Nutritional Stress in Head and Neck Cancer Originating Cell Lines: The Sensitivity of the NRF2-NQO1 Axis

 ,
, 
 , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Cell Culture Conditions
2.2. Cell Viability Assay
2.3. Cell Proliferation Assay
2.4. ROS Measurement
2.5. Nucleic Acids Extraction
2.6. Construction of Primers
2.7. Reverse Transcription, RT – PCR, and PCR
2.8. Densitometry, Purification of PCR Products from Agarose Gel and Sequencing
2.9. Real-Time RT-PCR and Rationale for Specific TaqMan’s Probe Selection
2.10. Protein Extraction and Western Blot Analyses
2.11. Statistical Analyses
3. Results
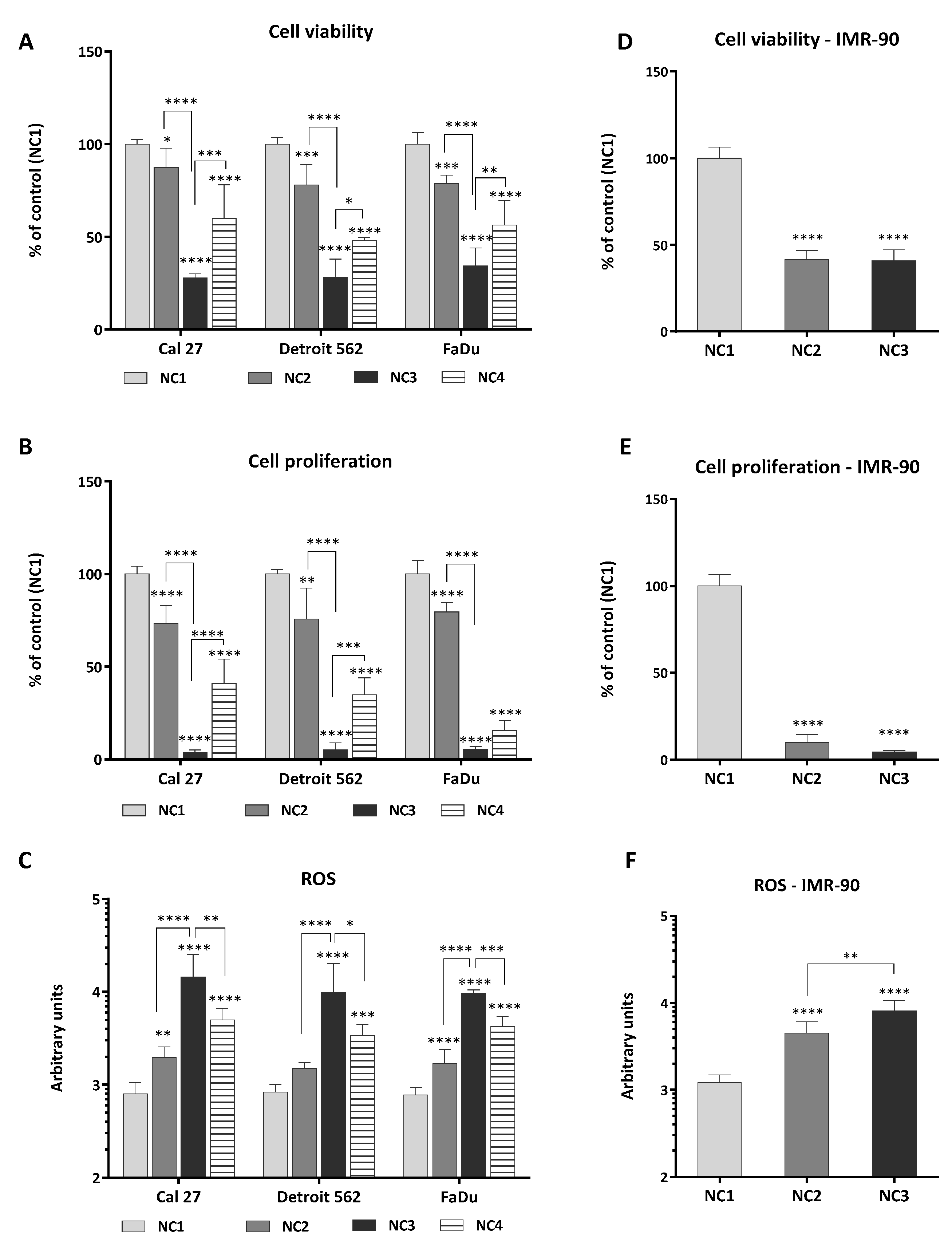
3.1. Viability, Proliferation and Generation of ROS
3.2. Quantification of NQO1 and NRF2 in a Real-Time
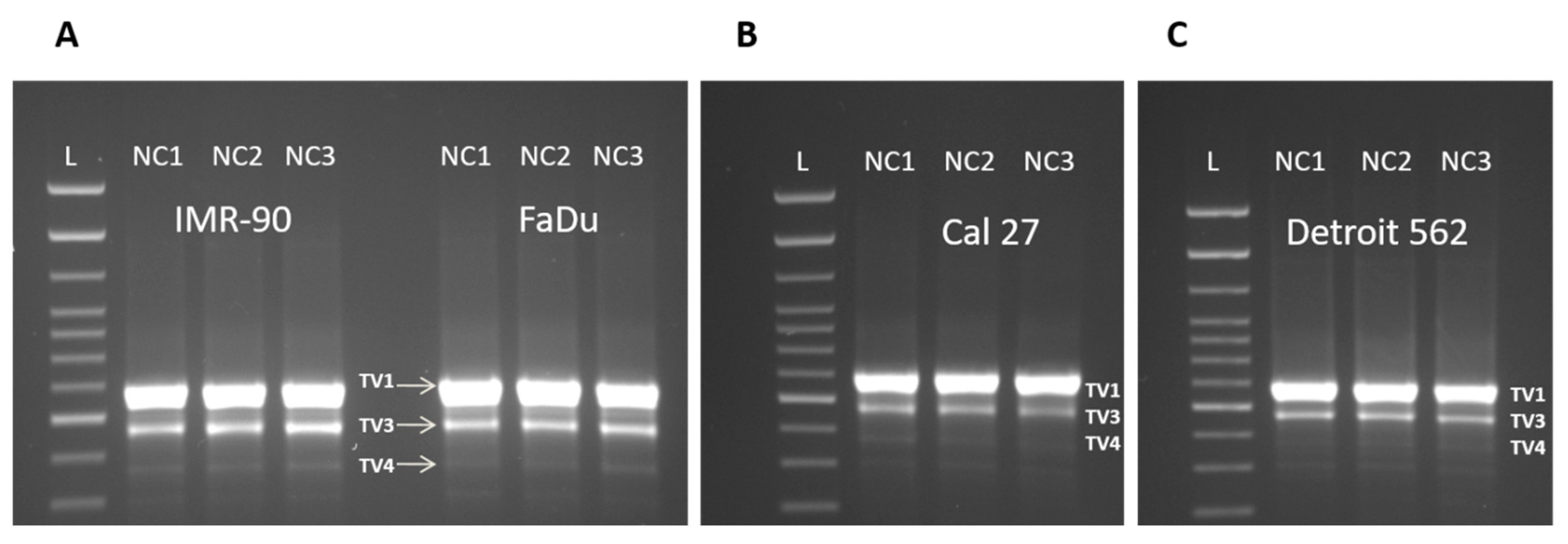
3.3. Three Splice Variants of NQO1 are Present in All Four Cell Lines, Notwithstanding the Experimental Conditions Applied
3.4. Detection of SNVs in Intron/Exon Boundaries
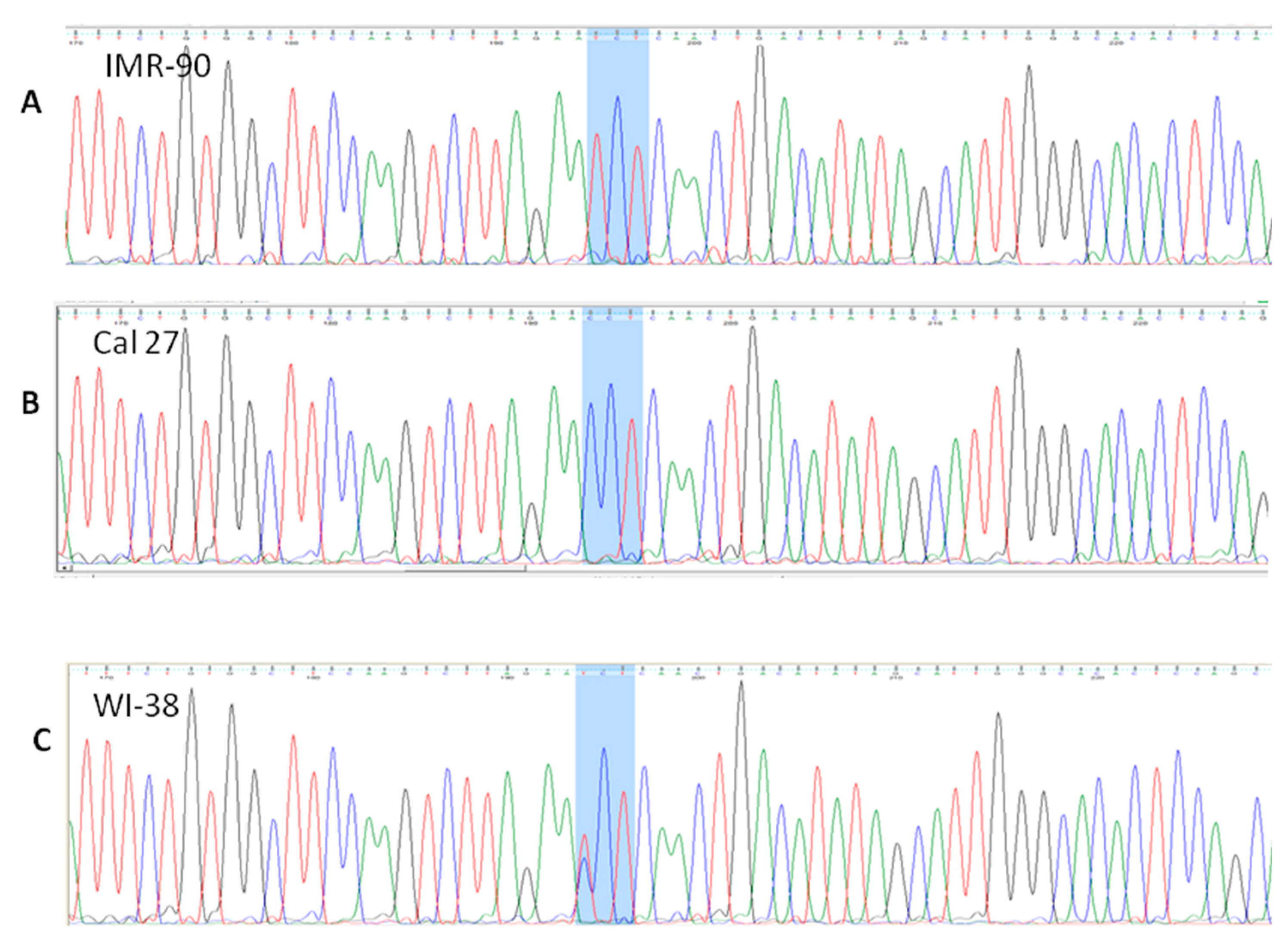
3.4.1. rs 689460, G+C, Is Present in IMR-90 and Does Not Influence the NQO1 Splicing
3.4.2. rs 689452, C+G, Is Present in Detroit 562 and Cal 27
3.4.3. Presence of rs1800566 in IMR-90 but Not in Cancer Cell Lines
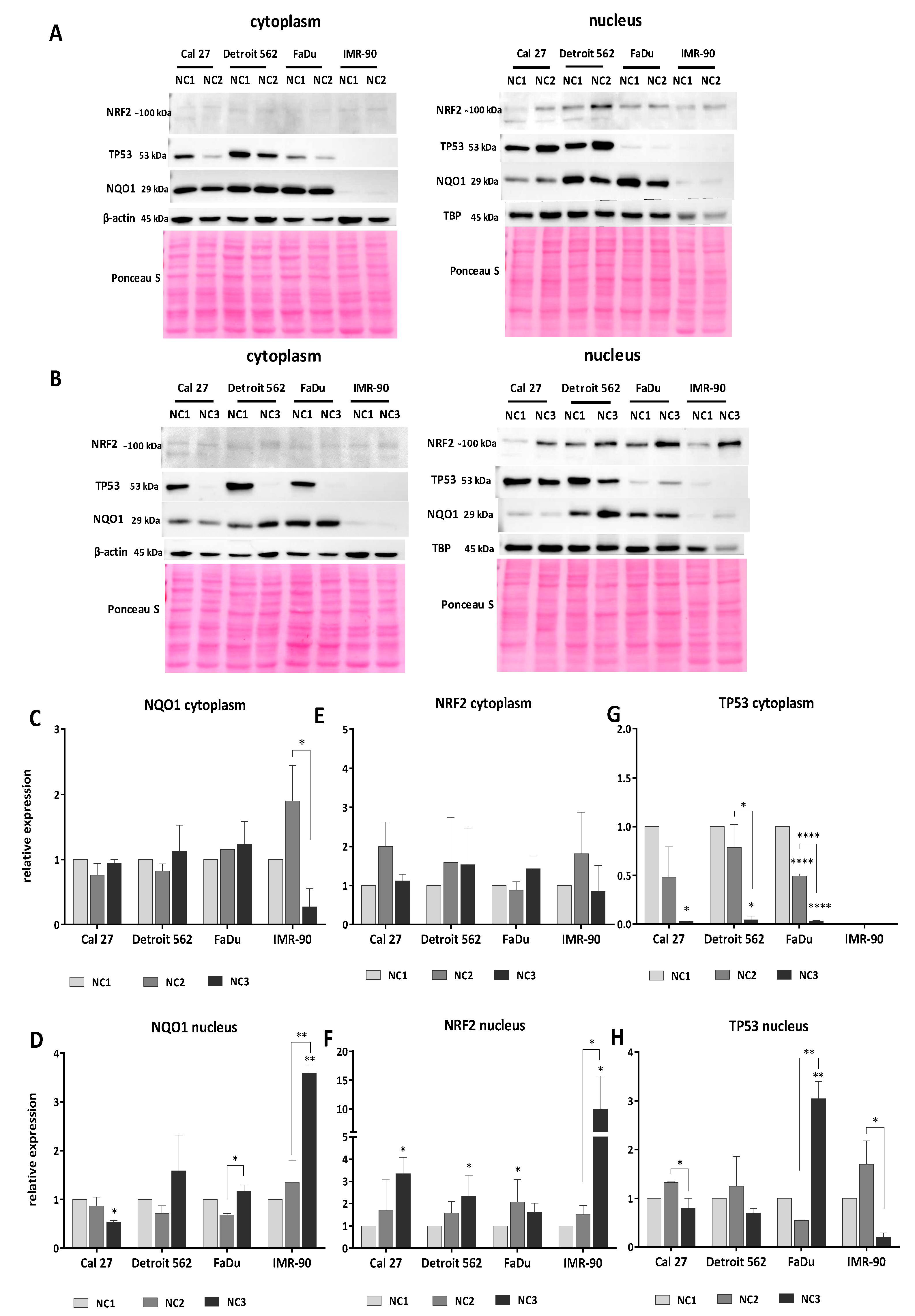
3.5. Western Blot Analyses
3.5.1. Analyses of NQO1
3.5.2. Analyses of NRF2
3.5.3. Analyses of TP53
3.5.4. The General Picture of the NRF2-NQO1 Axis and TP53 at the Protein Level, in the Experimental Model
4. Discussion
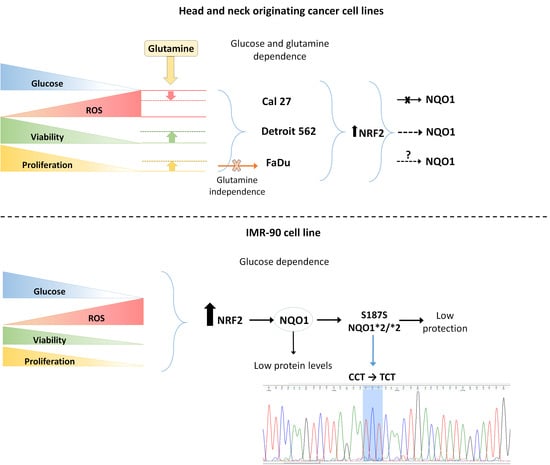
4.1. Cancer Cells and Fibroblasts IMR-90 Differ in Their Sensitivity to Nutrient Deprivation
4.2. Importance of Defining the SNVs, in a Given Experimental Model
4.3. FaDu Proliferative Potential with Respect to GLS1 and Decreased Sensitivity to Glutamine When Deprived of Glucose
4.4. NQO1 and Its Splice Variants—TaqMan Probes Validation
4.5. Activation of NRF2 and Sensitivity to Glutamine
4.6. TP53 and Its Potential to Influence Phenomena Observed
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AMPK | 5′AMP-Activated Protein Kinase |
| ARE | Antioxidant Response Element |
| ATCC | American Type Culture Collection |
| ATP | Adenosine Triphosphate |
| BrdU | 5-Bromo-2′-Deoxyuridine |
| COSMIC | Catalogue of Somatic Mutations In Cancer |
| DCFH-DA | 2ʹ,7ʹ-Dichlorofluorescin Diacetate |
| DMEM | Dulbecco’s Modified Eagle’s Medium |
| EDTA | Ethylenediaminetetraacetic Acid |
| ER | Endoplasmic Reticulum |
| EtdBr | Ethidium Bromide |
| FAD | Flavin Adenine Dinucleotide |
| FBS | Fetal Bovine Serum |
| G6PD | Glucose-6-Phosphate Dehydrogenase |
| GAPDH | Glyceraldehyde-3-Phosphate Dehydrogenase |
| gDNA | Genomic DNA |
| GLS1 | Glutaminase 1 |
| HCT116 | Human Colorectal Carcinoma |
| HIF-1α | Hypoxia-Inducible Factor, Alpha Subunit |
| HMOX-1 | Heme Oxygenase 1 |
| HNSCC | Head and Neck Squamous Cell Carcinoma |
| HPRT1 | Hypoxanthine Phosphoribosyltransferase 1 |
| HSP90 | Heat Shock Protein 90 |
| IR-C | Intron Retention Complex |
| IR-S | Intron Retention Simple |
| KEAP-1 | Kelch-Like ECH-Associated Protein 1 |
| LUAD | Lung Adenocarcinoma |
| Mdm-2 | Mouse Double Minute 2 Homolog |
| MT | Mutated |
| NADH/NAD+ | Nicotinamide Adenine Dinucleotide |
| NADPH/NADP+ | Nicotinamide Adenine Dinucleotide Phosphate |
| NC | Nutritional Condition |
| NC1 | Nutritional Condition 1 |
| NC2 | Nutritional Condition 2 |
| NC3 | Nutritional Condition 3 |
| NC4 | Nutritional Condition 4 |
| NFE2L2 (NRF2) | Nuclear Factor (Erythroid-Derived 2)-Like 2 |
| NHLF | Normal Human Lung Fibroblasts |
| NQO1 | NAD(P)H:Quinone Oxidoreductase 1 |
| PARP-1 | Poly (ADP-Ribose) Polymerase 1 |
| PCR | Polymerase Chain Reaction |
| PPP | Pentose Phosphate Pathway |
| ROS | Reactive Oxygene Species |
| RT-qPCR | Reverse Transcription-QuantitativePolymerase Chain Reaction |
| SIRT1 | Sirtuin 1 |
| SLC7A11 | Solute-Like Carrier Family 7, Member 11 |
| SLC7A3 | Solute-Like Carrier Family 7, Member 3 |
| SNP | Single-Nucleotide Polymorphism |
| SNV | Single Nucleotide Variant |
| TBP | TATA-Box Binding Protein |
| TBS | Tris-Buffered Saline |
| TBST | Tris Buffered Saline with Tween-20 |
| TE buffer | Tris-EDTA Buffer |
| TP53 | Tumor Protein P53 |
| TV | Transcript Variant |
| WOX1 | WUSCHEL Related Homeobox 1 |
| WT | Wild-Type |
References
- Bott, A.J.; Maimouni, S.; Zong, W.-X. The pleiotropic effects of glutamine metabolism in cancer. Cancers 2019, 11, 770. [Google Scholar] [CrossRef] [PubMed]
- Jeon, S.-M.; Chandel, N.S.; Hay, N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature 2012, 485, 661–665. [Google Scholar] [CrossRef]
- Wu, C.-A.; Chao, Y.; Shiah, S.-G.; Lin, W.-W. Nutrient deprivation induces the Warburg effect through ROS/AMPK-dependent activation of pyruvate dehydrogenase kinase. Biochim. Biophys. Acta 2013, 1833, 1147–1156. [Google Scholar] [CrossRef] [PubMed]
- Son, J.; Lyssiotis, C.A.; Ying, H.; Wang, X.; Hua, S.; Ligorio, M.; Perera, R.M.; Ferrone, C.R.; Mullarky, E.; Shyh-Chang, N.; et al. Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 2013, 496, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Lie, S.; Wang, T.; Forbes, B.; Proud, C.G.; Petersen, J. The ability to utilise ammonia as nitrogen source is cell type specific and intricately linked to GDH, AMPK and mTORC1. Sci. Rep. 2019, 9, 1461. [Google Scholar] [CrossRef] [PubMed]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Jiang, P.; Du, W.; Wang, X.; Mancuso, A.; Gao, X.; Wu, M.; Yang, X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat. Cell Biol. 2011, 13, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Oh, E.-T.; Choi, B.-H.; Park, M.-T.; Lee, J.-K.; Lee, J.-S.; Park, H.J. NQO1-induced activation of AMPK contributes to cancer cell death by oxygen-glucose deprivation. Sci. Rep. 2015, 5, 7769. [Google Scholar] [CrossRef]
- Jaiswal, A.K.; McBride, O.W.; Adesnik, M.; Nebert, D.W. Human dioxin-inducible cytosolic NAD(P)H:menadione oxidoreductase. cDNA sequence and localization of gene to chromosome 16. J. Biol. Chem. 1988, 263, 13572–13578. [Google Scholar] [PubMed]
- Lind, C.; Cadenas, E.; Hochstein, P.; Ernster, L. DT-diaphorase: Purification, properties, and function. Methods Enzymol. 1990, 186, 287–301. [Google Scholar] [CrossRef] [PubMed]
- Hosoda, S.; Nakamura, W.; Hayashi, K. Properties and reaction mechanism of DT diaphorase from rat liver. J. Biol. Chem. 1974, 249, 6416–6423. [Google Scholar] [PubMed]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef] [PubMed]
- Winski, S.L.; Koutalos, Y.; Bentley, D.L.; Ross, D. Subcellular localization of NAD(P)H:quinone oxidoreductase 1 in human cancer cells. Cancer Res. 2002, 62, 1420–1424. [Google Scholar] [PubMed]
- Criddle, D.N.; Gillies, S.; Baumgartner-Wilson, H.K.; Jaffar, M.; Chinje, E.C.; Passmore, S.; Chvanov, M.; Barrow, S.; Gerasimenko, O.V.; Tepikin, A.V.; et al. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. J. Biol. Chem. 2006, 281, 40485–40492. [Google Scholar] [CrossRef] [PubMed]
- Siegel, D.; Dehn, D.D.; Bokatzian, S.S.; Quinn, K.; Backos, D.S.; Di Francesco, A.; Bernier, M.; Reisdorph, N.; de Cabo, R.; Ross, D. Redox modulation of NQO1. PLoS ONE 2018, 13, e0190717. [Google Scholar] [CrossRef] [PubMed]
- Ross, D.; Siegel, D. NQO1 in protection against oxidative stress. Curr. Opin. Toxicol. 2018, 7, 67–72. [Google Scholar] [CrossRef]
- Ross, D.; Siegel, D. Functions of NQO1 in cellular protection and CoQ10 metabolism and its potential role as a redox sensitive molecular switch. Front. Physiol. 2017, 8, 595. [Google Scholar] [CrossRef]
- Asher, G.; Lotem, J.; Cohen, B.; Sachs, L.; Shaul, Y. Regulation of p53 stability and p53-dependent apoptosis by NADH quinone oxidoreductase 1. Proc. Natl. Acad. Sci. USA 2001, 98, 1188–1193. [Google Scholar] [CrossRef]
- Oh, E.-T.; Kim, J.-W.; Kim, J.M.; Kim, S.J.; Lee, J.-S.; Hong, S.-S.; Goodwin, J.; Ruthenborg, R.J.; Jung, M.G.; Lee, H.-J.; et al. NQO1 inhibits proteasome-mediated degradation of HIF-1α. Nat. Commun. 2016, 7, 13593. [Google Scholar] [CrossRef]
- Asher, G.; Lotem, J.; Tsvetkov, P.; Reiss, V.; Sachs, L.; Shaul, Y. P53 hot-spot mutants are resistant to ubiquitin-independent degradation by increased binding to NAD(P)H:quinone oxidoreductase 1. Proc. Natl. Acad. Sci. USA 2003, 100, 15065–15070. [Google Scholar] [CrossRef]
- Han, Y.; Shen, H.; Carr, B.I.; Wipf, P.; Lazo, J.S.; Pan, S. NAD(P)H:quinone oxidoreductase-1-dependent and -independent cytotoxicity of potent quinone Cdc25 phosphatase inhibitors. J. Pharmacol. Exp. Ther. 2004, 309, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Asher, G.; Lotem, J.; Kama, R.; Sachs, L.; Shaul, Y. NQO1 stabilizes p53 through a distinct pathway. Proc. Natl. Acad. Sci. USA 2002, 99, 3099–3104. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.A.; Drees, J.; Loggie, B.W.; Russell, G.B.; Geisinger, K.R.; Morris, R.T.; Sachs, D.; McQuellon, R.P. Clinical significance of a NAD(P)H: Quinone oxidoreductase 1 polymorphism in patients with disseminated peritoneal cancer receiving intraperitoneal hyperthermic chemotherapy with mitomycin C. Pharmacogenetics 2002, 12, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Oh, E.-T.; Park, H.J. Implications of NQO1 in cancer therapy. BMB Rep. 2015, 48, 609–617. [Google Scholar] [CrossRef] [PubMed]
- Itahana, Y.; Itahana, K. Emerging roles of p53 family members in glucose metabolism. Int. J. Mol. Sci. 2018, 19, 776. [Google Scholar] [CrossRef] [PubMed]
- Reid, M.A.; Wang, W.-I.; Rosales, K.R.; Welliver, M.X.; Pan, M.; Kong, M. The B55α subunit of PP2A drives a p53-dependent metabolic adaptation to glutamine deprivation. Mol. Cell 2013, 50, 200–211. [Google Scholar] [CrossRef] [PubMed]
- Lowman, X.H.; Hanse, E.A.; Yang, Y.; Ishak Gabra, M.B.; Tran, T.Q.; Li, H.; Kong, M. p53 promotes cancer cell adaptation to glutamine deprivation by upregulating Slc7a3 to increase arginine uptake. Cell Rep. 2019, 26, 3051–3060. [Google Scholar] [CrossRef] [PubMed]
- Krajka-Kuźniak, V.; Paluszczak, J.; Baer-Dubowska, W. The Nrf2-ARE signaling pathway: An update on its regulation and possible role in cancer prevention and treatment. Pharmacol. Rep. 2017, 69, 393–402. [Google Scholar] [CrossRef] [PubMed]
- Dhakshinamoorthy, S.; Porter, A.G. Nitric oxide-induced transcriptional up-regulation of protective genes by Nrf2 via the antioxidant response element counteracts apoptosis of neuroblastoma cells. J. Biol. Chem. 2004, 279, 20096–20107. [Google Scholar] [CrossRef]
- Gasdaska, P.Y.; Fisher, H.; Powis, G. An alternatively spliced form of NQO1 (DT-diaphorase) messenger RNA lacking the putative quinone substrate binding site is present in human normal and tumor tissues. Cancer Res. 1995, 55, 2542–2547. [Google Scholar]
- Pan, S.-S.; Han, Y.; Farabaugh, P.; Xia, H. Implication of alternative splicing for expression of a variant NAD(P)H:quinone oxidoreductase-1 with a single nucleotide polymorphism at 465C>T. Pharmacogenetics 2002, 12, 479–488. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.S.; Godwin, A.K.; Johnson, C.; O’Dwyer, P.J. Alternative splicing and differential expression of DT-diaphorase transcripts in human colon tumors and in peripheral mononuclear cells in response to mitomycin C treatment. Cancer Res. 1996, 56, 1731–1736. [Google Scholar] [PubMed]
- Parenteau, J.; Maignon, L.; Berthoumieux, M.; Catala, M.; Gagnon, V.; Abou Elela, S. Introns are mediators of cell response to starvation. Nature 2019, 565, 612–617. [Google Scholar] [CrossRef] [PubMed]
- Pleiss, J.A.; Whitworth, G.B.; Bergkessel, M.; Guthrie, C. Rapid, transcript-specific changes in splicing in response to environmental stress. Mol. Cell 2007, 27, 928–937. [Google Scholar] [CrossRef] [PubMed]
- Tsalikis, J.; Pan, Q.; Tattoli, I.; Maisonneuve, C.; Blencowe, B.J.; Philpott, D.J.; Girardin, S.E. The transcriptional and splicing landscape of intestinal organoids undergoing nutrient starvation or endoplasmic reticulum stress. BMC Genom. 2016, 17, 680. [Google Scholar] [CrossRef] [PubMed]
- Li, L.-S.; Reddy, S.; Lin, Z.-H.; Liu, S.; Park, H.; Chun, S.G.; Bornmann, W.G.; Thibodeaux, J.; Yan, J.; Chakrabarti, G.; et al. NQO1-mediated tumor-selective lethality and radiosensitization for head and neck cancer. Mol. Cancer Ther. 2016, 15, 1757–1767. [Google Scholar] [CrossRef] [PubMed]
- Punganuru, S.R.; Madala, H.R.; Arutla, V.; Zhang, R.; Srivenugopal, K.S. Characterization of a highly specific NQO1-activated near-infrared fluorescent probe and its application for in vivo tumor imaging. Sci. Rep. 2019, 9, 8577. [Google Scholar] [CrossRef] [PubMed]
- Green, M.R.; Sambrook, J. Isolation of high-molecular-weight DNA using organic solvents. Cold Spring Harb. Protoc. 2017, 2017, pdb.prot093450. [Google Scholar] [CrossRef] [PubMed]
- Rasband, W.S. ImageJ. 1997–2018; U. S. National Institutes of Health: Bethesda, MD, USA. Available online: https://imagej.nih.gov/ij/ (accessed on 11 July 2019).
- Krzystek-Korpacka, M.; Hotowy, K.; Czapinska, E.; Podkowik, M.; Bania, J.; Gamian, A.; Bednarz-Misa, I. Serum availability affects expression of common house-keeping genes in colon adenocarcinoma cell lines: Implications for quantitative real-time PCR studies. Cytotechnology 2016, 68, 2503–2517. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Lienhart, W.-D.; Gudipati, V.; Uhl, M.K.; Binter, A.; Pulido, S.A.; Saf, R.; Zangger, K.; Gruber, K.; Macheroux, P. Collapse of the native structure caused by a single amino acid exchange in human NAD(P)H:quinone oxidoreductase(1.). FEBS J. 2014, 281, 4691–4704. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, X.; Chang, S.; Lu, W.; Liu, M.; Pang, X. β-Lapachone induces NAD(P)H:quinone oxidoreductase-1- and oxidative stress-dependent heat shock protein 90 cleavage and inhibits tumor growth and angiogenesis. J. Pharmacol. Exp. Ther. 2016, 357, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Novak Kujundzić, R.; Grbesa, I.; Ivkić, M.; Katdare, M.; Gall-Troselj, K. Curcumin downregulates H19 gene transcription in tumor cells. J. Cell. Biochem. 2008, 104, 1781–1792. [Google Scholar] [CrossRef] [PubMed]
- Kanska, J.; Aspuria, P.-J.P.; Taylor-Harding, B.; Spurka, L.; Funari, V.; Orsulic, S.; Karlan, B.Y.; Wiedemeyer, W.R. Glucose deprivation elicits phenotypic plasticity via ZEB1-mediated expression of NNMT. Oncotarget 2017, 8, 26200–26220. [Google Scholar] [CrossRef]
- Bannai, S.; Kitamura, E. Adaptive enhancement of cystine and glutamate uptake in human diploid fibroblasts in culture. Biochim. Biophys. Acta 1982, 721, 1–10. [Google Scholar] [CrossRef]
- Kim, M.P.; Lozano, G. Mutant p53 partners in crime. Cell Death Differ. 2018, 25, 161–168. [Google Scholar] [CrossRef]
- Yuneva, M.; Zamboni, N.; Oefner, P.; Sachidanandam, R.; Lazebnik, Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J. Cell Biol. 2007, 178, 93–105. [Google Scholar] [CrossRef]
- Terashima, J.; Habano, W.; Gamou, T.; Ozawa, S. Induction of CYP1 family members under low-glucose conditions requires AhR expression and occurs through the nuclear translocation of AhR. Drug Metab. Pharmacokinet. 2011, 26, 577–583. [Google Scholar] [CrossRef]
- Liu, K.; Jin, B.; Wu, C.; Yang, J.; Zhan, X.; Wang, L.; Shen, X.; Chen, J.; Chen, H.; Mao, Z. NQO1 stabilizes p53 in response to oncogene-induced senescence. Int. J. Biol. Sci. 2015, 11, 762–771. [Google Scholar] [CrossRef]
- van den Heuvel, A.P.J.; Jing, J.; Wooster, R.F.; Bachman, K.E. Analysis of glutamine dependency in non-small cell lung cancer: GLS1 splice variant GAC is essential for cancer cell growth. Cancer Biol. Ther. 2012, 13, 1185–1194. [Google Scholar] [CrossRef] [PubMed]
- Okuno, S.; Sato, H.; Kuriyama-Matsumura, K.; Tamba, M.; Wang, H.; Sohda, S.; Hamada, H.; Yoshikawa, H.; Kondo, T.; Bannai, S. Role of cystine transport in intracellular glutathione level and cisplatin resistance in human ovarian cancer cell lines. Br. J. Cancer 2003, 88, 951–956. [Google Scholar] [CrossRef] [PubMed]
- Koppula, P.; Zhang, Y.; Shi, J.; Li, W.; Gan, B. The glutamate/cystine antiporter SLC7A11/xCT enhances cancer cell dependency on glucose by exporting glutamate. J. Biol. Chem. 2017, 292, 14240–14249. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Sun, X.; Song, B.; Qiu, X.; Zhao, J. MiR-375/SLC7A11 axis regulates oral squamous cell carcinoma proliferation and invasion. Cancer Med. 2017, 6, 1686–1697. [Google Scholar] [CrossRef] [PubMed]
- Probst, B.L.; McCauley, L.; Trevino, I.; Wigley, W.C.; Ferguson, D.A. Cancer cell growth is differentially affected by constitutive activation of NRF2 by KEAP1 deletion and pharmacological activation of NRF2 by the synthetic triterpenoid, RTA 405. PLoS ONE 2015, 10, e0135257. [Google Scholar] [CrossRef] [PubMed]
- Tsvetkov, P.; Adamovich, Y.; Elliott, E.; Shaul, Y. E3 ligase STUB1/CHIP regulates NAD(P)H:quinone oxidoreductase 1 (NQO1) accumulation in aged brain, a process impaired in certain Alzheimer disease patients. J. Biol. Chem. 2011, 286, 8839–8845. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Guo, Y.; Seo, W.; Zhang, R.; Lu, C.; Wang, Y.; Luo, L.; Paul, B.; Yan, W.; Saxena, D.; et al. Targeting cellular metabolism to reduce head and neck cancer growth. Sci. Rep. 2019, 9, 4995. [Google Scholar] [CrossRef]
- Lee, J.-S.; Kang, J.H.; Lee, S.-H.; Hong, D.; Son, J.; Hong, K.M.; Song, J.; Kim, S.-Y. Dual targeting of glutaminase 1 and thymidylate synthase elicits death synergistically in NSCLC. Cell Death Dis. 2016, 7, e2511. [Google Scholar] [CrossRef]
- Sandulache, V.C.; Ow, T.J.; Pickering, C.R.; Frederick, M.J.; Zhou, G.; Fokt, I.; Davis-Malesevich, M.; Priebe, W.; Myers, J.N. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer 2011, 117, 2926–2938. [Google Scholar] [CrossRef]
- Gall Trošelj, K.; Novak Kujundzic, R.; Ugarkovic, D. Polycomb repressive complex’s evolutionary conserved function: The role of EZH2 status and cellular background. Clin. Epigenetics 2016, 8, 55. [Google Scholar] [CrossRef]
- Romero, R.; Sayin, V.I.; Davidson, S.M.; Bauer, M.R.; Singh, S.X.; LeBoeuf, S.E.; Karakousi, T.R.; Ellis, D.C.; Bhutkar, A.; Sánchez-Rivera, F.J.; et al. Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat. Med. 2017, 23, 1362–1368. [Google Scholar] [CrossRef] [PubMed]
- Sayin, V.I.; LeBoeuf, S.E.; Singh, S.X.; Davidson, S.M.; Biancur, D.; Guzelhan, B.S.; Alvarez, S.W.; Wu, W.L.; Karakousi, T.R.; Zavitsanou, A.M.; et al. Activation of the NRF2 antioxidant program generates an imbalance in central carbon metabolism in cancer. eLife 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Lisek, K.; Campaner, E.; Ciani, Y.; Walerych, D.; Del Sal, G. Mutant p53 tunes the NRF2-dependent antioxidant response to support survival of cancer cells. Oncotarget 2018, 9, 20508–20523. [Google Scholar] [CrossRef] [PubMed]
- Kalo, E.; Kogan-Sakin, I.; Solomon, H.; Bar-Nathan, E.; Shay, M.; Shetzer, Y.; Dekel, E.; Goldfinger, N.; Buganim, Y.; Stambolsky, P.; et al. Mutant p53R273H attenuates the expression of phase 2 detoxifying enzymes and promotes the survival of cells with high levels of reactive oxygen species. J. Cell Sci. 2012, 125, 5578–5586. [Google Scholar] [CrossRef] [PubMed]
- Haupt, Y.; Maya, R.; Kazaz, A.; Oren, M. Mdm2 promotes the rapid degradation of p53. Nature 1997, 387, 296–299. [Google Scholar] [CrossRef] [PubMed]
- Barak, Y.; Juven, T.; Haffner, R.; Oren, M. Mdm2 expression is induced by wild type p53 activity. EMBO J. 1993, 12, 461–468. [Google Scholar] [CrossRef]
- Qie, S.; Liang, D.; Yin, C.; Gu, W.; Meng, M.; Wang, C.; Sang, N. Glutamine depletion and glucose depletion trigger growth inhibition via distinctive gene expression reprogramming. Cell Cycle 2012, 11, 3679–3690. [Google Scholar] [CrossRef]
- Kanai, M.; Hanashiro, K.; Kim, S.-H.; Hanai, S.; Boulares, A.H.; Miwa, M.; Fukasawa, K. Inhibition of Crm1-p53 interaction and nuclear export of p53 by poly(ADP-ribosyl)ation. Nat. Cell Biol. 2007, 9, 1175–1183. [Google Scholar] [CrossRef]
- Chiodi, I.; Picco, G.; Martino, C.; Mondello, C. Cellular response to glutamine and/or glucose deprivation in in vitro transformed human fibroblasts. Oncol. Rep. 2019, 41, 3555–3564. [Google Scholar] [CrossRef]
- Scovassi, A.I.; Poirier, G.G. Poly(ADP-ribosylation) and apoptosis. Mol. Cell. Biochem. 1999, 199, 125–137. [Google Scholar] [CrossRef]
- Whitacre, C.M.; Hashimoto, H.; Tsai, M.L.; Chatterjee, S.; Berger, S.J.; Berger, N.A. Involvement of NAD-poly(ADP-ribose) metabolism in p53 regulation and its consequences. Cancer Res. 1995, 55, 3697–3701. [Google Scholar] [PubMed]
- Chang, N.S.; Pratt, N.; Heath, J.; Schultz, L.; Sleve, D.; Carey, G.B.; Zevotek, N. Hyaluronidase induction of a WW domain-containing oxidoreductase that enhances tumor necrosis factor cytotoxicity. J. Biol. Chem. 2001, 276, 3361–3370. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primers | Primer Sequences | Ref. Seq. | Primer Position | Amplicon Size |
|---|---|---|---|---|
| GAPDH primers | ||||
| GAPDH1 | 5′AACGGATTTGGTCGTATTGGGC3′ | NM_002046.7 | 101–122 | 600 bps |
| GAPDH2 | 5′AGGGATGATGTTCTGGAGAGCC3′ | 679–700 | ||
| GAPDH2 | 5′AGGGATGATGTTCTGGAGAGCC3′ | NG_007073.2 | 3145–3166 | 644 bps |
| GAPDH3 | 5′AAGCTGACTCAGCCCGCAAAGG3′ | 2523–2545 | ||
| NQO1 primers | ||||
| cDNA | ||||
| NQO1 F | 5′GTCGGACCTCTATGCCATGA3′ | NM_000903.3 | 238–257 | TV1—685 bps |
| TV2—583 bps | ||||
| TV3—571 bps | ||||
| NQO1 R | 5′GTCAGTTGGGATGGACTTGC3′ | 905–922 | TV4—469 bps | |
| NQO1389460 F | 5′CAGCTCACCGAGAGCCTAGT3′ | 3756 | ||
| NQO1389460 R | 5′CATGGCATAGAGGTCCGACT3′ | 237–257 | 221 bps | |
| Genomic DNA | ||||
| NQO1 g1F | 5′CACACACACCCCTACAATCCCC3′ | (−246)–(−225) | 509 bps | |
| NQO1 g1R | 5′CCAGGTCCCTAATCTCTTCCC3′ | 243263 | ||
| NQO1 g2F | 5′ACATTTCTGGCTACAGGAGATGGA3′ | 78827905 | 704 bps | |
| NQO1 g3 | 5′GTCAGTTGGGATGGACTTGC3′ | 8573–8594 | ||
| NQO1 g4F | 5′CAGCTCACCGAGAGCCTAGT3′ | 11299–11318 | 361 bps | |
| NQO1 g4R | 5′GAAATCCATGTAATACTGCACCT3′ | NG_011504.2 | 11641–11659 | |
| NQO1 g5F | 5′AGTTGGCTGACCAAGGACAA3′ | 13285–13304 | 591 bps | |
| NQO1 g5R | 5′CCCTGCATCAGGACAGACC3′ | 13855–13875 | ||
| NQO1 g6F | 5′TAGCTCAGGGGAGCCAAAGT3′ | 15104–15124 | 693 bps | |
| NQO1 g6R | 5′TGAATTCCCCTGAAGGTTCGT3′ | 1577715796 | ||
| NQO1 g1F | 5′TGGTAACGGCTAGGTAGAGGG3′ | (−246)–(−225) | 509 bps | |
| NQO1 g1R | 5′AGCCCAGTCGGATTTTGGTT3′ | 243–263 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milković, L.; Tomljanović, M.; Čipak Gašparović, A.; Novak Kujundžić, R.; Šimunić, D.; Konjevoda, P.; Mojzeš, A.; Đaković, N.; Žarković, N.; Gall Trošelj, K. Nutritional Stress in Head and Neck Cancer Originating Cell Lines: The Sensitivity of the NRF2-NQO1 Axis. Cells 2019, 8, 1001. https://doi.org/10.3390/cells8091001
Milković L, Tomljanović M, Čipak Gašparović A, Novak Kujundžić R, Šimunić D, Konjevoda P, Mojzeš A, Đaković N, Žarković N, Gall Trošelj K. Nutritional Stress in Head and Neck Cancer Originating Cell Lines: The Sensitivity of the NRF2-NQO1 Axis. Cells. 2019; 8(9):1001. https://doi.org/10.3390/cells8091001
Chicago/Turabian StyleMilković, Lidija, Marko Tomljanović, Ana Čipak Gašparović, Renata Novak Kujundžić, Dina Šimunić, Paško Konjevoda, Anamarija Mojzeš, Nikola Đaković, Neven Žarković, and Koraljka Gall Trošelj. 2019. "Nutritional Stress in Head and Neck Cancer Originating Cell Lines: The Sensitivity of the NRF2-NQO1 Axis" Cells 8, no. 9: 1001. https://doi.org/10.3390/cells8091001
APA StyleMilković, L., Tomljanović, M., Čipak Gašparović, A., Novak Kujundžić, R., Šimunić, D., Konjevoda, P., Mojzeš, A., Đaković, N., Žarković, N., & Gall Trošelj, K. (2019). Nutritional Stress in Head and Neck Cancer Originating Cell Lines: The Sensitivity of the NRF2-NQO1 Axis. Cells, 8(9), 1001. https://doi.org/10.3390/cells8091001