, 28, 221–232. [Google Scholar] [CrossRef]
- Mar, J.C.; Matigian, N.A.; Mackay-Sim, A.; Mellick, G.D.; Sue, C.M.; Silburn, P.A.; McGrath, J.J.; Quackenbush, J.; Wells, C.A. Variance of gene expression identifies altered network constraints in neurological disease. PLoS Genet. 2011, 7, e1002207. [Google Scholar] [CrossRef]
- Dar, R.D.; Hosmane, N.N.; Arkin, M.R.; Siliciano, R.F.; Weinberger, L.S. Screening for noise in gene expression identifies drug synergies. Science 2014, 344, 1392–1396. [Google Scholar] [CrossRef]
- Ecker, S.; Pancaldi, V.; Valencia, A.; Beck, S.; Paul, D.S. Epigenetic and Transcriptional Variability Shape Phenotypic Plasticity. Bioessays 2018, 40. [Google Scholar] [CrossRef]
- Dueck, H.; Khaladkar, M.; Kim, T.K.; Spaethling, J.M.; Francis, C.; Suresh, S.; Fisher, S.A.; Seale, P.; Beck, S.G.; Bartfai, T.; et al. Deep sequencing reveals cell-type-specific patterns of single-cell transcriptome variation. Genome Biol. 2015, 16, 122. [Google Scholar] [CrossRef]
- Neitzel, H. A routine method for the establishment of permanent growing lymphoblastoid cell lines. Hum. Genet. 1986, 73, 320–326. [Google Scholar] [CrossRef] [PubMed]
- Tonegawa, S. Somatic generation of antibody diversity. Nature 1983, 302, 575–581. [Google Scholar] [CrossRef] [PubMed]
- Papavasiliou, F.; Casellas, R.; Suh, H.; Qin, X.F.; Besmer, E.; Pelanda, R.; Nemazee, D.; Rajewsky, K.; Nussenzweig, M.C. V(D)J recombination in mature B cells: A mechanism for altering antibody responses. Science 1997, 278, 298–301. [Google Scholar] [CrossRef] [PubMed]
- Ryan, J.L.; Kaufmann, W.K.; Raab-Traub, N.; Oglesbee, S.E.; Carey, L.A.; Gulley, M.L. Clonal evolution of lymphoblastoid cell lines. Lab. Invest. 2006, 86, 1193–1200. [Google Scholar] [CrossRef][Green Version]
- Altan-Bonnet, G.; Mukherjee, R. Cytokine-mediated communication: A quantitative appraisal of immune complexity. Nat. Rev. Immunol. 2019. [Google Scholar] [CrossRef]
- Hiemstra, P.S.; McCray, P.B., Jr.; Bals, R. The innate immune function of airway epithelial cells in inflammatory lung disease. Eur Respir J. 2015, 45, 1150–1162. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, J.; Phatnani, H.; Scheu, S.; Maniatis, T. Stochastic expression of the interferon-beta gene. PLoS Biol. 2012, 10, e1001249. [Google Scholar] [CrossRef]
- Sacco, O.; Silvestri, M.; Sabatini, F.; Sale, R.; Defilippi, A.C.; Rossi, G.A. Epithelial cells and fibroblasts: Structural repair and remodelling in the airways. Paediatr Respir Rev. 2004, 5 (Suppl. A), S35–S40. [Google Scholar] [CrossRef]
- Huang, G.; Osorio, D.; Guan, J.; Ji, G.; Cai, J.J. Overdispersed gene expression characterizes schizophrenic brains. bioRxiv 2018. [Google Scholar] [CrossRef]
- Guan, J.; Yang, E.; Yang, J.; Zeng, Y.; Ji, G.; Cai, J.J. Exploiting aberrant mRNA expression in autism for gene discovery and diagnosis. Hum. Genet. 2016, 135, 797–811. [Google Scholar] [CrossRef]
- Ecker, S.; Pancaldi, V.; Rico, D.; Valencia, A. Higher gene expression variability in the more aggressive subtype of chronic lymphocytic leukemia. Genome Med. 2015, 7, 8. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Liu, Y.; Kim, T.; Min, R.; Zhang, Z. Gene expression variability within and between human populations and implications toward disease susceptibility. PLoS Comput. Biol. 2010, 6. [Google Scholar] [CrossRef] [PubMed]
- Spreizer, S.; Aertsen, A.; Kumar, A. From space to time: Spatial inhomogeneities lead to the emergence of spatiotemporal sequences in spiking neuronal networks. PLoS Computat. Biol. 2019, 15. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.W.; Stefani, M.; dos Remedios, C.G.; Charleston, M.A. Differential variability analysis of gene expression and its application to human diseases. Bioinformatics 2008, 24, i390–i398. [Google Scholar] [CrossRef] [PubMed]
- Bendall, S.C.; Nolan, G.P. From single cells to deep phenotypes in cancer. Nat. Biotechnol. 2012, 30, 639–647. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Type | Highly Variable Genes (HVGs), Top 50 | Enriched GO Terms, Top 10 | Enriched Reactome Pathways, Top 10 |
|---|---|---|---|
| Lymphoblastoid Cell line (LCL) | ANKRD37 ATF3 BIN1 BMP4 CAMP CCL22 CCL3 CCL3L3 CCL4 CCL4L2 CCR7 CD69 CD7 CD83 CDKN1A CTSC CYP1B1 DHRS9 DUSP2 FSCN1 HIST1H1C IER3 IFI27 IGHG1 IGHG3 IGHM IGKC ITM2A KCNMA1 LINC00176 LINC01588 LMNA LTA LTB MAL MIER2 MIR155HG MYC NFKBIA PMCH PRSS2 RGS1 RGS16 RGS2 RP11-291B21.2 S100A4 SFN TNFAIP2 TUBB4B WFDC2 |
|
|
| Lung airway epithelial cell (LAEC) | AMTN ANKRD1 AREG CCL2 CCL5 CCL7 COL1A1 COL1A2 COL3A1 COL6A1 COL6A3 CRCT1 CTGF CXCL5 CXCL6 FBXO32 GREM1 HAS2 IFNL1 IFNL2 IFNL3 IGFBP5 IGFL1 IL17C IL23A KRT14 KRT6B KRT81 LY6D MEG3 MMP1 MSMB OVOS2 PI3 POSTN PPBP RP11-338I21.1 S100A7 S100A8 S100A9 SERPINB2 SERPINB3 SERPINB4 SLC15A2 SPARC SUGCT SULF1 TEX26-AS1 TNFAIP6 TSLP |
|
|
| Dermal fibroblast (DF) | ACAN ACTA2 ACTC1 CEMIP CLU COMP CTSC CXCL1 DCN DKK1 FLG G0S2 GAL HIST1H4C IGFBP5 IGFBP7 IL1RL1 KCNMA1 KRT14 KRT17 KRT19 KRT34 KRT81 KRTAP1-5 KRTAP2-3 LCE1F LUM MGP MMP1 MMP3 MT1X NMB OLFM2 PCP4 PENK PGF PI16 POSTN PPP1R14A PTTG1 PTX3 RARRES2 RGCC SCG5 SERPINE2 SFRP2 SFRP4 STMN2 TFPI2 TNFRSF11B |
|
|
| Shared Highly Variable Genes (HVGs) | Enriched GO Terms, Top 5 | Enriched Reactome Pathways |
|---|---|---|
| ID3 MARCKSL1 DPT ID2 CYP1B1 IGFBP2 IGFBP5 APOD IGFBP7 SFRP2 PLK2 SOX4 PI16 CTGF SGK1 CITED2 IGFBP3 SERPINE1 TIMP1 OSR2 HAS2 MYC B4GALT1 PTGDS CRYAB BAMBI MFAP5 CSRP2 LUM PGF THBS1 FGF7 MT2A MT1X WISP2 ADAMTS1 |
|
|
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osorio, D.; Yu, X.; Zhong, Y.; Li, G.; Serpedin, E.; Huang, J.Z.; Cai, J.J. Single-Cell Expression Variability Implies Cell Function. Cells 2020, 9, 14. https://doi.org/10.3390/cells9010014
Osorio D, Yu X, Zhong Y, Li G, Serpedin E, Huang JZ, Cai JJ. Single-Cell Expression Variability Implies Cell Function. Cells. 2020; 9(1):14. https://doi.org/10.3390/cells9010014
Chicago/Turabian StyleOsorio, Daniel, Xue Yu, Yan Zhong, Guanxun Li, Erchin Serpedin, Jianhua Z. Huang, and James J. Cai. 2020. "Single-Cell Expression Variability Implies Cell Function" Cells 9, no. 1: 14. https://doi.org/10.3390/cells9010014
APA StyleOsorio, D., Yu, X., Zhong, Y., Li, G., Serpedin, E., Huang, J. Z., & Cai, J. J. (2020). Single-Cell Expression Variability Implies Cell Function. Cells, 9(1), 14. https://doi.org/10.3390/cells9010014