Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge



Abstract

1. Triple-Negative Breast Cancer
2. TNBC Chemotherapy Basics
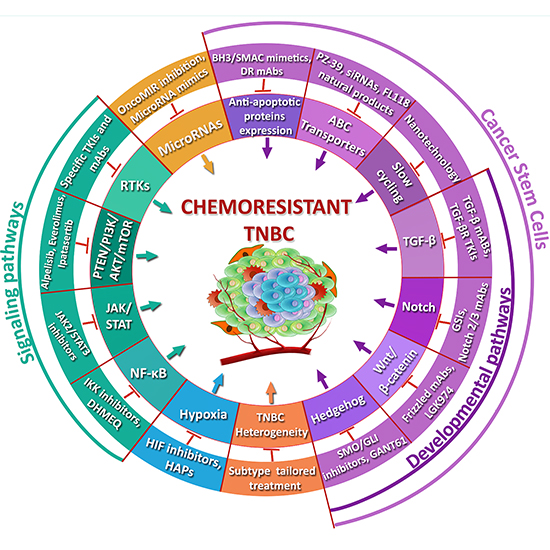
3. TNBC Chemoresistance
3.1. ABC Transporters
3.2. Cancer Stem Cells
3.2.1. TGF-β Pathway
3.2.2. Notch Pathway
3.2.3. Wnt/β-Catenin Pathway
3.2.4. Hedgehog Pathway
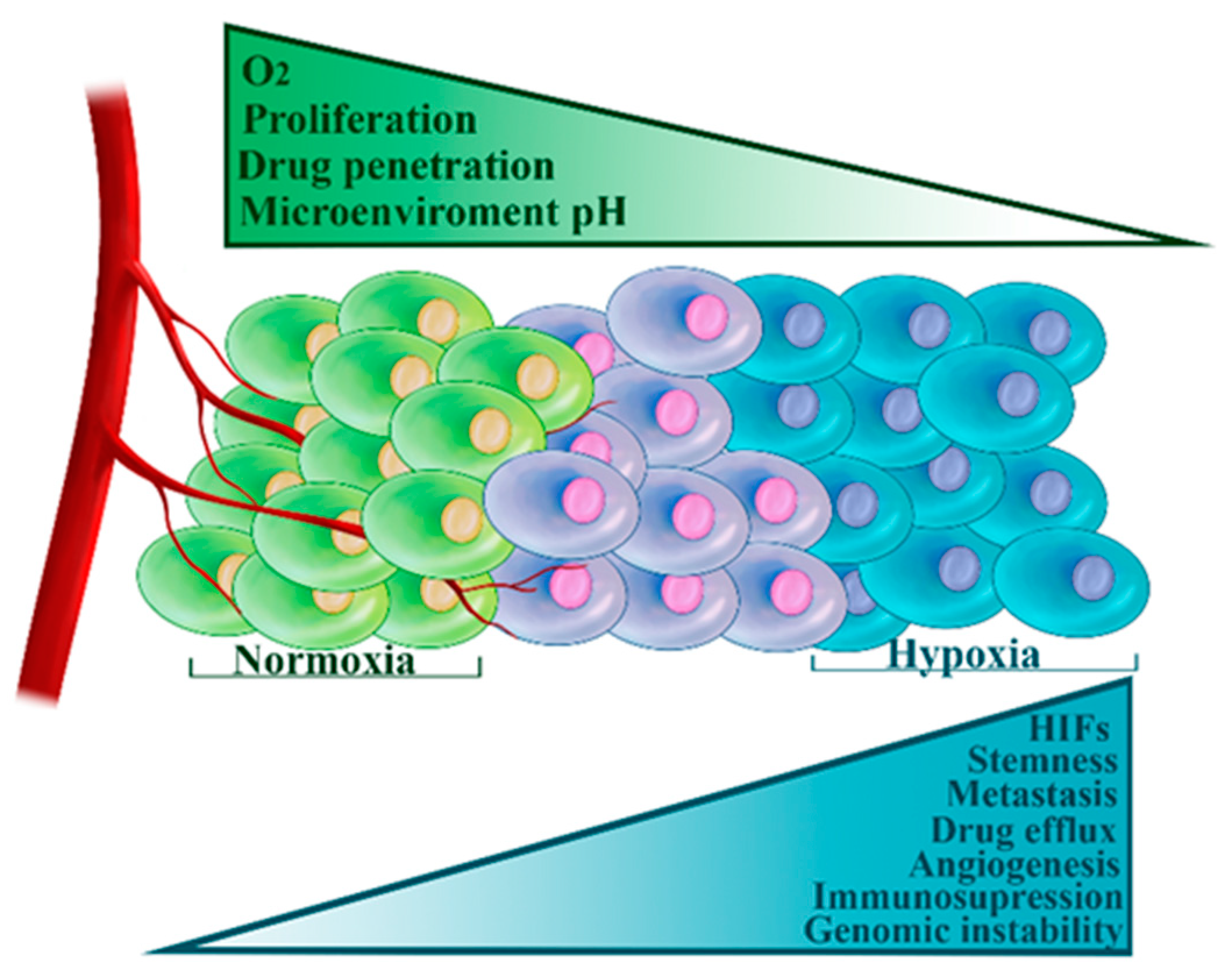
3.3. Hypoxia
3.4. Avoidance of Apoptosis
3.5. Role of Signaling Pathways in TNBC Chemoresistance
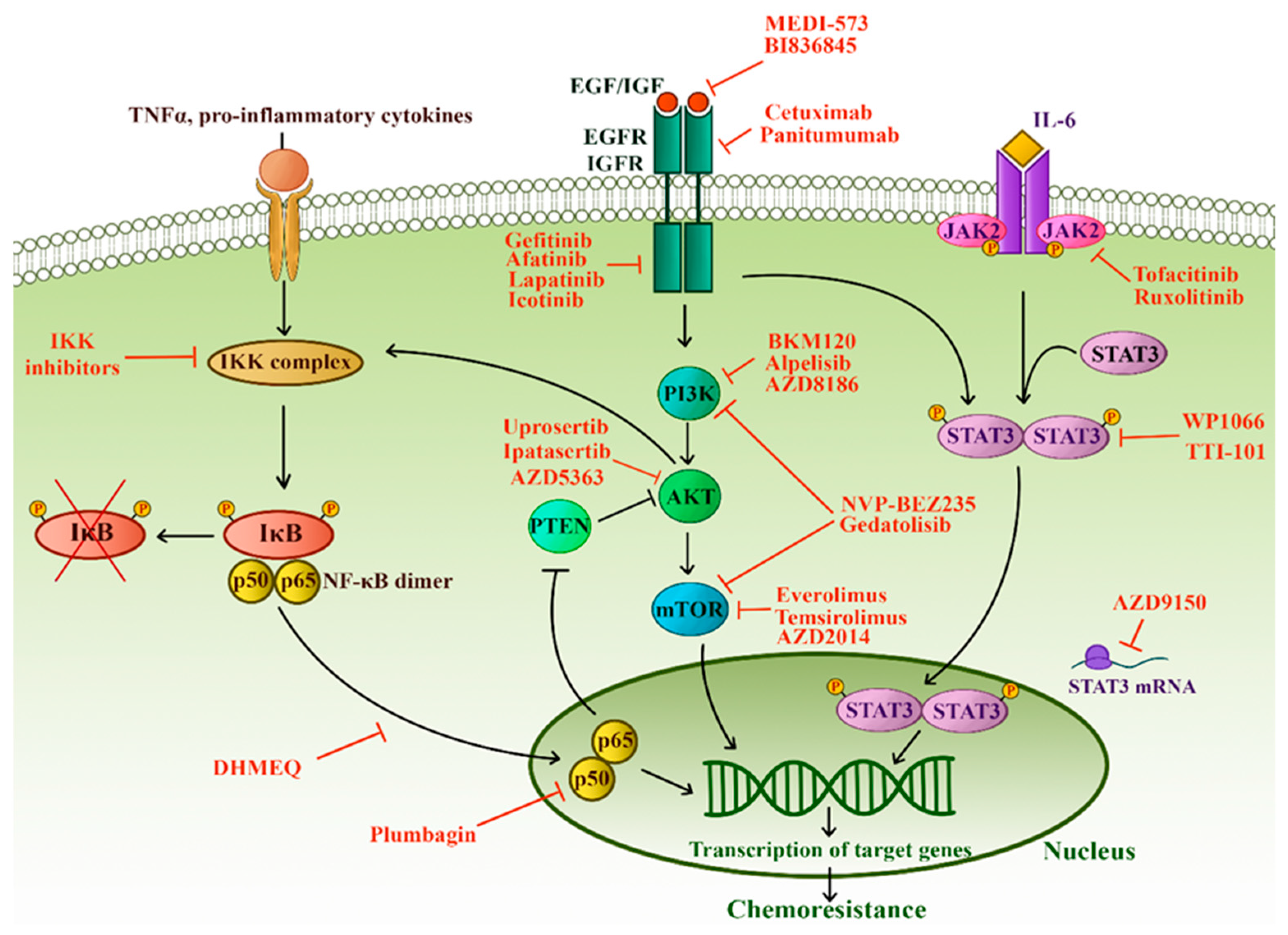
3.5.1. NF-κB
3.5.2. PTEN and PI3K-AKT-mTOR Pathway
3.5.3. JAK/STAT Pathway
3.5.4. Receptor Tyrosine Kinases
3.6. MicroRNAs
3.7. TNBC Heterogeneity
4. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Siddharth, S.; Sharma, D. Racial Disparity and Triple-Negative Breast Cancer in African-American Women: A Multifaceted Affair between Obesity, Biology, and Socioeconomic Determinants. Cancers 2018, 10, 514. [Google Scholar] [CrossRef] [PubMed]
- Dent, R.; Trudeau, M.; Pritchard, K.I.; Hanna, W.M.; Kahn, H.K.; Sawka, C.A.; Lickley, L.A.; Rawlinson, E.; Sun, P.; Narod, S.A. Triple-negative breast cancer: Clinical features and patterns of recurrence. Clin. Cancer Res. 2007, 13, 4429–4434. [Google Scholar] [CrossRef] [PubMed]
- Kennecke, H.; Yerushalmi, R.; Woods, R.; Cheang, M.C.U.; Voduc, D.; Speers, C.H.; Nielsen, T.O.; Gelmon, K. Metastatic behavior of breast cancer subtypes. J. Clin. Oncol. 2010, 28, 3271–3277. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef]
- Prat, A.; Adamo, B.; Cheang, M.C.U.; Anders, C.K.; Carey, L.A.; Perou, C.M. Molecular Characterization of Basal-Like and Non-Basal-Like Triple-Negative Breast Cancer. Oncologist 2013, 18, 123–133. [Google Scholar] [CrossRef]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef]
- Lebert, J.M.; Lester, R.; Powell, E.; Seal, M.; McCarthy, J. Advances in the systemic treatment of triple-negative breast cancer. Curr. Oncol. 2018, 25, S142–S150. [Google Scholar] [CrossRef]
- Liedtke, C.; Mazouni, C.; Hess, K.R.; André, F.; Tordai, A.; Mejia, J.A.; Symmans, W.F.; Gonzalez-Angulo, A.M.; Hennessy, B.; Green, M.; et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J. Clin. Oncol. 2008, 26, 1275–1281. [Google Scholar] [CrossRef]
- Carey, L.A.; Dees, E.C.; Sawyer, L.; Gatti, L.; Moore, D.T.; Collichio, F.; Ollila, D.W.; Sartor, C.I.; Graham, M.L.; Perou, C.M. The triple negative paradox: Primary tumor chemosensitivity of breast cancer subtypes. Clin. Cancer Res. 2007, 13, 2329–2334. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Sissung, T.M.; Baum, C.E.; Kirkland, C.T.; Gao, R.; Gardner, E.R.; Figg, W.D. Pharmacogenetics of membrane transporters: An update on current approaches. Mol. Biotechnol. 2010, 44, 152–167. [Google Scholar] [CrossRef] [PubMed]
- Sharom, F.J. ABC multidrug transporters: Structure, function and role in chemoresistance. Pharmacogenomics 2008, 9, 105–127. [Google Scholar] [CrossRef] [PubMed]
- Yamada, A.; Ishikawa, T.; Ota, I.; Kimura, M.; Shimizu, D.; Tanabe, M.; Chishima, T.; Sasaki, T.; Ichikawa, Y.; Morita, S.; et al. High expression of ATP-binding cassette transporter ABCC11 in breast tumors is associated with aggressive subtypes and low disease-free survival. Breast Cancer Res. Treat. 2013, 137, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Zhao, Z.; Wang, K.; Zhou, H.; Xing, C. Expression of aldehyde dehydrogenase 1 and ATP-binding cassette superfamily G member 2 is enhanced in primary foci and metastatic lymph node from patients with triple-negative breast cancer. Biomed. Res. 2017, 28, 5078–5083. [Google Scholar]
- Guestini, F.; Ono, K.; Miyashita, M.; Ishida, T.; Ohuchi, N.; Nakagawa, S.; Hirakawa, H.; Tamaki, K.; Ohi, Y.; Rai, Y.; et al. Impact of Topoisomerase IIα, PTEN, ABCC1/MRP1, and KI67 on triple-negative breast cancer patients treated with neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2019, 173, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Das, S.; Samant, R.S.; Shevde, L.A. Nonclassical activation of Hedgehog signaling enhances multidrug resistance and makes cancer cells refractory to Smoothened-targeting Hedgehog inhibition. J. Biol. Chem. 2013, 288, 11824–11833. [Google Scholar] [CrossRef] [PubMed]
- Britton, K.M.; Eyre, R.; Harvey, I.J.; Stemke-Hale, K.; Browell, D.; Lennard, T.W.J.; Meeson, A.P. Breast cancer, side population cells and ABCG2 expression. Cancer Lett. 2012, 323, 97–105. [Google Scholar] [CrossRef]
- Arumugam, A.; Subramani, R.; Nandy, S.B.; Terreros, D.; Dwivedi, A.K.; Saltzstein, E.; Lakshmanaswamy, R. Silencing growth hormone receptor inhibits estrogen receptor negative breast cancer through ATP-binding cassette sub-family G member 2. Exp. Mol. Med. 2019, 51, 2. [Google Scholar] [CrossRef]
- Oguri, T.; Bessho, Y.; Achiwa, H.; Ozasa, H.; Maeno, K.; Maeda, H.; Sato, S.; Ueda, R. MRP8/ABCC11 directly confers resistance to 5-fluorouracil. Mol. Cancer Ther. 2007, 6, 122–127. [Google Scholar] [CrossRef] [PubMed]
- Hamed, A.R.; Abdel-Azim, N.S.; Shams, K.A.; Hammouda, F.M. Targeting multidrug resistance in cancer by natural chemosensitizers. Bull. Natl. Res. Cent. 2019, 43, 8. [Google Scholar] [CrossRef]
- O’Connor, R.; Heenan, M.; Connolly, L.; Larkin, A.; Clynes, M. Increased Anti-tumour Efficacy of Doxorubicin when Combined with Sulindac in a Xenograft Model of an MRP-1-positive Human Lung Cancer. Anticancer Res. 2004, 24, 457–464. [Google Scholar] [PubMed]
- O’Connor, R.; O’Leary, M.; Ballot, J.; Collins, C.D.; Kinsella, P.; Mager, D.E.; Arnold, R.D.; O’Driscoll, L.; Larkin, A.; Kennedy, S.; et al. A phase I clinical and pharmacokinetic study of the multi-drug resistance protein-1 (MRP-1) inhibitor sulindac, in combination with epirubicin in patients with advanced cancer. Cancer Chemother. Pharmacol. 2007, 59, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Dong, Z.; Qi, J.; Yang, Y.; Liu, Y.; Li, Z.; Xu, J.; Zhang, J.-T. A novel two mode-acting inhibitor of ABCG2-mediated multidrug transport and resistance in cancer chemotherapy. PLoS ONE 2009, 4, e5676. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Fu, L. Tyrosine kinase inhibitors enhanced the efficacy of conventional chemotherapeutic agent in multidrug resistant cancer cells. Mol. Cancer 2018, 17, 25. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Hung, M.-C.; Yamaguchi, H. A perspective on anti-EGFR therapies targeting triple-negative breast cancer. Am. J. Cancer Res. 2016, 6, 1609–1623. [Google Scholar]
- Zhu, X.; Zhou, W. The Emerging Regulation of VEGFR-2 in Triple-Negative Breast Cancer. Front. Endocrinol. 2015, 6, 159. [Google Scholar] [CrossRef]
- Wang, S.; Tan, X.; Li, S.; Zhou, Y.; Geng, P.; Hua, A.; Deng, A.; Yu, Z. Indomethacin-based stimuli-responsive micelles combined with paclitaxel to overcome multidrug resistance. Oncotarget 2017, 8, 111281–111294. [Google Scholar] [CrossRef]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-humans trial of an RNA interference therapeutic targeting VEGF and KSP in cancer patients with liver involvement. Cancer Discov. 2013, 3, 406–417. [Google Scholar] [CrossRef]
- Deng, T.; Xie, N.; Mou, L.; Jin, Y.; Hu, Z.; Li, Z.; Liu, W.; Yuan, J. Modulating Drug Resistance by Targeting BCRP/ABCG2 Using Retrovirus-Mediated RNA Interference. PLoS ONE 2014, 9, e103463. [Google Scholar]
- Hu, J.; Li, J.; Yue, X.; Wang, J.-c.; Wang, J.-f.; Liu, J.-z.; Kong, D.-l. Targeting BCRP/ABCG2 by RNA interference enhances the chemotherapy sensitivity of human colon cancer side population cells. J. Huazhong Univ. Sci. Technol.-Med. Sci. 2017, 37, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Liu, G.; Fang, T.; Wang, Y.; Zhang, H.; Yang, S.; Wei, J.; Lv, Z.; Tan, L.; Liu, J. Silencing MRP1-4 genes by RNA interference enhances sensitivity of human hepatoma cells to chemotherapy. Am. J. Transl. Res. 2016, 8, 2790–2802. [Google Scholar] [PubMed]
- Wang, Y.; Zhao, L.; Xiao, Q.; Jiang, L.; He, M.; Bai, X.; Ma, M.; Jiao, X.; Wei, M. miR-302a/b/c/d cooperatively inhibit BCRP expression to increase drug sensitivity in breast cancer cells. Gynecol. Oncol. 2016, 141, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Westover, D.; Ling, X.; Lam, H.; Welch, J.; Jin, C.; Gongora, C.; Del Rio, M.; Wani, M.; Li, F. FL118, a novel camptothecin derivative, is insensitive to ABCG2 expression and shows improved efficacy in comparison with irinotecan in colon and lung cancer models with ABCG2-induced resistance. Mol. Cancer 2015, 14, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ling, X.; Liu, X.; Zhong, K.; Smith, N.; Prey, J.; Li, F. Fl118, a novel camptothecin analogue, overcomes irinotecan and topotecan resistance in human tumor xenograft models. Am. J. Transl. Res. 2015, 7, 1765–1781. [Google Scholar]
- Shibata, M.; Hoque, M.O. Targeting Cancer Stem Cells: A Strategy for Effective Eradication of Cancer. Cancers 2019, 11. [Google Scholar] [CrossRef]
- Khan, A.Q.; Ahmed, E.I.; Elareer, N.R.; Junejo, K.; Steinhoff, M.; Uddin, S. Role of miRNA-Regulated Cancer Stem Cells in the Pathogenesis of Human Malignancies. Cells 2019, 8, 840. [Google Scholar] [CrossRef]
- Creighton, C.J.; Li, X.; Landis, M.; Dixon, J.M.; Neumeister, V.M.; Sjolund, A.; Rimm, D.L.; Wong, H.; Rodriguez, A.; Herschkowitz, J.I.; et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc. Natl. Acad. Sci. USA 2009, 106, 13820–13825. [Google Scholar] [CrossRef]
- Lee, H.E.; Kim, J.H.; Kim, Y.J.; Choi, S.Y.; Kim, S.W.; Kang, E.; Chung, I.Y.; Kim, I.A.; Kim, E.J.; Choi, Y.; et al. An increase in cancer stem cell population after primary systemic therapy is a poor prognostic factor in breast cancer. Br. J. Cancer 2011, 104, 1730–1738. [Google Scholar] [CrossRef]
- Park, S.Y.; Lee, H.E.; Li, H.; Shipitsin, M.; Gelman, R.; Polyak, K. Heterogeneity for Stem Cell-Related Markers According to Tumor Subtype and Histologic Stage in Breast Cancer. Clin. Cancer Res. 2010, 16, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, H.; Wang, H.; Shi, X.; Fan, Y.; Ding, X.; Lin, C.; Zhan, Q.; Qian, H.; Xu, B. Enriched CD44(+)/CD24(-) population drives the aggressive phenotypes presented in triple-negative breast cancer (TNBC). Cancer Lett. 2014, 353, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Ma, F.; Li, H.; Li, Y.; Ding, X.; Wang, H.; Fan, Y.; Lin, C.; Qian, H.; Xu, B. Aldehyde dehydrogenase 1 (ALDH1) expression is an independent prognostic factor in triple negative breast cancer (TNBC). Medicine 2017, 96, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Wang, L.; Song, Y.; Wang, S.; Huang, X.; Xuan, Q.; Kang, X.; Zhang, Q. CD44+/CD24- phenotype predicts a poor prognosis in triple-negative breast cancer. Oncol. Lett. 2017, 14, 5890–5898. [Google Scholar] [CrossRef] [PubMed]
- Bhola, N.E.; Balko, J.M.; Dugger, T.C.; Kuba, M.G.; Sánchez, V.; Sanders, M.; Stanford, J.; Cook, R.S.; Arteaga, C.L. TGF-β inhibition enhances chemotherapy action against triple-negative breast cancer. J. Clin. Invest. 2013, 123, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Samanta, D.; Gilkes, D.M.; Chaturvedi, P.; Xiang, L.; Semenza, G.L. Hypoxia-inducible factors are required for chemotherapy resistance of breast cancer stem cells. Proc. Natl. Acad. Sci. USA 2014, 111, E5429–E5438. [Google Scholar] [CrossRef] [PubMed]
- He, J.; Lee, H.-J.; Saha, S.; Ruan, D.; Guo, H.; Chan, C.-H. Inhibition of USP2 eliminates cancer stem cells and enhances TNBC responsiveness to chemotherapy. Cell Death Dis. 2019, 10, 285. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Schuetz, J.D.; Bunting, K.D.; Colapietro, A.M.; Sampath, J.; Morris, J.J.; Lagutina, I.; Grosveld, G.C.; Osawa, M.; Nakauchi, H.; et al. The ABC transporter Bcrp1/ABCG2 is expressed in a wide variety of stem cells and is a molecular determinant of the side-population phenotype. Nat. Med. 2001, 7, 1028–1034. [Google Scholar] [CrossRef] [PubMed]
- Shishido, Y.; Ueno, S.; Yamazaki, R.; Nagaoka, M.; Matsuzaki, T. ABCG2 inhibitor YHO-13351 sensitizes cancer stem/Initiating-like side population cells to irinotecan. Anticancer Res. 2013, 33, 1379–1386. [Google Scholar]
- Yang, C.; He, Y.; Zhang, H.; Liu, Y.; Wang, W.; Du, Y.; Gao, F. Selective killing of breast cancer cells expressing activated CD44 using CD44 ligand-coated nanoparticles in vitro and in vivo. Oncotarget 2015, 6. [Google Scholar] [CrossRef]
- He, L.; Gu, J.; Lim, L.Y.; Yuan, Z.X.; Mo, J. Nanomedicine-mediated therapies to target breast cancer stem cells. Front. Pharmacol. 2016, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.L.; Robin, T.P.; Ford, H.L. Molecular Pathways: Targeting the TGF-β Pathway for Cancer Therapy. Clin. Cancer Res. 2012, 18, 4514–4521. [Google Scholar] [CrossRef] [PubMed]
- Neuzillet, C.; Tijeras-Raballand, A.; Cohen, R.; Cros, J.; Faivre, S.; Raymond, E.; de Gramont, A. Targeting the TGFβ pathway for cancer therapy. Pharmacol. Ther. 2015, 147, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Asiedu, M.K.; Ingle, J.N.; Behrens, M.D.; Radisky, D.C.; Knutson, K.L. TGFβ/TNFα-Mediated Epithelial-Mesenchymal Transition Generates Breast Cancer Stem Cells with a Claudin-Low Phenotype. Cancer Res. 2011, 71, 4707–4719. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Zhang, L.; He, X.; Zhang, P.; Sun, C.; Xu, X.; Lu, Y.; Li, F. TGF-β plays a vital role in triple-negative breast cancer (TNBC) drug-resistance through regulating stemness, EMT and apoptosis. Biochem. Biophys. Res. Commun. 2018, 502, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Giaccone, G.; Bazhenova, L.A.; Nemunaitis, J.; Tan, M.; Juhász, E.; Ramlau, R.; van den Heuvel, M.M.; Lal, R.; Kloecker, G.; Eaton, K.D.; et al. A phase III study of belagenpumatucel-L, an allogeneic tumour cell vaccine, as maintenance therapy for non-small cell lung cancer. Eur. J. Cancer 2015, 51, 2321–2329. [Google Scholar] [CrossRef] [PubMed]
- Bogdahn, U.; Hau, P.; Stockhammer, G.; Venkataramana, N.K.; Mahapatra, A.K.; Suri, A.; Balasubramaniam, A.; Nair, S.; Oliushine, V.; Parfenov, V.; et al. Targeted therapy for high-grade glioma with the TGF- 2 inhibitor trabedersen: Results of a randomized and controlled phase IIb study. Neuro. Oncol. 2011, 13, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Shi, M.; Zhu, J.; Wang, R.; Chen, X.; Mi, L.; Walz, T.; Springer, T.A. Latent TGF-β structure and activation. Nature 2011, 474, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Aster, J.C.; Pear, W.S.; Blacklow, S.C. The Varied Roles of Notch in Cancer. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 245–275. [Google Scholar] [CrossRef]
- Rustighi, A.; Zannini, A.; Tiberi, L.; Sommaggio, R.; Piazza, S.; Sorrentino, G.; Nuzzo, S.; Tuscano, A.; Eterno, V.; Benvenuti, F.; et al. Prolyl-isomerase Pin1 controls normal and cancer stem cells of the breast. EMBO Mol. Med. 2014, 6, 99–119. [Google Scholar] [CrossRef]
- Harrison, H.; Farnie, G.; Howell, S.J.; Rock, R.E.; Stylianou, S.; Brennan, K.R.; Bundred, N.J.; Clarke, R.B. Regulation of Breast Cancer Stem Cell Activity by Signaling through the Notch4 Receptor. Cancer Res. 2010, 70, 709–718. [Google Scholar] [CrossRef] [PubMed]
- Nagamatsu, I.; Onishi, H.; Matsushita, S.; Kubo, M.; Kai, M.; Imaizumi, A.; Nakano, K.; Hattori, M.; Oda, Y.; Tanaka, M.; et al. NOTCH4 is a potential therapeutic target for triple-negative breast cancer. Anticancer Res. 2014, 34, 69–80. [Google Scholar] [PubMed]
- Diluvio, G.; Del Gaudio, F.; Giuli, M.V.; Franciosa, G.; Giuliani, E.; Palermo, R.; Besharat, Z.M.; Pignataro, M.G.; Vacca, A.; D’Amati, G.; et al. NOTCH3 inactivation increases triple negative breast cancer sensitivity to gefitinib by promoting EGFR tyrosine dephosphorylation and its intracellular arrest. Oncogenesis 2018, 7, 42. [Google Scholar] [CrossRef] [PubMed]
- Zang, S.; Chen, F.; Dai, J.; Guo, D.; Tse, W.; Qu, X.; Ma, D.; Ji, C. RNAi-mediated knockdown of Notch-1 leads to cell growth inhibition and enhanced chemosensitivity in human breast cancer. Oncol. Rep. 2010, 23. [Google Scholar]
- Choy, L.; Hagenbeek, T.J.; Solon, M.; French, D.; Finkle, D.; Shelton, A.; Venook, R.; Brauer, M.J.; Siebel, C.W. Constitutive NOTCH3 Signaling Promotes the Growth of Basal Breast Cancers. Cancer Res. 2017, 77, 1439–1452. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Liu, X.; Luo, J.; Xiao, W.; Ye, X.; Chen, M.; Li, Y.; Zhang, G.-J. Notch3 inhibits epithelial–mesenchymal transition by activating Kibra-mediated Hippo/YAP signaling in breast cancer epithelial cells. Oncogenesis 2016, 5, e269. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.; Stephen, S.L.; Hanby, A.M.; Horgan, K.; Perry, S.L.; Richardson, J.; Roundhill, E.A.; Valleley, E.M.A.; Verghese, E.T.; Williams, B.J.; et al. Chemotherapy induces Notch1-dependent MRP1 up-regulation, inhibition of which sensitizes breast cancer cells to chemotherapy. BMC Cancer 2015, 15, 634. [Google Scholar] [CrossRef]
- Li, Z.-L.; Chen, C.; Yang, Y.; Wang, C.; Yang, T.; Yang, X.; Liu, S.-C. Gamma secretase inhibitor enhances sensitivity to doxorubicin in MDA-MB-231 cells. Int. J. Clin. Exp. Pathol. 2015, 8, 4378–4387. [Google Scholar]
- Qiu, M.; Peng, Q.; Jiang, I.; Carroll, C.; Han, G.; Rymer, I.; Lippincott, J.; Zachwieja, J.; Gajiwala, K.; Kraynov, E.; et al. Specific inhibition of Notch1 signaling enhances the antitumor efficacy of chemotherapy in triple negative breast cancer through reduction of cancer stem cells. Cancer Lett. 2013, 328, 261–270. [Google Scholar] [CrossRef]
- Locatelli, M.A.; Aftimos, P.; Dees, E.C.; LoRusso, P.M.; Pegram, M.D.; Awada, A.; Huang, B.; Cesari, R.; Jiang, Y.; Shaik, M.N.; et al. Phase I study of the gamma secretase inhibitor PF-03084014 in combination with docetaxel in patients with advanced triple-negative breast cancer. Oncotarget 2017, 8, 2320–2328. [Google Scholar] [CrossRef]
- Schott, A.F.; Landis, M.D.; Dontu, G.; Griffith, K.A.; Layman, R.M.; Krop, I.; Paskett, L.A.; Wong, H.; Dobrolecki, L.E.; Lewis, M.T.; et al. Preclinical and clinical studies of gamma secretase inhibitors with docetaxel on human breast tumors. Clin. Cancer Res. 2013, 19, 1512–1524. [Google Scholar] [CrossRef] [PubMed]
- Broner, E.C.; Alpert, G.; Gluschnaider, U.; Mondshine, A.; Solomon, O.; Sloma, I.; Rauch, R.; Izumchenko, E.; Aster, J.C.; Davis, M. AL101 mediated tumor inhibition in notch-altered TNBC PDX models. J. Clin. Oncol. 2019, 37, 1064. [Google Scholar] [CrossRef]
- Smith, D.C.; Chugh, R.; Patnaik, A.; Papadopoulos, K.P.; Wang, M.; Kapoun, A.M.; Xu, L.; Dupont, J.; Stagg, R.J.; Tolcher, A. A phase 1 dose escalation and expansion study of Tarextumab (OMP-59R5) in patients with solid tumors. Invest. New Drugs 2019, 37, 722–730. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Kaur, P.; Bunnag, N.; Suresh, J.; Sung, I.C.H.; Tan, Q.H.; Gruber, J.; Tolwinski, N.S. WNT Signaling in Disease. Cells 2019, 8, 826. [Google Scholar] [CrossRef] [PubMed]
- Duchartre, Y.; Kim, Y.-M.; Kahn, M. The Wnt signaling pathway in cancer. Crit. Rev. Oncol. Hematol. 2016, 99, 141–149. [Google Scholar] [CrossRef]
- Dey, N.; Barwick, B.G.; Moreno, C.S.; Ordanic-Kodani, M.; Chen, Z.; Oprea-Ilies, G.; Tang, W.; Catzavelos, C.; Kerstann, K.F.; Sledge, G.W.; et al. Wnt signaling in triple negative breast cancer is associated with metastasis. BMC Cancer 2013, 13, 537. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Prosperi, J.R.; Choudhury, N.; Olopade, O.I.; Goss, K.H. β-Catenin is required for the tumorigenic behavior of triple-negative breast cancer cells. PLoS ONE 2015, 10, e0117097. [Google Scholar] [CrossRef]
- Shen, H.; Yan, W.; Yuan, J.; Wang, Z.; Wang, C. Nek2B activates the wnt pathway and promotes triple-negative breast cancer chemothezrapy-resistance by stabilizing β-catenin. J. Exp. Clin. Cancer Res. 2019, 38, 243. [Google Scholar] [CrossRef]
- Corda, G.; Sala, G.; Lattanzio, R.; Iezzi, M.; Sallese, M.; Fragassi, G.; Lamolinara, A.; Mirza, H.; Barcaroli, D.; Ermler, S.; et al. Functional and prognostic significance of the genomic amplification of frizzled 6 (FZD6) in breast cancer. J. Pathol. 2017, 241, 350–361. [Google Scholar] [CrossRef]
- Yin, S.; Xu, L.; Bonfil, R.D.; Banerjee, S.; Sarkar, F.H.; Sethi, S.; Reddy, K.B. Tumor-initiating cells and FZD8 play a major role in drug resistance in triple-negative breast cancer. Mol. Cancer Ther. 2013, 12, 491–498. [Google Scholar] [CrossRef]
- Ma, J.; Lu, W.; Chen, D.; Xu, B.; Li, Y. Role of Wnt Co-Receptor LRP6 in Triple Negative Breast Cancer Cell Migration and Invasion. J. Cell. Biochem. 2017, 118, 2968–2976. [Google Scholar] [CrossRef] [PubMed]
- Gangrade, A.; Pathak, V.; Augelli-Szafran, C.E.; Wei, H.-X.; Oliver, P.; Suto, M.; Buchsbaum, D.J. Preferential Inhibition of Wnt/β-Catenin Signaling by Novel Benzimidazole Compounds in Triple-Negative Breast Cancer. Int. J. Mol. Sci. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Yang, H.; Li, X.; Han, L.; Xu, N.; Shi, A. Signaling pathway inhibitors target breast cancer stem cells in triple-negative breast cancer. Oncol. Rep. 2019, 41, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Jang, G.-B.; Hong, I.-S.; Kim, R.-J.; Lee, S.-Y.; Park, S.-J.; Lee, E.-S.; Park, J.H.; Yun, C.-H.; Chung, J.-U.; Lee, K.-J.; et al. Wnt/-Catenin Small-Molecule Inhibitor CWP232228 Preferentially Inhibits the Growth of Breast Cancer Stem-like Cells. Cancer Res. 2015, 75, 1691–1702. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, K.; Koval, A.; Xu, J.; Bodmer, A.; Katanaev, V.L. Towards the first targeted therapy for triple-negative breast cancer: Repositioning of clofazimine as a chemotherapy-compatible selective Wnt pathway inhibitor. Cancer Lett. 2019, 449, 45–55. [Google Scholar] [CrossRef] [PubMed]
- Ha, G.-H.; Kim, D.-Y.; Breuer, E.-K.; Kim, C.K. Combination Treatment of Polo-Like Kinase 1 and Tankyrase-1 Inhibitors Enhances Anticancer Effect in Triple-negative Breast Cancer Cells. Anticancer Res. 2018, 38, 1303–1310. [Google Scholar]
- Xie, W.; Zhang, Y.; He, Y.; Zhang, K.; Wan, G.; Huang, Y.; Zhou, Z.; Huang, G.; Wang, J. A novel recombinant human Frizzled-7 protein exhibits anti-tumor activity against triple negative breast cancer via abating Wnt/β-catenin pathway. Int. J. Biochem. Cell Biol. 2018, 103, 45–55. [Google Scholar] [CrossRef]
- Skoda, A.M.; Simovic, D.; Karin, V.; Kardum, V.; Vranic, S.; Serman, L. The role of the Hedgehog signaling pathway in cancer: A comprehensive review. Bosn. J. basic Med. Sci. 2018, 18, 8–20. [Google Scholar] [CrossRef]
- Harris, L.G.; Pannell, L.K.; Singh, S.; Samant, R.S.; Shevde, L.A. Increased vascularity and spontaneous metastasis of breast cancer by hedgehog signaling mediated upregulation of cyr61. Oncogene 2012, 31, 3370–3380. [Google Scholar] [CrossRef]
- Kwon, Y.-J.; Hurst, D.R.; Steg, A.D.; Yuan, K.; Vaidya, K.S.; Welch, D.R.; Frost, A.R. Gli1 enhances migration and invasion via up-regulation of MMP-11 and promotes metastasis in ERα negative breast cancer cell lines. Clin. Exp. Metastasis 2011, 28, 437–449. [Google Scholar] [CrossRef]
- Di Mauro, C.; Rosa, R.; D’Amato, V.; Ciciola, P.; Servetto, A.; Marciano, R.; Orsini, R.C.; Formisano, L.; De Falco, S.; Cicatiello, V.; et al. Hedgehog signalling pathway orchestrates angiogenesis in triple-negative breast cancers. Br. J. Cancer 2017, 116, 1425–1435. [Google Scholar] [CrossRef] [PubMed]
- Bhateja, P.; Cherian, M.; Majumder, S.; Ramaswamy, B. The Hedgehog Signaling Pathway: A Viable Target in Breast Cancer? Cancers 2019, 11, 1126. [Google Scholar] [CrossRef] [PubMed]
- Han, B.; Qu, Y.; Jin, Y.; Yu, Y.; Deng, N.; Wawrowsky, K.; Zhang, X.; Li, N.; Bose, S.; Wang, Q.; et al. FOXC1 Activates Smoothened-Independent Hedgehog Signaling in Basal-like Breast Cancer. Cell Rep. 2015, 13, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006, 66, 6063–6071. [Google Scholar] [CrossRef]
- Sims-Mourtada, J.; Opdenaker, L.M.; Davis, J.; Arnold, K.M.; Flynn, D. Taxane-induced hedgehog signaling is linked to expansion of breast cancer stem-like populations after chemotherapy. Mol. Carcinog. 2015, 54, 1480–1493. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Fan, L.; Wei, G.; Chen, X.; Duan, W.; Xu, Q.; Sheng, W.; Wang, K.; Li, X. Gli-1 is crucial for hypoxia-induced epithelial-mesenchymal transition and invasion of breast cancer. Tumour Biol. 2015, 36, 3119–3126. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.M.; Pohlig, R.T.; Sims-Mourtada, J. Co-activation of Hedgehog and Wnt signaling pathways is associated with poor outcomes in triple negative breast cancer. Oncol. Lett. 2017, 14, 5285–5292. [Google Scholar] [CrossRef]
- O’Toole, S.A.; Machalek, D.A.; Shearer, R.F.; Millar, E.K.A.; Nair, R.; Schofield, P.; McLeod, D.; Cooper, C.L.; McNeil, C.M.; McFarland, A.; et al. Hedgehog overexpression is associated with stromal interactions and predicts for poor outcome in breast cancer. Cancer Res. 2011, 71, 4002–4014. [Google Scholar] [CrossRef]
- Koike, Y.; Ohta, Y.; Saitoh, W.; Yamashita, T.; Kanomata, N.; Moriya, T.; Kurebayashi, J. Anti-cell growth and anti-cancer stem cell activities of the non-canonical hedgehog inhibitor GANT61 in triple-negative breast cancer cells. Breast Cancer 2017, 24, 683–693. [Google Scholar] [CrossRef]
- Vaupel, P. Hypoxia and Aggressive Tumor Phenotype: Implications for Therapy and Prognosis. Oncologist 2008, 13, 21–26. [Google Scholar] [CrossRef]
- Gerweck, L.E.; Vijayappa, S.; Kozin, S. Tumor pH controls the in vivo efficacy of weak acid and base chemotherapeutics. Mol. Cancer Ther. 2006, 5, 1275–1279. [Google Scholar] [CrossRef] [PubMed]
- Cosse, J.-P.; Michiels, C. Tumour hypoxia affects the responsiveness of cancer cells to chemotherapy and promotes cancer progression. Anticancer. Agents Med. Chem. 2008, 8, 790–797. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Lin, Q.; Glazer, P.M.; Yun, Z. The hypoxic tumor microenvironment in vivo selects the cancer stem cell fate of breast cancer cells. Breast Cancer Res. 2018, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Chouaib, S.; Noman, M.Z.; Kosmatopoulos, K.; Curran, M.A. Hypoxic stress: Obstacles and opportunities for innovative immunotherapy of cancer. Oncogene 2017, 36, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Xiang, L.; Liu, Z.H.; Huan, Q.; Su, P.; Du, G.J.; Wang, Y.; Gao, P.; Zhou, G.Y. Hypoxia-inducible factor-2a is associated with ABCG2 expression, histology-grade and Ki67 expression in breast invasive ductal carcinoma. Diagn. Pathol. 2012, 7, 32. [Google Scholar] [CrossRef] [PubMed]
- Lv, Y.; Zhao, S.; Han, J.; Zheng, L.; Yang, Z.; Zhao, L. Hypoxia-inducible factor-1α induces multidrug resistance protein in colon cancer. Onco. Targets. Ther. 2015, 8, 1941–1948. [Google Scholar] [CrossRef]
- Daskalaki, I.; Gkikas, I.; Tavernarakis, N. Hypoxia and Selective Autophagy in Cancer Development and Therapy. Front. Cell Dev. Biol. 2018, 6, 1–22. [Google Scholar] [CrossRef]
- Livasy, C.A.; Karaca, G.; Nanda, R.; Tretiakova, M.S.; Olopade, O.I.; Moore, D.T.; Perou, C.M. Phenotypic evaluation of the basal-like subtype of invasive breast carcinoma. Mod. Pathol. 2006, 19, 264–271. [Google Scholar] [CrossRef]
- Tan, E.Y.; Yan, M.; Campo, L.; Han, C.; Takano, E.; Turley, H.; Candiloro, I.; Pezzella, F.; Gatter, K.C.; Millar, E.K.A.; et al. The key hypoxia regulated gene CAIX is upregulated in basal-like breast tumours and is associated with resistance to chemotherapy. Br. J. Cancer 2009, 100, 405–411. [Google Scholar] [CrossRef]
- Montagner, M.; Enzo, E.; Forcato, M.; Zanconato, F.; Parenti, A.; Rampazzo, E.; Basso, G.; Leo, G.; Rosato, A.; Bicciato, S.; et al. SHARP1 suppresses breast cancer metastasis by promoting degradation of hypoxia-inducible factors. Nature 2012, 487, 380–384. [Google Scholar] [CrossRef]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, R.; Gianni, L. Hallmarks of triple negative breast cancer emerging at last? Cell Res. 2014, 24, 904–905. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.S.; Lee, H.; Park, I.A.; Chung, Y.R.; Im, S.A.; Lee, K.H.; Moon, H.G.; Han, W.; Kim, K.; Kim, T.Y.; et al. Overexpression of HIF1α and CAXI predicts poor outcome in early-stage triple negative breast cancer. Virchows Arch. 2016, 469, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Bharti, S.K.; Mironchik, Y.; Wildes, F.; Penet, M.-F.; Goggins, E.; Krishnamachary, B.; Bhujwalla, Z.M. Metabolic consequences of HIF silencing in a triple negative human breast cancer xenograft. Oncotarget 2018, 9, 15326–15339. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.R.; Hay, M.P. Targeting hypoxia in cancer therapy. Nat. Rev. Cancer 2011, 11, 393–410. [Google Scholar] [CrossRef] [PubMed]
- Borad, M.J.; Reddy, S.G.; Bahary, N.; Uronis, H.E.; Sigal, D.; Cohn, A.L.; Schelman, W.R.; Stephenson, J.; Chiorean, E.G.; Rosen, P.J.; et al. Randomized phase II trial of gemcitabine plus TH-302 versus gemcitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2015, 33, 1475–1481. [Google Scholar] [CrossRef] [PubMed]
- Baran, N.; Konopleva, M. Molecular Pathways: Hypoxia-Activated Prodrugs in Cancer Therapy. Clin. Cancer Res. 2017, 23, 2382–2390. [Google Scholar] [CrossRef] [PubMed]
- Ban, H.S.; Kim, B.K.; Lee, H.; Kim, H.M.; Harmalkar, D.; Nam, M.; Park, S.K.; Lee, K.; Park, J.T.; Kim, I.; et al. The novel hypoxia-inducible factor-1α inhibitor IDF-11774 regulates cancer metabolism, thereby suppressing tumor growth. Cell Death Dis. 2017, 8, e2843. [Google Scholar] [CrossRef] [PubMed]
- Soni, S.; Padwad, Y.S. HIF-1 in cancer therapy: Two decade long story of a transcription factor. Acta Oncol. 2017, 56, 503–515. [Google Scholar] [CrossRef]
- Courtney, K.D.; Infante, J.R.; Lam, E.T.; Figlin, R.A.; Rini, B.I.; Brugarolas, J.; Zojwalla, N.J.; Lowe, A.M.; Wang, K.; Wallace, E.M.; et al. Phase I dose-escalation trial of PT2385, a first-in-class hypoxia-inducible factor-2a antagonist in patients with previously treated advanced clear cell renal cell carcinoma. J. Clin. Oncol. 2018, 36, 867–874. [Google Scholar] [CrossRef]
- Simões-Wüst, A.P.; Schürpf, T.; Hall, J.; Stahel, R.A.; Zangemeister-Wittke, U. Bcl-2/bcl-xL bispecific antisense treatment sensitizes breast carcinoma cells to doxorubicin, paclitaxel and cyclophosphamide. Breast Cancer Res. Treat. 2002, 76, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Inao, T.; Iida, Y.; Moritani, T.; Okimoto, T.; Tanino, R.; Kotani, H.; Harada, M. Bcl-2 inhibition sensitizes triple-negative human breast cancer cells to doxorubicin. Oncotarget 2018, 9, 25545–25556. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.J.; Dhayade, S.; Ferrari, N.; Sims, A.H.; Johnson, E.; Mason, S.M.; Dickson, A.; Ryan, K.M.; Kalna, G.; Edwards, J.; et al. MCL-1 is a prognostic indicator and drug target in breast cancer. Cell Death Dis. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Ozretic, P.; Alvir, I.; Sarcevic, B.; Vujaskovic, Z.; Rendic-Miocevic, Z.; Roguljic, A.; Beketic-Oreskovic, L. Apoptosis regulator bcl-2 is an independent prognostic marker for worse overall survival in triple-negative breast cancer patients. Int. J. Biol. Markers 2018, 33, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sánchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245. [Google Scholar] [CrossRef] [PubMed]
- Wertz, I.E.; Kusam, S.; Lam, C.; Okamoto, T.; Sandoval, W.; Anderson, D.J.; Helgason, E.; Ernst, J.A.; Eby, M.; Liu, J.; et al. Sensitivity to antitubulin chemotherapeutics is regulated by MCL1 and FBW7. Nature 2011, 471, 110–114. [Google Scholar] [CrossRef] [PubMed]
- Koff, J.L.; Ramachandiran, S.; Bernal-Mizrachi, L. A time to kill: Targeting apoptosis in cancer. Int. J. Mol. Sci. 2015, 16, 2942–2955. [Google Scholar] [CrossRef] [PubMed]
- Merino, D.; Kelly, G.L.; Lessene, G.; Wei, A.H.; Roberts, A.W.; Strasser, A. BH3-Mimetic Drugs: Blazing the Trail for New Cancer Medicines. Cancer Cell 2018, 34, 879–891. [Google Scholar] [CrossRef]
- Lemke, J.; von Karstedt, S.; Zinngrebe, J.; Walczak, H. Getting TRAIL back on track for cancer therapy. Cell Death Differ. 2014, 21, 1350–1364. [Google Scholar] [CrossRef]
- Greer, Y.E.; Gilbert, S.F.; Gril, B.; Narwal, R.; Peacock Brooks, D.L.; Tice, D.A.; Steeg, P.S.; Lipkowitz, S. MEDI3039, a novel highly potent tumor necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) receptor 2 agonist, causes regression of orthotopic tumors and inhibits outgrowth of metastatic triple-negative breast cancer. Breast Cancer Res. 2019, 21, 27. [Google Scholar] [CrossRef]
- Wang, S.; Bai, L.; Lu, J.; Liu, L.; Yang, C.-Y.; Sun, H. Targeting inhibitors of apoptosis proteins (IAPs) for new breast cancer therapeutics. J. Mammary Gland Biol. Neoplasia 2012, 17, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Bardia, A.; Parton, M.; Kümmel, S.; Estévez, L.G.; Huang, C.-S.; Cortés, J.; Ruiz-Borrego, M.; Telli, M.L.; Martin-Martorell, P.; López, R.; et al. Paclitaxel With Inhibitor of Apoptosis Antagonist, LCL161, for Localized Triple-Negative Breast Cancer, Prospectively Stratified by Gene Signature in a Biomarker-Driven Neoadjuvant Trial. J. Clin. Oncol. 2018, 36, 3126–3133. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Dutta, J.; Gupta, N.; Fan, G.; Gélinas, C. Regulation of programmed cell death by NF-κB and its role in tumorigenesis and therapy. Adv. Exp. Med. Biol. 2008, 615, 223–250. [Google Scholar] [PubMed]
- Ossovskaya, V.; Wang, Y.; Budoff, A.; Xu, Q.; Lituev, A.; Potapova, O.; Vansant, G.; Monforte, J.; Daraselia, N. Exploring Molecular Pathways of Triple-Negative Breast Cancer. Genes Cancer 2011, 2, 870–879. [Google Scholar] [CrossRef] [PubMed]
- Fusella, F.; Seclì, L.; Busso, E.; Krepelova, A.; Moiso, E.; Rocca, S.; Conti, L.; Annaratone, L.; Rubinetto, C.; Mello-Grand, M.; et al. The IKK/NF-κB signaling pathway requires Morgana to drive breast cancer metastasis. Nat. Commun. 2017, 8, 1636. [Google Scholar] [CrossRef] [PubMed]
- D’Ignazio, L.; Rocha, S. Hypoxia Induced NF-κB. Cells 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed]
- Begalli, F.; Bennett, J.; Capece, D.; Verzella, D.; D’Andrea, D.; Tornatore, L.; Franzoso, G. Unlocking the NF-κB Conundrum: Embracing Complexity to Achieve Specificity. Biomedicines 2017, 5. [Google Scholar]
- Messeha, S.S.; Zarmouh, N.O.; Mendonca, P.; Alwagdani, H.; Kolta, M.G.; Soliman, K.F.A. The inhibitory effects of plumbagin on the NF-κB pathway and CCL2 release in racially different triple-negative breast cancer cells. PLoS ONE 2018, 13, 1–25. [Google Scholar] [CrossRef]
- Pan, H.; Zhou, W.; He, W.; Liu, X.; Ding, Q.; Ling, L.; Zha, X.; Wang, S. Genistein inhibits MDA-MB-231 triple-negative breast cancer cell growth by inhibiting NF-κB activity via the Notch-1 pathway. Int. J. Mol. Med. 2012, 30, 337–343. [Google Scholar] [CrossRef]
- Labbozzetta, M.; Poma, P.; Vivona, N.; Gulino, A.; D’Alessandro, N.; Notarbartolo, M. Epigenetic changes and nuclear factor-κB activation, but not microRNA-224, downregulate Raf-1 kinase inhibitor protein in triple-negative breast cancer SUM 159 cells. Oncol. Lett. 2015, 10, 3807–3815. [Google Scholar] [CrossRef]
- Li, L.; Ross, A.H. Why is PTEN an important tumor suppressor? J. Cell. Biochem. 2007, 102, 1368–1374. [Google Scholar] [CrossRef] [PubMed]
- Inanc, M.; Ozkan, M.; Karaca, H.; Berk, V.; Bozkurt, O.; Duran, A.O.; Ozaslan, E.; Akgun, H.; Tekelioglu, F.; Elmali, F. Cytokeratin 5/6, c-Met expressions, and PTEN loss prognostic indicators in triple-negative breast cancer. Med. Oncol. 2014, 31. [Google Scholar] [CrossRef]
- Ueng, S.H.; Chen, S.C.; Chang, Y.S.; Hsueh, S.; Lin, Y.C.; Chien, H.P.; Lo, Y.F.; Shen, S.C.; Hsueh, C. Phosphorylated mTOR expression correlates with poor outcome in early-stage triple negative breast carcinomas. Int. J. Clin. Exp. Pathol. 2012, 5, 806–813. [Google Scholar] [PubMed]
- Steelman, L.S.; Navolanic, P.M.; Sokolosky, M.L.; Taylor, J.R.; Lehmann, B.D.; Chappell, W.H.; Abrams, S.L.; Wong, E.W.T.; Stadelman, K.M.; Terrian, D.M.; et al. Suppression of PTEN function increases breast cancer chemotherapeutic drug resistance while conferring sensitivity to mTOR inhibitors. Oncogene 2008, 27, 4086–4095. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.J.; Heo, J.H.; Park, J.Y.; Jeong, J.Y.; Cho, H.J.; Park, K.S.; Kim, S.H.; Moon, Y.W.; Kim, J.S.; An, H.J. A novel PI3K/mTOR dual inhibitor, CMG002, overcomes the chemoresistance in ovarian cancer. Gynecol. Oncol. 2019, 153, 135–148. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Shyr, Y.; Pietenpol, J.A.; Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Invest. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-pi3k-inhibitor-breast-cancer (accessed on 7 August 2019).
- Dent, R.; Im, S.-A.; Espie, M.; Blau, S.; Tan, A.R.; Isakoff, S.J.; Oliveira, M.; Saura, C.; Wongchenko, M.; Kapp, A.V.; et al. Overall survival (OS) update of the double-blind placebo (PBO)-controlled randomized phase 2 LOTUS trial of first-line ipatasertib (IPAT) + paclitaxel (PAC) for locally advanced/metastatic triple-negative breast cancer (mTNBC). J. Clin. Oncol. 2018, 36, 1008. [Google Scholar] [CrossRef]
- Schmid, P.; Abraham, J.; Chan, S.; Wheatley, D.; Brunt, M.; Nemsadze, G.; Baird, R.; Park, Y.H.; Hall, P.; Perren, T.; et al. AZD5363 plus paclitaxel versus placebo plus paclitaxel as first-line therapy for metastatic triple-negative breast cancer (PAKT): A randomised, double-blind, placebo-controlled, phase II trial. J. Clin. Oncol. 2018, 36, 1007. [Google Scholar] [CrossRef]
- Guanizo, A.C.; Fernando, C.D.; Garama, D.J.; Gough, D.J. STAT3: A multifaceted oncoprotein. Growth Factors 2018, 36, 1–14. [Google Scholar] [CrossRef]
- Hartman, Z.C.; Poage, G.M.; den Hollander, P.; Tsimelzon, A.; Hill, J.; Panupinthu, N.; Zhang, Y.; Mazumdar, A.; Hilsenbeck, S.G.; Mills, G.B.; et al. Growth of Triple-Negative Breast Cancer Cells Relies upon Coordinate Autocrine Expression of the Proinflammatory Cytokines IL-6 and IL-8. Cancer Res. 2013, 73, 3470–3480. [Google Scholar] [CrossRef]
- Wei, W.; Tweardy, D.J.; Zhang, M.; Zhang, X.; Landua, J.; Petrovic, I.; Bu, W.; Roarty, K.; Hilsenbeck, S.G.; Rosen, J.M.; et al. STAT3 signaling is activated preferentially in tumor-initiating cells in claudin-low models of human breast cancer. Stem Cells 2014, 32, 2571–2582. [Google Scholar] [CrossRef] [PubMed]
- Sirkisoon, S.R.; Carpenter, R.L.; Rimkus, T.; Anderson, A.; Harrison, A.; Lange, A.M.; Jin, G.; Watabe, K.; Lo, H.W. Interaction between STAT3 and GLI1/tGLI1 oncogenic transcription factors promotes the aggressiveness of triple-negative breast cancers and HER2-enriched breast cancer. Oncogene 2018, 37, 2502–2514. [Google Scholar] [CrossRef] [PubMed]
- Kuo, W.Y.; Hwu, L.; Wu, C.Y.; Lee, J.S.; Chang, C.W.; Liu, R.S. STAT3/NF-κB-regulated lentiviral TK/GCV suicide gene therapy for cisplatin-resistant triple-negative breast cancer. Theranostics 2017, 7, 647–663. [Google Scholar] [CrossRef] [PubMed]
- Moreira, M.P.; da Conceição Braga, L.; Cassali, G.D.; Silva, L.M. STAT3 as a promising chemoresistance biomarker associated with the CD44 +/high/CD24 -/low/ALDH + BCSCs-like subset of the triple-negative breast cancer (TNBC) cell line. Exp. Cell Res. 2018, 363, 283–290. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Zhu, X.; Zhang, K.; Yin, Y.; Chen, Y.; Zhang, T. Interleukin-6 contributes to chemoresistance in MDA-MB-231 cells via targeting HIF-1α. J. Biochem. Mol. Toxicol. 2018, 32, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.J.; Yan, L.; Zhang, J.; Zhang, W.D. STAT3 as a potential therapeutic target in triple negative breast cancer: A systematic review. J. Exp. Clin. Cancer Res. 2019, 38, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.-C.; Shi, L.-H.; Wang, X.-J.; Wang, S.-X.; Wan, X.-Q.; Liu, S.-R.; Wang, Y.-F.; Lu, Z.; Wang, L.-H.; Ding, Y. Stat3/Oct-4/c-Myc signal circuit for regulating stemness-mediated doxorubicin resistance of triple-negative breast cancer cells and inhibitory effects of WP1066. Int. J. Oncol. 2018, 53, 339–348. [Google Scholar] [CrossRef]
- Abyaneh, H.S.; Soleimani, A.H.; Vakili, M.R.; Soudy, R.; Kaur, K.; Cuda, F.; Tavassoli, A.; Lavasanifar, A. Modulation of hypoxia-induced chemoresistance to polymeric micellar cisplatin: The effect of ligand modification of micellar carrier versus inhibition of the mediators of drug resistance. Pharmaceutics 2018, 10. [Google Scholar]
- Balko, J.M.; Schwarz, L.J.; Luo, N.; Estrada, M.V.; Giltnane, J.M.; Dávila-González, D.; Wang, K.; Sánchez, V.; Dean, P.T.; Combs, S.E.; et al. Triple-negative breast cancers with amplification of JAK2 at the 9p24 locus demonstrate JAK2-specific dependence. Sci. Transl. Med. 2016, 8, ra53–ra334. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.S.; Peres, L.L.; Fari, A.V.S.; Milani, R.; Silva, R.A.; da Costa Fernandes, C., Jr.; Peppelenbosch, M.P.; Ferreira-Halder, C.V.; Zambuzzi, W.F. Phosphoproteome profiling reveals critical role of JAK-STAT signaling in maintaining chemoresistance in breast cancer. Oncotarget 2017, 8, 114756–114768. [Google Scholar] [CrossRef] [PubMed]
- Park, H.S.; Jang, M.H.; Kim, E.J.; Kim, H.J.; Lee, H.J.; Kim, Y.J.; Kim, J.H.; Kang, E.; Kim, S.-W.; Kim, I.A.; et al. High EGFR gene copy number predicts poor outcome in triple-negative breast cancer. Mod. Pathol. 2014, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zhang, X.; Zhao, S.; Wang, Y.; Di, W.; Zhao, G.; Yang, M.; Zhang, Q. Prognostic value of survivin and EGFR protein expression in triple-negative breast cancer (TNBC) patients. Target. Oncol. 2014, 9, 349–357. [Google Scholar] [CrossRef] [PubMed]
- Porcelli, L.; Giovannetti, E.; Assaraf, Y.G.; Jansen, G.; Scheffer, G.L.; Kathman, I.; Azzariti, A.; Paradiso, A.; Peters, G.J. The EGFR pathway regulates BCRP expression in NSCLC cells: Role of erlotinib. Curr. Drug Targets 2014, 15, 1322–1330. [Google Scholar] [CrossRef] [PubMed]
- Meyer zu Schwabedissen, H.E.M.; Grube, M.; Dreisbach, A.; Jedlitschky, G.; Meissner, K.; Linnemann, K.; Fusch, C.; Ritter, C.A.; Völker, U.; Kroemer, H.K. EPIDERMAL GROWTH FACTOR-MEDIATED ACTIVATION OF THE MAP KINASE CASCADE RESULTS IN ALTERED EXPRESSION AND FUNCTION OF ABCG2 (BCRP). Drug Metab. Dispos. 2006, 34, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.-N.; Zhang, Y.-K.; Wang, Y.-J.; Gupta, P.; Ashby, C.R.; Alqahtani, S.; Deng, T.; Bates, S.E.; Kaddoumi, A.; Wurpel, J.N.D.; et al. Epidermal growth factor receptor (EGFR) inhibitor PD153035 reverses ABCG2-mediated multidrug resistance in non-small cell lung cancer: In vitro and in vivo. Cancer Lett. 2018, 424, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Baselga, J.; Gómez, P.; Greil, R.; Braga, S.; Climent, M.A.; Wardley, A.M.; Kaufman, B.; Stemmer, S.M.; Pêgo, A.; Chan, A.; et al. Randomized Phase II Study of the Anti–Epidermal Growth Factor Receptor Monoclonal Antibody Cetuximab With Cisplatin Versus Cisplatin Alone in Patients With Metastatic Triple-Negative Breast Cancer. J. Clin. Oncol. 2013, 31, 2586–2592. [Google Scholar] [CrossRef] [PubMed]
- Carey, L.A.; Rugo, H.S.; Marcom, P.K.; Mayer, E.L.; Esteva, F.J.; Ma, C.X.; Liu, M.C.; Storniolo, A.M.; Rimawi, M.F.; Forero-Torres, A.; et al. TBCRC 001: Randomized Phase II Study of Cetuximab in Combination With Carboplatin in Stage IV Triple-Negative Breast Cancer. J. Clin. Oncol. 2012, 30, 2615–2623. [Google Scholar] [CrossRef]
- Matsuda, N.; Wang, X.; Lim, B.; Krishnamurthy, S.; Alvarez, R.H.; Willey, J.S.; Parker, C.A.; Song, J.; Shen, Y.; Hu, J.; et al. Safety and Efficacy of Panitumumab Plus Neoadjuvant Chemotherapy in Patients With Primary HER2-Negative Inflammatory Breast Cancer. JAMA Oncol. 2018, 4, 1207. [Google Scholar] [CrossRef]
- Cowherd, S.; Miller, L.D.; Melin, S.A.; Akman, S.; Isom, S.; Cole, J.; Pullikuth, A.; Lawrence, J.A. A phase II clinical trial of weekly paclitaxel and carboplatin in combination with panitumumab in metastatic triple negative breast cancer. Cancer Biol. Ther. 2015, 16, 678–683. [Google Scholar] [CrossRef]
- Nabholtz, J.M.; Abrial, C.; Mouret-Reynier, M.A.; Dauplat, M.M.; Weber, B.; Gligorov, J.; Forest, A.M.; Tredan, O.; Vanlemmens, L.; Petit, T.; et al. Multicentric neoadjuvant phase II study of panitumumab combined with an anthracycline/taxane-based chemotherapy in operable triple-negative breast cancer: Identification of biologically defined signatures predicting treatment impact. Ann. Oncol. 2014, 25, 1570–1577. [Google Scholar] [CrossRef]
- Ferraro, D.A.; Gaborit, N.; Maron, R.; Cohen-Dvashi, H.; Porat, Z.; Pareja, F.; Lavi, S.; Lindzen, M.; Ben-Chetrit, N.; Sela, M.; et al. Inhibition of triple-negative breast cancer models by combinations of antibodies to EGFR. Proc. Natl. Acad. Sci. USA 2013, 110, 1815–1820. [Google Scholar] [CrossRef] [PubMed]
- El Guerrab, A.; Bamdad, M.; Kwiatkowski, F.; Bignon, Y.-J.; Penault-Llorca, F.; Aubel, C. Anti-EGFR monoclonal antibodies and EGFR tyrosine kinase inhibitors as combination therapy for triple-negative breast cancer. Oncotarget 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.H.-F.; Tan, W.-J.; Thike, A.-A.; Cheok, P.-Y.; Tse, G.M.-K.; Wong, N.-S.; Yip, G.W.-C.; Bay, B.-H.; Tan, P.-H. Mutations in the epidermal growth factor receptor (EGFR) gene in triple negative breast cancer: Possible implications for targeted therapy. Breast Cancer Res. 2011, 13, R35. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Jang, M.H.; Lee, S.J.; Bae, Y.K. Mutations of the Epidermal Growth Factor Receptor Gene in Triple-Negative Breast Cancer. J. Breast Cancer 2017, 20, 150–159. [Google Scholar] [CrossRef] [PubMed]
- Ali, R.; Wendt, M.K. The paradoxical functions of EGFR during breast cancer progression. Signal. Transduct. Target. Ther. 2017, 2, 16042. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Yin, Z.; Tao, K.; Wang, G.; Gao, J. Function of insulin-like growth factor 1 receptor in cancer resistance to chemotherapy (Review). Oncol. Lett. 2017, 15, 41–47. [Google Scholar] [CrossRef] [PubMed]
- Farabaugh, S.M.; Boone, D.N.; Lee, A.V. Role of IGF1R in Breast Cancer Subtypes, Stemness, and Lineage Differentiation. Front. Endocrinol. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Heskamp, S.; Boerman, O.C.; Molkenboer-Kuenen, J.D.M.; Wauters, C.A.; Strobbe, L.J.A.; Mandigers, C.M.P.W.; Bult, P.; Oyen, W.J.G.; van der Graaf, W.T.A.; van Laarhoven, H.W.M. Upregulation of IGF-1R Expression during Neoadjuvant Therapy Predicts Poor Outcome in Breast Cancer Patients. PLoS ONE 2015, 10, e0117745. [Google Scholar] [CrossRef]
- Litzenburger, B.C.; Creighton, C.J.; Tsimelzon, A.; Chan, B.T.; Hilsenbeck, S.G.; Wang, T.; Carboni, J.M.; Gottardis, M.M.; Huang, F.; Chang, J.C.; et al. High IGF-IR Activity in Triple-Negative Breast Cancer Cell Lines and Tumorgrafts Correlates with Sensitivity to Anti-IGF-IR Therapy. Clin. Cancer Res. 2011, 17, 2314–2327. [Google Scholar] [CrossRef]
- Yee, D. Insulin-like Growth Factor Receptor Inhibitors: Baby or the Bathwater? JNCI J. Natl. Cancer Inst. 2012, 104, 975–981. [Google Scholar] [CrossRef]
- Ekyalongo, R.C.; Yee, D. Revisiting the IGF-1R as a breast cancer target. NPJ Precis. Oncol. 2017, 1, 14. [Google Scholar] [CrossRef] [PubMed]
- Bai, X.; Han, G.; Liu, Y.; Jiang, H.; He, Q. MiRNA-20a-5p promotes the growth of triple-negative breast cancer cells through targeting RUNX3. Biomed. Pharmacother. 2018, 103, 1482–1489. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Zhang, X.; Wang, X.; He, M.; Qiao, S. MicroRNA-224 Promotes Tumorigenesis through Downregulation of Caspase-9 in Triple-Negative Breast Cancer. Dis. Markers 2019, 2019, 7378967. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Wang, Z.; Liu, Z.; Shi, S.; Zhang, Z.; Zhang, J.; Lin, H. miR-221/222 activate the Wnt/β-catenin signaling to promote triple-negative breast cancer. J. Mol. Cell Biol. 2018, 10, 302–315. [Google Scholar] [CrossRef] [PubMed]
- Lü, L.; Mao, X.; Shi, P.; He, B.; Xu, K.; Zhang, S.; Wang, J. MicroRNAs in the prognosis of triple-negative breast cancer. Medicine 2017, 96, e7085. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Li, D.; Wu, T.; Xie, D.; Hua, K.; Hu, J.; Deng, X.; Ji, C.; Deng, Y.; Fang, L. MicroRNA-301b promotes cell proliferation and apoptosis resistance in triple-negative breast cancer by targeting CYLD. BMB Rep. 2018, 51, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-Y.; Liang, J.-L.; Kuo, Y.-L.; Lee, H.-H.; Calkins, M.J.; Chang, H.-T.; Lin, F.-C.; Chen, Y.-C.; Hsu, T.-I.; Hsiao, M.; et al. miR-105/93-3p promotes chemoresistance and circulating miR-105/93-3p acts as a diagnostic biomarker for triple negative breast cancer. Breast Cancer Res. 2017, 19, 133. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Zhao, A.; Tan, T.; Wang, Y.; Shen, Z. Overexpression of microRNA-620 facilitates the resistance of triple negative breast cancer cells to gemcitabine treatment by targeting DCTD. Exp. Ther. Med. 2019, 18, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Piasecka, D.; Braun, M.; Kordek, R.; Sadej, R.; Romanska, H. MicroRNAs in regulation of triple-negative breast cancer progression. J. Cancer Res. Clin. Oncol. 2018, 144, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Tormo, E.; Ballester, S.; Adam-Artigues, A.; Burgués, O.; Alonso, E.; Bermejo, B.; Menéndez, S.; Zazo, S.; Madoz-Gúrpide, J.; Rovira, A.; et al. The miRNA-449 family mediates doxorubicin resistance in triple-negative breast cancer by regulating cell cycle factors. Sci. Rep. 2019, 9, 5316. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Gong, C.; Xu, R.; Chen, Y.; Wang, X. MicroRNA-5195-3p enhances the chemosensitivity of triple-negative breast cancer to paclitaxel by downregulating EIF4A2. Cell. Mol. Biol. Lett. 2019, 24, 47. [Google Scholar] [CrossRef] [PubMed]
- García-Vazquez, R.; Ruiz-García, E.; Meneses García, A.; Astudillo-de la Vega, H.; Lara-Medina, F.; Alvarado-Miranda, A.; Maldonado-Martínez, H.; González-Barrios, J.A.; Campos-Parra, A.D.; Rodríguez Cuevas, S.; et al. A microRNA signature associated with pathological complete response to novel neoadjuvant therapy regimen in triple-negative breast cancer. Tumor Biol. 2017, 39, 101042831770289. [Google Scholar] [CrossRef] [PubMed]
- Petrovic, N.; Ergun, S. miRNAs as Potential Treatment Targets and Treatment Options in Cancer. Mol. Diagn. Ther. 2018, 22, 157–168. [Google Scholar] [CrossRef] [PubMed]
- Devulapally, R.; Sekar, N.M.; Sekar, T.V.; Foygel, K.; Massoud, T.F.; Willmann, J.K.; Paulmurugan, R. Polymer nanoparticles mediated codelivery of antimiR-10b and antimiR-21 for achieving triple negative breast cancer therapy. ACS Nano 2015, 9, 2290–2302. [Google Scholar] [CrossRef]
- Hashemi, Z.S.; Moghadam, M.F.; Farokhimanesh, S.; Rajabibazl, M.; Sadroddiny, E. Inhibition of breast cancer metastasis by co-transfection of miR-31/193b-mimics. Iran. J. Basic Med. Sci. 2018, 21, 427–433. [Google Scholar] [PubMed]
- Lehmann, B.D.; Jovanović, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef] [PubMed]
- Burstein, M.D.; Tsimelzon, A.; Poage, G.M.; Covington, K.R.; Contreras, A.; Fuqua, S.A.W.; Savage, M.I.; Kent Osborne, C.; Hilsenbeck, S.G.; Chang, J.C.; et al. Comprehensive Genomic Analysis Identifies Novel Subtypes and Targets of Triple-negative Breast Cancer Analysis and interpretation of data: HHS Public Access. Clin Cancer Res. 2015, 21, 1688–1698. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Yu, K.-D.; Jin, X.; Di, G.-H.; Li, D.-Q.; He, X.-H.; Jiang, Y.-Z.; Liu, Y.-R.; Liu, G.-Y.; Hu, W.-G.; et al. Comprehensive transcriptome analysis identifies novel molecular subtypes and subtype-specific RNAs of triple-negative breast cancer. Breast Cancer Res. 2016, 18, 1–10. [Google Scholar]
- Echavarria, I.; López-Tarruella, S.; Picornell, A.; García-Saenz, J.Á.; Jerez, Y.; Hoadley, K.; Gómez, H.L.; Moreno, F.; Del Monte-Millan, M.; Márquez-Rodas, I.; et al. Pathological Response in a Triple-Negative Breast Cancer Cohort Treated with Neoadjuvant Carboplatin and Docetaxel According to Lehmann’s Refined Classification. Clin. Cancer Res. 2018, 24, 1845–1852. [Google Scholar] [CrossRef] [PubMed]
- Loibl, S.; Müller, B.M.; von Minckwitz, G.; Schwabe, M.; Roller, M.; Darb-Esfahani, S.; Ataseven, B.; du Bois, A.; Fissler-Eckhoff, A.; Gerber, B.; et al. Androgen receptor expression in primary breast cancer and its predictive and prognostic value in patients treated with neoadjuvant chemotherapy. Breast Cancer Res. Treat. 2011, 130, 477–487. [Google Scholar] [CrossRef]
- Asghar, U.S.; Barr, A.R.; Cutts, R.; Beaney, M.; Babina, I.; Sampath, D.; Giltnane, J.; Lacap, J.A.; Crocker, L.; Young, A.; et al. Single-Cell Dynamics Determines Response to CDK4/6 Inhibition in Triple-Negative Breast Cancer. Clin. Cancer Res. 2017, 23, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, B.D.; Bauer, J.A.; Schafer, J.M.; Pendleton, C.S.; Tang, L.; Johnson, K.C.; Chen, X.; Balko, J.M.; Gómez, H.; Arteaga, C.L.; et al. PIK3CA mutations in androgen receptor-positive triple negative breast cancer confer sensitivity to the combination of PI3K and androgen receptor inhibitors. Breast Cancer Res. 2014, 16, 406. [Google Scholar] [CrossRef] [PubMed]
- Tutt, A.; Tovey, H.; Cheang, M.C.U.; Kernaghan, S.; Kilburn, L.; Gazinska, P.; Owen, J.; Abraham, J.; Barrett, S.; Barrett-Lee, P.; et al. Carboplatin in BRCA1/2-mutated and triple-negative breast cancer BRCAness subgroups: The TNT Trial. Nat. Med. 2018, 24, 628–637. [Google Scholar] [CrossRef] [PubMed]
- Robson, M.; Im, S.-A.; Senkus, E.; Xu, B.; Domchek, S.M.; Masuda, N.; Delaloge, S.; Li, W.; Tung, N.; Armstrong, A.; et al. Olaparib for Metastatic Breast Cancer in Patients with a Germline BRCA Mutation. N. Engl. J. Med. 2017, 377, 523–533. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-talazoparib-gbrcam-her2-negative-locally-advanced-or-metastatic-breast-cancer (accessed on 22 August 2019).
- Dieci, M.V.; Criscitiello, C.; Goubar, A.; Viale, G.; Conte, P.; Guarneri, V.; Ficarra, G.; Mathieu, M.C.; Delaloge, S.; Curigliano, G.; et al. Prognostic value of tumor-infiltrating lymphocytes on residual disease after primary chemotherapy for triple-negative breast cancer: A retrospective multicenter study. Ann. Oncol. 2014, 25, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Diéras, V.; Hegg, R.; Im, S.-A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef]
- U.S. Food and Drug Administration. Available online: https://www.fda.gov/drugs/drug-approvals-and-databases/fda-approves-atezolizumab-pd-l1-positive-unresectable-locally-advanced-or-metastatic-triple-negative (accessed on 7 August 2019).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TNBC Subtype and Its Characteristics [147,198] | Promising Therapy | Clinical Studies |
|---|---|---|
| Basal-like 1 Cell cycle, cell division, DNA damage response. High pCR rate | Platinum | NCT02441933, Phase III, Recruiting, Carboplatin in combination with standard chemotherapy in early TNBC |
| NCT01881230, phase II/III, superior efficacy of Nab Paclitaxel/Carboplatin combination in metastatic TNBC | ||
| PARP inhibitors | NCT02000622, Phase III, Olaparib as monotherapy significantly increased PFS and reduced the risk of progression and death in metastatic breast cancer including TNBC | |
| NCT01945775, Phase III, Talazoparib as monotherapy significantly increased PFS in advanced breast cancer including TNBC | ||
| Aurora inhibitors | NCT01639248, Phase II, ENMD-2076 in previously treated, advanced or metastatic TNBC | |
| NCT00858377, Phase I, AMG 900 in taxane-resistant TNBC failed to provide clinical benefit | ||
| CHK1 inhibitors | NCT01359696, Phase I, GDC-0425 in combination with gemcitabine showed preliminary anti-tumor activity in TP53-mutated TNBC | |
| NCT00779584, phase I, MK-8776 demonstrated preliminary clinical efficacy with acceptable toxicity in advanced solid tumors | ||
| Basal-like 2 Growth factor signaling, myoepithelial markers, activated glycolysis and gluconeogenesis pathways. Moderate pCR rate Shares susceptibility to platinum and PARP inhibitors with BL1 | EGFR pathway inhibitors | NCT00463788, Phase II, Cetuximab in combination with cisplatin increased PFS and OS in metastatic TNBC |
| NCT01036087, Phase II, Panitumumab in combination with nab-paclitaxel and carboplatin demonstrated highest ever reported pCR rates in inflammatory TNBC | ||
| NCT02362230, Phase II, Recruiting, Icotinib is being evaluated in previously treated metastatic TNBC | ||
| IGF1R pathway inhibitors | NCT01340040, Phase I, MEDI-573 was well tolerated but showed limited activity as monotherapy in advanced solid tumors | |
| NCT00626106, Phase II, Ganitumab in combination with endocrine therapy failed to improve outcome in patients with previously treated HR+ advanced or metastatic breast cancer | ||
| Mesenchymal EMT, stemness, growth factor signaling, angiogenesis, high rate of PAM pathway aberrations. Low pCR rates | TGF-β inhibitors | NCT02672475, Phase I, Recruiting, Galunisertib in combination with paclitaxel in metastatic TNBC |
| Notch inhibitors | NCT01876251, Phase I, PF-03084014 in combination with docetaxel was well tolerated and showed clinical benefit in patients with advanced TNBC | |
| NCT00645333, phase I/II, MK-0752 together with docetaxel had limited anti-tumor activity but reduced breast CSC burden in advanced or metastatic breast cancer | ||
| Wnt/β-catenin inhibitors | NCT01351103, Phase I, Recruiting, LGK974 in patients with Wnt-ligand dependent malignancies including TNBC | |
| Hedgehog inhibitors | NCT02027376, Phase I, Sonidegib in combination with docetaxel was well tolerated and showed preliminary anti-tumor activity in advanced, pre-treated TNBC | |
| NCT02694224, Phase II, Recruiting, Vismodegib in combination with standard neoadjuvant chemotherapy in TNBC | ||
| PI3K inhibitors | NCT01629615, Phase II, BKM120 as monotherapy in metastatic TNBC | |
| NCT01884285, Phase I, AZD8186 as monotherapy or in combination with other agents in advanced solid tumors including TNBC, demonstrated preliminary anti-tumor activity with serious adverse events | ||
| mTOR inhibitors | NCT02531932, Phase II, Recruiting, Everolimus together with carboplatin in advanced TNBC | |
| NCT01920061, Phase I, Recruiting, Gedatolisib in combination with either cisplatin or docetaxel in TNBC | ||
| AKT inhibitors | NCT02162719, Phase II, Ipatasertib in combination with paclitaxel, increased PFS and OS of patients with metastatic TNBC compared to paclitaxel alone | |
| NCT02423603, Phase II, AZD5363 in combination with paclitaxel, prolonged the PFS and OS of patients with metastatic TNBC compared to paclitaxel alone | ||
| Anti-angiogenic therapy | NCT01176669, Phase II, Apatinib as a single agent demonstrated clinical benefit in pre-treated metastatic TNBC | |
| NCT03348098, Phase II, Recruiting, Apatinib in combination with paclitaxel as neoadjuvant treatment for locally advanced TNBC | ||
| NCT01234337, Phase III, Sorafenib in combination with capecitabine failed to show clinical benefit compared to capecitabine alone in advanced or metastatic HER2-negative breast cancer including TNBC | ||
| Luminal androgen receptor Hormonal-mediated signaling, androgen receptor, PI3KCA mutations. Very low pCR rate | Androgen receptor | NCT03055312, Phase III, Recruiting, Bicalutamide as single agent compared to the efficacy of standard chemotherapy in metastatic, AR+ TNBC |
| NCT01889238, Phase II, Enzalutamide as monotherapy was well tolerated and showed clinical benefit in patients with advanced AR+ pre-treated TNBC | ||
| PI3K inhibitors | NCT02457910, Phase I/II, Enzalutamide in combination with Taselisib (PI3K inhibitor) in patients with AR+, metastatic TNBC | |
| Heat shock protein 90 | NCT01677455, Phase II, Ganetespib as a single agent in TNBC patients who were not subjected to prior systemic treatment in the metastatic setting | |
| NCT02474173, Phase I, Recruiting, Onalespib in combination with paclitaxel in patients with metastatic TNBC | ||
| Immunomodulatory Immune-mediated signaling | Checkpoint inhibitors | NCT02425891, Phase III, Atezolizumab in combination with nab–paclitaxel prolonged both PFS and OS of patients with metastatic TNBC, especially in patients with PD-L1 expressing tumors |
| NCT02926196, Phase III, Recruiting, Avelumab as adjuvant treatment for high-risk TNBC | ||
| JAK/STAT pathway | NCT02876302, Phase II, Recruiting, Ruxolitinib, in combination with standard neoadjuvant chemotherapy in triple-negative inflammatory breast cancer | |
| NCT03195699, Phase I, Recruiting, TTI-101 as monotherapy in advanced cancers including breast cancer |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nedeljković, M.; Damjanović, A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells 2019, 8, 957. https://doi.org/10.3390/cells8090957
Nedeljković M, Damjanović A. Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells. 2019; 8(9):957. https://doi.org/10.3390/cells8090957
Chicago/Turabian StyleNedeljković, Milica, and Ana Damjanović. 2019. "Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge" Cells 8, no. 9: 957. https://doi.org/10.3390/cells8090957
APA StyleNedeljković, M., & Damjanović, A. (2019). Mechanisms of Chemotherapy Resistance in Triple-Negative Breast Cancer—How We Can Rise to the Challenge. Cells, 8(9), 957. https://doi.org/10.3390/cells8090957