IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β–Nrf2–BNIP3 Pathway



Abstract
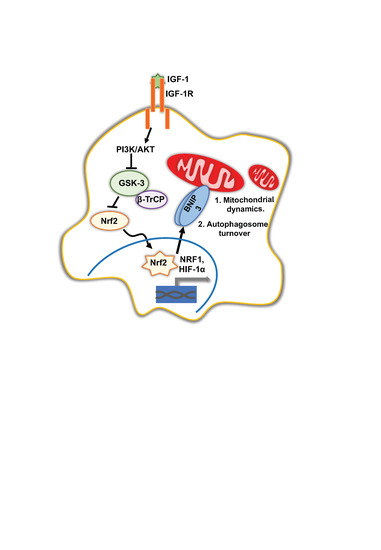
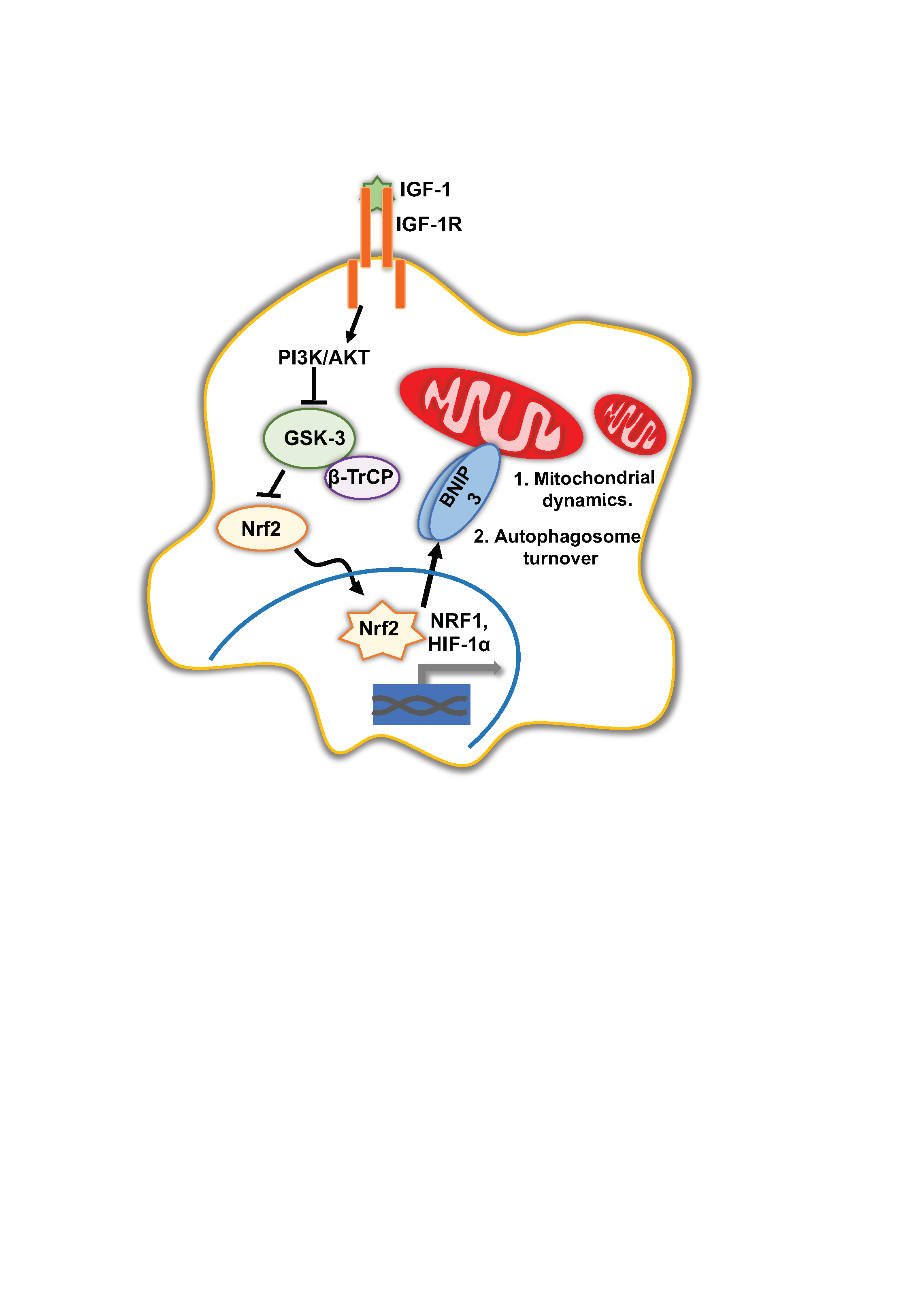{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. List of Abbreviations
2.2. Antibodies
2.3. Cell Lines and Cell Culture
2.4. SiRNA Transfection
2.5. Plasmid Transfection
2.6. Immunofluorescence
2.7. Cell Lysis, SDS-PAGE and Western Blotting
2.8. Subcellular Fractionation
2.9. Quantitative Real-Time PCR
2.10. Flow Cytometry
2.11. Analysis of Transcription Factor Binding Sites in the BNIP3 Promoter Region
3. Results
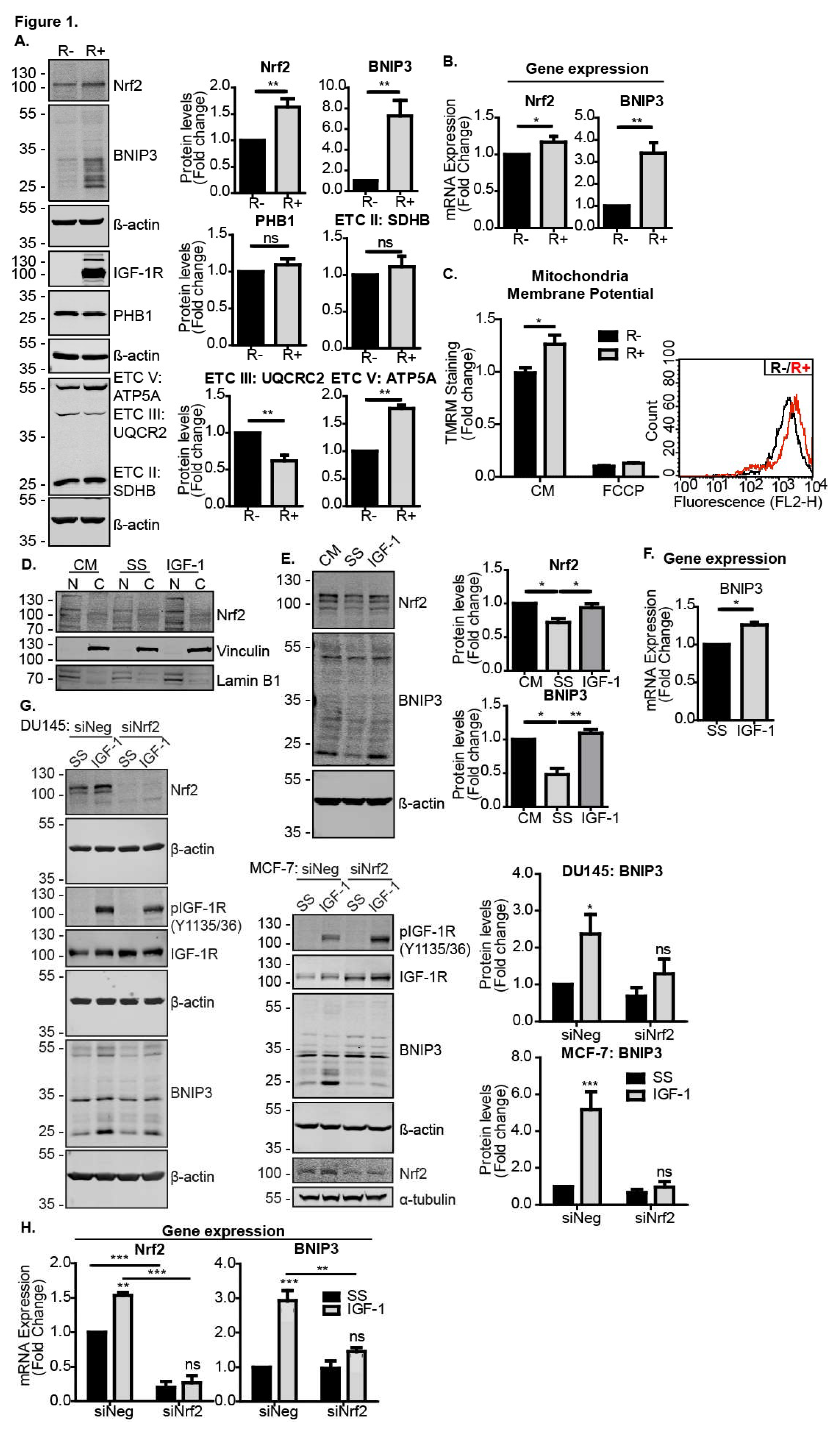
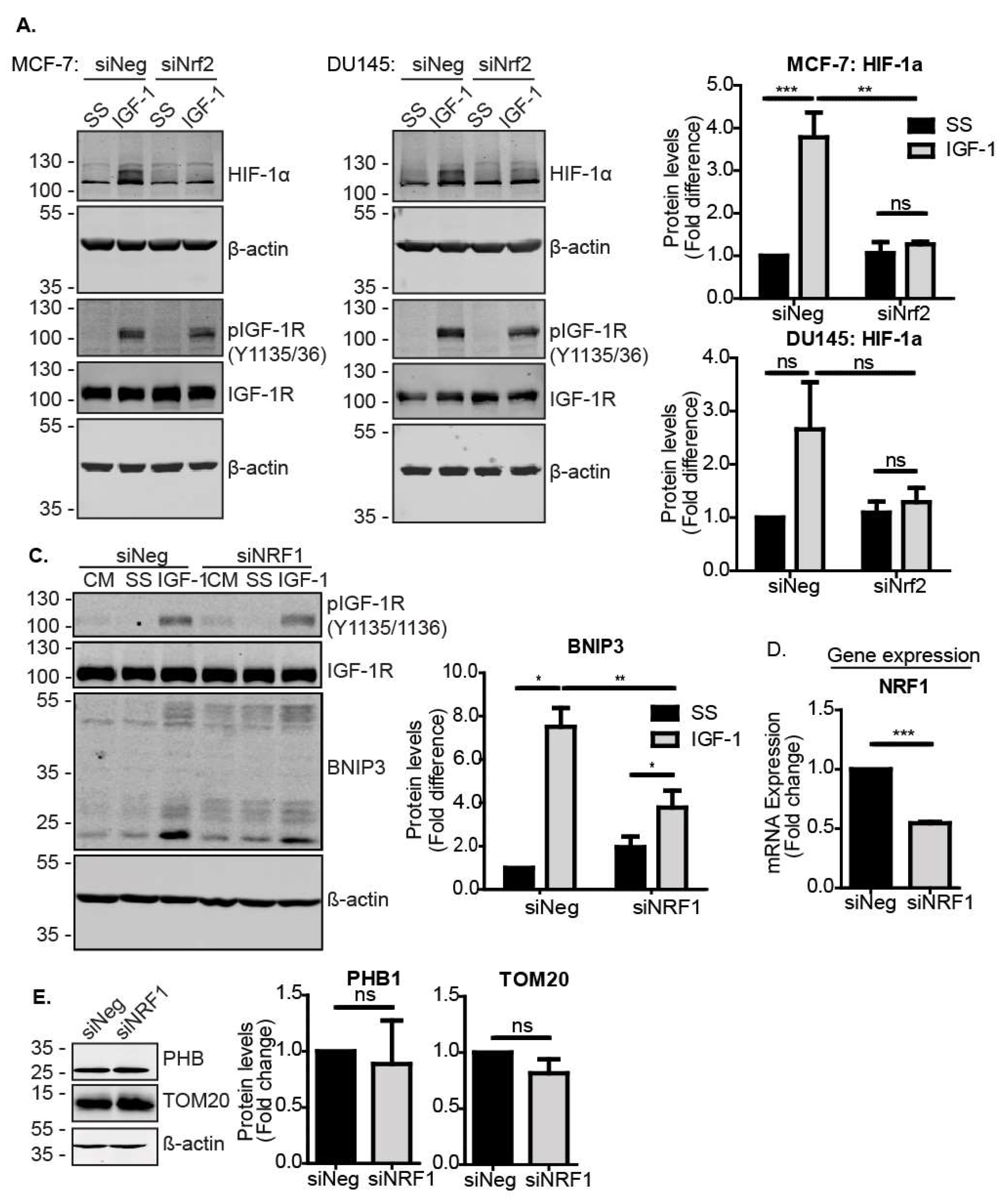
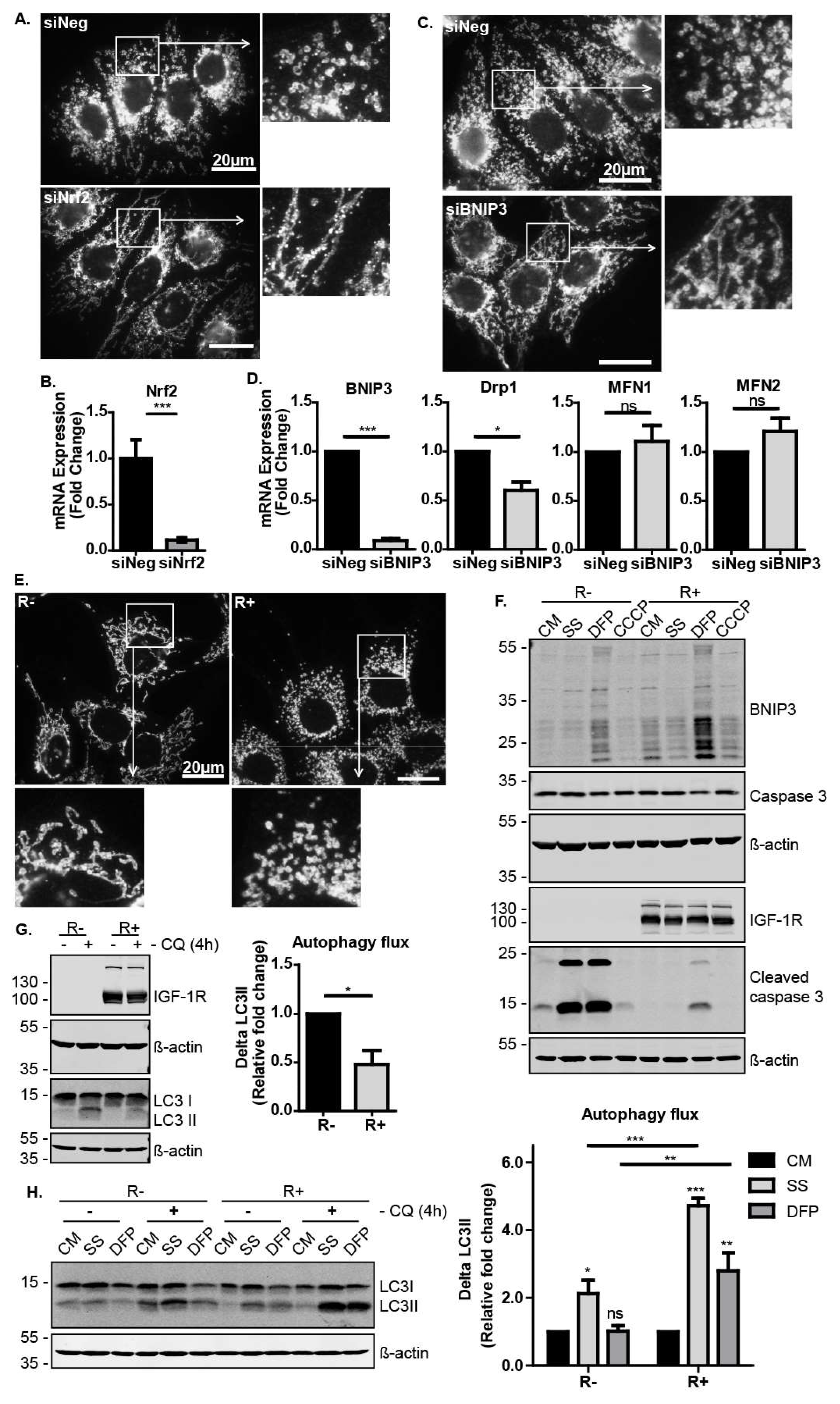
3.1. Nrf2 Is Required for IGF-1-Mediated Induction of the Mitophagy Receptor BNIP3
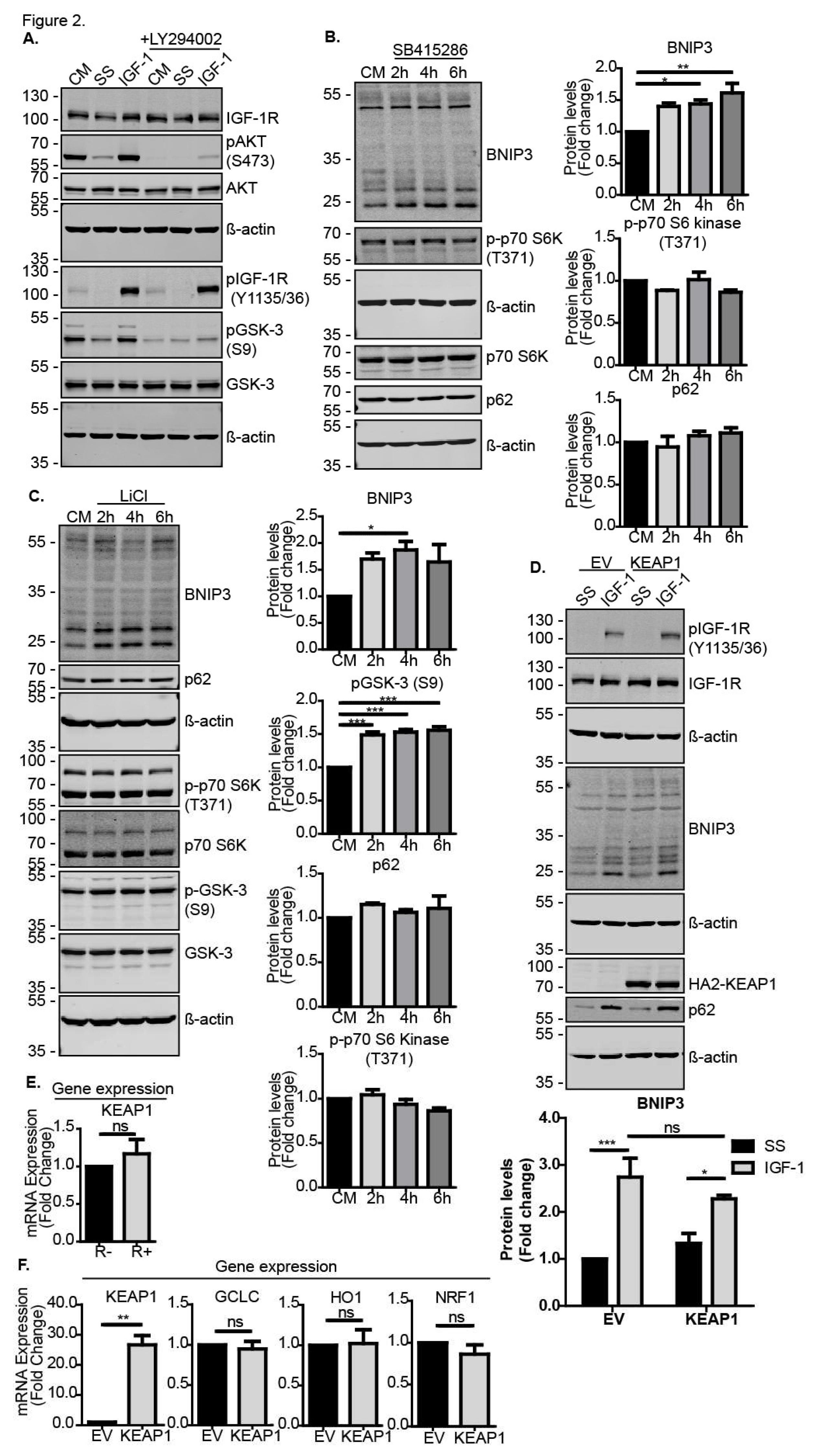
3.2. IGF-1 Induced BNIP3 through an Inhibitory Phosphorylation of GSK-3β
3.3. Nrf2 Induced BNIP3 Expression through HIF-1α and NRF1
3.4. The IGF-1-Nrf2-BNIP3 Pathway Regulates Mitochondrial Dynamics and Cellular Capacity for Autophagosomal Turnover in Response to Metabolic and Mitochondrial Stress
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hakuno, F.; Takahashi, S.I. IGF1 Receptor Signaling Pathways. J. Mol. Endocrinol. 2018, 61, T69–T86. [Google Scholar] [CrossRef] [PubMed]
- Denduluri, S.K.; Idowu, O.; Wang, Z.; Liao, Z.; Yan, Z.; Mohammed, M.K.; Ye, J.; Wei, Q.; Wang, J.; Zhao, L.; et al. Insulin-like growth factor (IGF) signaling in tumorigenesis and the development of cancer drug resistance. Genes Dis. 2015, 2, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Guevara, A.; Palacios, I.; Pérez, M.; Prócel, P.; Terán, E. GH and GHR signaling in human disease. Growth Horm. IGF Res. 2018, 38, 34–38. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Wu, Z.; Dong, W.; Zhang, T.; Wang, L.; Pang, Z.; Ma, W.; Du, J. Update of IGF-1 receptor inhibitor (ganitumab, dalotuzumab, cixutumumab, teprotumumab and figitumumab) effects on cancer therapy. Oncotarget 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Osher, E.; Macaulay, V.M. Therapeutic Targeting of the IGF Axis. Cells 2019, 8, 895. [Google Scholar] [CrossRef] [PubMed]
- Vyas, S.; Zaganjor, E.; Haigis, M.C. Mitochondria and Cancer. Cell 2016, 166, 555–566. [Google Scholar] [CrossRef]
- Drake, L.E.; Springer, M.Z.; Poole, L.P.; Kim, C.J.; Macleod, K.F. Expanding perspectives on the significance of mitophagy in cancer. Semin. Cancer Biol. 2017, 47, 110–124. [Google Scholar] [CrossRef]
- Pickles, S.; Vigie, P.; Youle, R.J. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr. Biol. 2018, 28, R170–R185. [Google Scholar] [CrossRef]
- Sádaba, M.C.; Martín-Estal, I.; Puche, J.E.; Castilla-Cortázar, I. Insulin-like growth factor 1 (IGF-1) therapy: Mitochondrial dysfunction and diseases. Biochimica et Biophysica Acta (BBA) Mol. Basis Dis. 2016, 1862, 1267–1278. [Google Scholar] [CrossRef]
- Favre, C.; Zhdanov, A.; Leahy, M.; Papkovsky, D.; O’Connor, R. Mitochondrial pyrimidine nucleotide carrier (PNC1) regulates mitochondrial biogenesis and the invasive phenotype of cancer cells. Oncogene 2010, 29, 3964–3976. [Google Scholar] [CrossRef]
- Ribeiro, M.; Rosenstock, T.R.; Oliveira, A.M.; Oliveira, C.R.; Rego, A.C. Insulin and IGF-1 improve mitochondrial function in a PI-3K/Akt-dependent manner and reduce mitochondrial generation of reactive oxygen species in Huntington’s disease knock-in striatal cells. Free Radic. Biol. Med. 2014, 74, 129–144. [Google Scholar] [CrossRef] [PubMed]
- Lyons, A.; Coleman, M.; Riis, S.; Favre, C.; O’Flanagan, C.H.; Zhdanov, A.V.; Papkovsky, D.B.; Hursting, S.D.; O’Connor, R. Insulin-like Growth Factor-1 signalling is essential for Mitochondrial Biogenesis and Mitophagy in cancer cells. J. Biol. Chem. 2017, 292. [Google Scholar] [CrossRef]
- Choe, S.C.; Hamacher-Brady, A.; Brady, N.R. Autophagy capacity and sub-mitochondrial heterogeneity shape Bnip3-induced mitophagy regulation of apoptosis. Cell Commun. Signal. CCS 2015, 13, 37. [Google Scholar] [CrossRef] [PubMed]
- Chourasia, A.H.; Boland, M.L.; Macleod, K.F. Mitophagy and cancer. Cancer Metabol. 2015, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Niu, Y.; Lin, Z.; Wan, A.; Chen, H.; Liang, H.; Sun, L.; Wang, Y.; Li, X.; Xiong, X.; Wei, B.; et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol. Cancer 2019, 18, 46. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Li, J.; Xu, T.; Zhou, D.D.; Zhang, L.; Wang, X. MicroRNA-145 induces apoptosis of glioma cells by targeting BNIP3 and Notch signaling. Oncotarget 2017, 8, 61510–61527. [Google Scholar]
- Palikaras, K.; Lionaki, E.; Tavernarakis, N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 2015, 521, 525–528. [Google Scholar] [CrossRef]
- Palikaras, K.; Lionaki, E.N.; Tavernarakis, N. Coupling mitogenesis and mitophagy for longevity. Autophagy 2015, 11, 1428–1430. [Google Scholar] [CrossRef]
- Mellor, H.R.; Rouschop, K.M.; Wigfield, S.M.; Wouters, B.G.; Harris, A.L. Synchronised phosphorylation of BNIP3, Bcl-2 and Bcl-xL in response to microtubule-active drugs is JNK-independent and requires a mitotic kinase. Biochem. Pharmacol. 2010, 79, 1562–1572. [Google Scholar] [CrossRef]
- Graham, R.M.; Thompson, J.W.; Wei, J.; Bishopric, N.H.; Webster, K.A. Regulation of Bnip3 death pathways by calcium, phosphorylation, and hypoxia-reoxygenation. Antioxid. Redox. Signal. 2007, 9, 1309–1315. [Google Scholar] [CrossRef]
- Sell, C.; Deveaud, C.; Miura, M.; Coppola, D.; DeAngelis, T.; Rubin, R.; Efstratiadis, A.; Baserga, R. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol. Cell. Biol. 1994, 14, 3604–3612. [Google Scholar] [CrossRef] [PubMed]
- Lau, L.; Hansford, L.M.; Cheng, L.S.; Hang, M.; Baruchel, S.; Kaplan, D.R.; Irwin, M.S. Cyclooxygenase inhibitors modulate the p53/HDM2 pathway and enhance chemotherapy-induced apoptosis in neuroblastoma. Oncogene 2007, 26, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Dreos, R.; Ambrosini, G.; Groux, R.; Cavin Périer, R.; Bucher, P. The eukaryotic promoter database in its 30th year: Focus on non-vertebrate organisms. Nucleic Acids Res. 2017, 45, D51–D55. [Google Scholar] [CrossRef] [PubMed]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- Lesurf, R.; Cotto, K.C.; Wang, G.; Griffith, M.; Kasaian, K.; Jones, S.J.M.; Montgomery, S.B.; Griffith, O.L.; The Open Regulatory Annotation Consortium. ORegAnno 3.0: A community-driven resource for curated regulatory annotation. Nucleic Acid Res. 2016, 44, D126–D132. [Google Scholar]
- Fornes, O.; Castro-Mondragon, J.A.; Khan, A.; van der Lee, R.; Zhang, X.; Richmond, P.A.; Modi, B.P.; Correard, S.; Gheorghe, M.; Baranašić, D.; et al. JASPAR 2020: Update of the open-access database of transcription factor binding profiles. Nucleic Acid Res. 2019. [CrossRef]
- Kansanen, E.; Kuosmanen, S.M.; Leinonen, H.; Levonen, A.L. The Keap1-Nrf2 pathway: Mechanisms of activation and dysregulation in cancer. Redox Biol. 2013, 1, 45–49. [Google Scholar] [CrossRef]
- Chowdhry, S.; Zhang, Y.; McMahon, M.; Sutherland, C.; Cuadrado, A.; Hayes, J.D. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 2013, 32, 3765–3781. [Google Scholar] [CrossRef]
- Tebay, L.E.; Robertson, H.; Durant, S.T.; Vitale, S.R.; Penning, T.M.; Dinkova-Kostova, A.T.; Hayes, J.D. Mechanisms of activation of the transcription factor Nrf2 by redox stressors, nutrient cues, and energy status and the pathways through which it attenuates degenerative disease. Free Radic. Biol. Med. 2015, 88 Pt B, 108–146. [Google Scholar] [CrossRef]
- Sutherland, C.; Leighton, I.A.; Cohen, P. Inactivation of glycogen synthase kinase-3 beta by phosphorylation: New kinase connections in insulin and growth-factor signalling. Biochem. J. 1993, 296 Pt 1, 15–19. [Google Scholar] [CrossRef]
- Stambolic, V.; Ruel, L.; Woodgett, J.R. Lithium inhibits glycogen synthase kinase-3 activity and mimics Wingless signalling in intact cells. Curr. Biol. 1996, 6, 1664–1669. [Google Scholar] [CrossRef]
- Salazar, M.; Rojo, A.I.; Velasco, D.; de Sagarra, R.M.; Cuadrado, A. Glycogen Synthase Kinase-3β Inhibits the Xenobiotic and Antioxidant Cell Response by Direct Phosphorylation and Nuclear Exclusion of the Transcription Factor Nrf2. J. Biol. Chem. 2006, 281, 14841–14851. [Google Scholar] [CrossRef] [PubMed]
- Korolchuk, V.I.; Mansilla, A.; Menzies, F.M.; Rubinsztein, D.C. Autophagy Inhibition Compromises Degradation of Ubiquitin-Proteasome Pathway Substrates. Mol. Cell 2009, 33, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell. Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef]
- Guo, K.; Searfoss, G.; Krolikowski, D.; Pagnoni, M.; Franks, C.; Clark, K.; Yu, K.T.; Jaye, M.; Ivashchenko, Y. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ. 2001, 8, 367–376. [Google Scholar] [CrossRef]
- Bellot, G.; Garcia-Medina, R.; Gounon, P.; Chiche, J.; Roux, D.; Pouysségur, J.; Mazure, N.M. Hypoxia-Induced Autophagy Is Mediated through Hypoxia-Inducible Factor Induction of BNIP3 and BNIP3L via Their BH3 Domains. Mol. Cell. Biol. 2009, 29, 2570–2581. [Google Scholar] [CrossRef]
- Lee, S.; Hallis, S.P.; Jung, K.; Ryu, D.; Kwak, M. Impairment of HIF-1α-mediated metabolic adaption by NRF2-silencing in breast cancer cells. Redox Biol. 2019, 24, 101210. [Google Scholar] [CrossRef]
- Gugneja, S.; Scarpulla, R.C. Serine Phosphorylation within a Concise Amino-terminal Domain in Nuclear Respiratory Factor 1 Enhances DNA Binding. J. Biol. Chem. 1997, 272, 18732–18739. [Google Scholar] [CrossRef]
- Ma, Q. Role of Nrf2 in Oxidative Stress and Toxicity. Ann. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef]
- Wilson, L.A.; Gemin, A.; Espiritu, R.; Singh, G. ets-1 is transcriptionally up-regulated by H2O2 via an antioxidant response element. FASEB J. 2005, 19, 2085–2087. [Google Scholar] [CrossRef]
- Gleyzer, N.; Vercauteren, K.; Scarpulla, R.C. Control of Mitochondrial Transcription Specificity Factors (TFB1M and TFB2M) by Nuclear Respiratory Factors (NRF-1 and NRF-2) and PGC-1 Family Coactivators. Mol. Cell Biol. 2005, 25, 1354–1366. [Google Scholar] [PubMed]
- Shao, D.; Liu, Y.; Liu, X.; Zhu, L.; Cui, Y.; Cui, A.; Qiao, A.; Kong, X.; Liu, Y.; Chen, Q.; et al. PGC-1β-Regulated mitochondrial biogenesis and function in myotubes is mediated by NRF-1 and ERRα. Mitochondrion 2010, 10, 516–527. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, H.; Hanna, R.A.; Gustafsson, Å.B. Mitochondrial autophagy by Bnip3 involves Drp1-mediated mitochondrial fission and recruitment of Parkin in cardiac myocytes. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1924–H1931. [Google Scholar] [CrossRef] [PubMed]
- Burman, J.L.; Pickles, S.; Wang, C.; Sekine, S.; Vargas, J.N.S.; Zhang, Z.; Youle, A.M.; Nezich, C.L.; Wu, X.; Hammer, J.A.; et al. Mitochondrial fission facilitates the selective mitophagy of protein aggregates. J. Cell. Biol. 2017, 216, 3231–3247. [Google Scholar] [CrossRef] [PubMed]
- Rojo de la Vega, M.; Chapman, E.; Zhang, D.D. NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34, 21–43. [Google Scholar] [CrossRef]
- Wang, X.-J.; Sun, Z.; Villeneuve, N.F.; Zhang, S.; Zhao, F.; Li, Y.; Chen, W.; Yi, X.; Zheng, W.; Wondrak, G.T.; et al. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis 2008, 29, 1235–1243. [Google Scholar] [CrossRef]
- Zhang, H.-S.; Du, G.-Y.; Zhang, Z.-G.; Zhou, Z.; Sun, H.-L.; Yu, X.-Y.; Shi, Y.-T.; Xiong, D.-N.; Li, H.; Huang, Y.-H. NRF2 facilitates breast cancer cell growth via HIF1ɑ-mediated metabolic reprogramming. Int. J. Biochem. Cell Biol. 2018, 95, 85–92. [Google Scholar] [CrossRef]
- Um, J.-H.; Yun, J. Emerging role of mitophagy in human diseases and physiology. BMB Rep. 2017, 50, 299–307. [Google Scholar] [CrossRef]
- Fu, W.; Liu, Y.; Yin, H. Mitochondrial Dynamics: Biogenesis, Fission, Fusion, and Mitophagy in the Regulation of Stem Cell Behaviors. Stem Cells Int. 2019, 2019, 9757201. [Google Scholar] [CrossRef]
- Esteban-Martinez, L.; Boya, P. BNIP3L/NIX-dependent mitophagy regulates cell differentiation via metabolic reprogramming. Autophagy 2017, 14, 915–917. [Google Scholar] [CrossRef]
- Lee, H.J.; Jung, Y.H.; Choi, G.E.; Ko, S.H.; Lee, S.-J.; Lee, S.H.; Han, H.J. BNIP3 induction by hypoxia stimulates FASN-dependent free fatty acid production enhancing therapeutic potential of umbilical cord blood-derived human mesenchymal stem cells. Redox Biol. 2017, 13, 426–443. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, M.; Moriyama, H.; Uda, J.; Kubo, H.; Nakajima, Y.; Goto, A.; Morita, T.; Hayakawa, T. BNIP3 upregulation via stimulation of ERK and JNK activity is required for the protection of keratinocytes from UVB-induced apoptosis. Cell Death Dis. 2017, 8, e2576. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Morrison, S.J.; Clarke, M.F.; Weissman, I.L. Stem cells, cancer, and cancer stem cells. Nature 2001, 414, 105–111. [Google Scholar] [CrossRef]
- Teng, C.-F.; Jeng, L.-B.; Shyu, W.-C. Role of Insulin-like Growth Factor 1 Receptor Signaling in Stem Cell Stemness and Therapeutic Efficacy. Cell Transplant. 2018, 27, 1313–1319. [Google Scholar] [CrossRef] [PubMed]
- Huat, T.J.; Khan, A.A.; Pati, S.; Mustafa, Z.; Abdullah, J.M.; Jaafar, H. IGF-1 enhances cell proliferation and survival during early differentiation of mesenchymal stem cells to neural progenitor-like cells. BMC Neurosci. 2014, 15, 91. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ney, P.A. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ. 2009, 16, 939–946. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Azad, M.; Shymko, M.D.; Henson, E.S.; Katyal, S.; Eisenstat, D.D.; Gibson, S.B. The BH3 only Bcl-2 family member BNIP3 regulates cellular proliferation. PLoS ONE 2018, 13, e0204792. [Google Scholar] [CrossRef] [PubMed]
- Jung, C.H.; Ro, S.-H.; Cao, J.; Otto, N.M.; Kim, D.-H. mTOR regulation of autophagy. FEBS Lett. 2010, 584, 1287–1295. [Google Scholar] [CrossRef] [PubMed]
- Macher-Goeppinger, S.; Keith, M.; Hatiboglu, G.; Hohenfellner, M.; Schirmacher, P.; Roth, W.; Tagscherer, K.E. Expression and Functional Characterization of the BNIP3 Protein in Renal Cell Carcinomas. Transl. Oncol. 2017, 10, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, N.; Sato, S.; Shiba, K.; Okatsu, K.; Saisho, K.; Gautier, C.A.; Sou, Y.-S.; Saiki, S.; Kawajiri, S.; Sato, F.; et al. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J. Cell Biol. 2010, 189, 211–221. [Google Scholar] [CrossRef]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open. Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Riis, S.; Murray, J.B.; O’Connor, R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β–Nrf2–BNIP3 Pathway. Cells 2020, 9, 147. https://doi.org/10.3390/cells9010147
Riis S, Murray JB, O’Connor R. IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β–Nrf2–BNIP3 Pathway. Cells. 2020; 9(1):147. https://doi.org/10.3390/cells9010147
Chicago/Turabian StyleRiis, Sarah, Joss B. Murray, and Rosemary O’Connor. 2020. "IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β–Nrf2–BNIP3 Pathway" Cells 9, no. 1: 147. https://doi.org/10.3390/cells9010147
APA StyleRiis, S., Murray, J. B., & O’Connor, R. (2020). IGF-1 Signalling Regulates Mitochondria Dynamics and Turnover through a Conserved GSK-3β–Nrf2–BNIP3 Pathway. Cells, 9(1), 147. https://doi.org/10.3390/cells9010147