Abstract
One of the crucial systems severely affected in several neuromuscular diseases is the loss of effective connection between muscle and nerve, leading to a pathological non-communication between the two tissues. The neuromuscular junction (NMJ) represents the critical region at the level of which muscle and nerve communicate. Defects in signal transmission between terminal nerve endings and muscle membrane is a common feature of several physio-pathologic conditions including aging and Amyotrophic Lateral Sclerosis (ALS). Nevertheless, controversy exists on whether pathological events beginning at the NMJ precede or follow loss of motor units. In this review, the role of NMJ in the physio-pathologic interplay between muscle and nerve is discussed.
1. Introduction
1.1. Muscle and Nerve Communication: A Peer to Peer Dialogue at the Neuromuscular Junction
Muscle and nerve communicate at the level of a specialized region, namely the neuromuscular junction (NMJ), a synaptic connection where the peripheral nervous system contacts skeletal muscle fibers, governing crucial vital processes, such as body voluntary movements and breathing []. Nerve activity guarantees not only muscle contraction but can induce myoblast orientation [] and strictly influences fiber type specification and myosin isoforms expression []. Skeletal muscle fibers can be generally classified as fast or slow twitch, based on their contractile and metabolic properties []. These properties are dependent on the pattern of motor nerve stimulation. Tonic motor neuron activity promotes the slow fiber phenotype, while infrequent motor neuron firing results in fast fibers generation []. Cross reinnervation experiments demonstrated that fast muscles turn into slow ones when reinnervated by a slow nerve, whereas slow muscles turn into fast ones when reinnervated by a fast nerve [,]. Of note, skeletal muscle is also a source of signals that influence neuron survival, axonal growth, and maintenance of synaptic connections. Indeed, the development in the absence of skeletal muscle results in the sequential ablation of motor neurons in the spinal cord and brain []. In addition, it has been demonstrated that during the stage of reinnervation the inducible depletion of adult satellite cells (SCs), the classical muscle stem cell compartment, can impair NMJ regeneration and affect NMJ morphology []. This suggests that skeletal muscle and nerve influence each other in a functional dialogue that is fundamental for their survival and mechanism of action. The loss of effective connection between muscle and nerve leads to a pathological non-communication between the two tissues. In this context, NMJ is the central player of the physiopathologic interplay between muscle and nerve.
1.2. Perisynaptic Schwann Cells, a Third Speaker in the Dialogue
NMJ is composed by three major elements: the presynaptic region containing the nerve terminal, the synaptic cleft, and the postsynaptic surface referred to as the endplate. Several studies have recently pointed out a critical cellular component in the neuromuscular synapse, namely the synapse-associated glial cells also called Perisynaptic Schwann Cells (PSCs).
PSCs play an active role in NMJ development and in the maintenance and remodeling of adult neuromuscular endplate. It has been demonstrated that during development, Schwann cells migrate in association with growing axons and are located close to NMJ just before muscle-nerve contacts []. Moreover, in vivo experiments demonstrated that PSCs increase their number before the period of synaptic growth and play a guidance role during axon growth and innervation. In contrast, lacks of PSCs can induce a complete loss of synapses during development or determine pre- and post-synaptic defects in developing NMJ [,].
In adult organisms, PSCs ensure NMJ stability, changing their properties according to the state of innervation []. PSCs provide several trophic molecules, such as agrin, transforming growth factor beta 1 (Tgf-β1), and Matrix Metalloproteinases (MMPs) that can support muscle-nerve contact and promote AChR clustering [,]. In turn, PSCs are influenced by muscle derived factors, such as NT-3, Neuregulin-1, or by neuronal released factors, such as ATP, Ach, and neuronal agrin [].
1.3. Kranocytes Cells, Capping Cells at the NMJ
An additional junctional cell type element, along with skeletal muscle fibers, motor neuron terminals, perisynaptic terminal Schwann cells, is a fibroblast-like cell, named kranocyte []. Kranocyte, lying outside the synaptic basal lamina, caps the NMJ above the perisynaptic Schwann cells and extends its cytoplasmic processes over the end-plate area [,]. In newborn mouse muscle, kranocytes are equally distributed, whereas during mouse postnatal development they become restricted at the endplate zone []. Whole mount immunostaining experiments demonstrated an unequivocal disposition of this subpopulation and elucidated that kranocytes are not Schwann cells. They express a specific immunocytochemical profile, including collagen-synthetizing enzyme and neuregulin []. In this context, kranocytes may interact with terminal Schwann cells through neuregulin mediated signaling [].
Although in adulthood kranocytes remain confined at the NMJ region, they play a role in nerve repair and regeneration after denervation or paralysis. Within 24 h of denervation, kranocytes proliferate and spread throughout the NMJ area, even before the sprouting of Schwann cells [,], suggesting that this different time of reaction may promote and trigger terminal Schwann cells outgrowth and activation []. Thus, mammalian kranocytes capping cells are integral player of neuromuscular functionality.
2. ALS and Aging as Paradigmatic Examples of Altered Nerve-Muscle Communication
The impaired neuromuscular transmission represents a critical feature of several pathological conditions in which structural changes in NMJ might contribute to muscle weakness, altered motor neuron activity, and loss of muscle fibers.
In this review, we will analyse two extreme physio-pathologic conditions, namely aging and Amyotrophic Lateral Sclerosis (ALS).
Aging represents a physiologic and progressive decay of the homeostatic processes of the entire organism and it is broadly defined as the time-dependent functional decline that affects most living organisms. Common denominators of aging in different organisms include genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication [].
ALS is a complex and severe disease associated with numerous pathologic mechanisms, including oxidative stress, mitochondrial dysfunction, axonal damage, microglial activation, inflammation, excitotoxicity, and protein aggregation [].
Interestingly, although aging and ALS display different progressive loss of physiological integrity, they share some common pathologic feature, including high levels of oxidative damage, decreased number of synaptic vesicles, reduced and altered mitochondria in the plaque region [,,,,] (Figure 1). Moreover, an early pathologic sign observed in both ALS and aging is the morphologic alteration of NMJ. However, whether changes in the NMJ precede or follow loss of motor units remains unresolved. Here we detail the NMJ degenerative features in ALS and aging and discuss the paradigm that retrograde signaling, from muscle to nerve, might represent an early event preceding motor neuron degeneration.
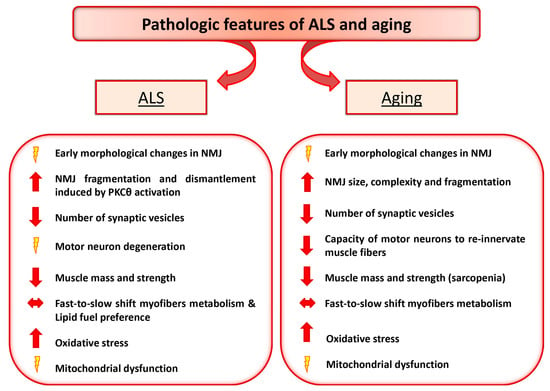
Figure 1.
Amyotrophic Lateral Sclerosis (ALS) and Aging share some common pathologic features. The diagram depicts the relevant pathologic changes observed in ALS and aging.
2.1. Motor Neuron Diseases: ALS
One of the best examples of impaired interplay between nerve and muscle is ALS, a fatal disease characterized by motor neurons degeneration, muscle atrophy, weakness and, ultimately, muscle paralysis with respiratory failure. During ALS progression muscle denervation is accompanied by changes in muscle fiber profile with a preference loss of fast-twitch fiber [,], due to higher vulnerability of fast-fatigable innervating motor neurons and to specific signals released by PSCs []. ALS is epidemiologically classified into two forms: sporadic (90–95%) and familial (5–10%) form. Among the familial cases, approximately 20% correlate with mutations in the sequence of the Superoxide Dismutase 1 gene (SOD1), which encodes for an important antioxidant enzyme. In addition to SOD1 mutations, other gene defects have been reported to cause ALS, including senataxin (SETX) [], alsin [,], dynactin [], synaptobrevin/VAMP (vesicle-associated membrane protein)-associated protein B (VAPB) [], TDP-43, FUS/TLS [,], profilin (PFN1) [], MATR3, CHCHD10, TBK1, TUBA4A, NEK1, C21orf2, and CCNF []. However, even in these cases, where a well-defined mutation has been linked to the disease, a clear correlation between the genetic defect and the pathophysiology of the disease has not yet been disclosed.
Much of what we presently know about the role of mutant genes in ALS is based on studies of transgenic animals in which the potential genes involved in ALS are overexpressed under the control of specific promoters. Nevertheless, the failure to translate the positive results obtained in animal models into successful trials in human has cooled the enthusiasms and raised important questions on the validity of either animal models or methodological approaches. Thus, robust criteria and guidelines for preclinical animal research in ALS are necessary []. One of the experimental models that has been widely used in ALS-related studies is the transgenic mutant SOD1 mouse. Although the SOD1 mutant mice present some limitations compared to ALS patients it remains, along with other ALS-related mice, an ideal model for preclinical tests and proof-of-concept studies []. The obvious loss of motor neurons in the spinal cord initially focused attention on how mutant SOD1 may act within motor neurons to provoke neuronal degeneration and death. Among pathogenic events, glutamate-induced excitotoxicity, oxidative stress, protein aggregation, and mitochondrial dysfunction within motor neurons have been proposed. However, the mutant gene products are widely expressed, raising the possibility that the toxic cascade may be achieved wholly or in part by mutant SOD1 action in non-neuronal cells. Notably, restriction of SOD1 mutant expression selectively to post-natal motor neurons failed to produce detectable sign of pathology or motor-neuron disease [], suggesting that other cell types may be involved in ALS-associated neurodegeneration. Indeed, analysis of chimeras generated between wild type and SOD1 mutant mouse embryonic cells revealed that wild type non neuronal cells in adult chimeric animals extended the survival of SOD1 mutant motor neurons [].
The generation of mouse models expressing human mutated SOD1 gene (SOD1G93A), exclusively in skeletal muscle, added new insights into the potential primary targets of the mutant SOD1 toxic protein. Muscle-specific expression of mutant SOD1G93A caused accumulation of reactive oxygen species (ROS), mitochondria dysfunction, muscle atrophy, NMJ dismantlement, microgliosis [,], and neuron degeneration []. All together this evidence suggests that local toxic effect of SOD1G93A is a primary determinant of ALS-associated muscle pathology and that retrograde signals from muscle to nerve may contribute, in a sort of dying back phenomenon, to synapse and axon damage [,]. In fact, several other recent evidences confirmed the hypothesis that motor neurons are not the only primary targets of SOD1G93A-mediated toxicity and revealed that early changes of neuromuscular transmission start long before motor symptoms onset. Fischer and colleagues demonstrated that pathological changes of motor neuron disease begin at the distal axon and they observed denervation and reinnervation changes in muscle tissue without any pathological signs in neurons cells []. Furthermore, Schafer’s group characterized two different types of motor neurons, the “losers” or denervated branches, and the “compensators”, or reinnervating branches, which display different “susceptibility” to the toxic properties of SOD1G93A mutant gene product []. Although a conclusive link is still missing, it is intriguing to speculate that loser and compensator neurons are subject to different influences from neighboring interneurons, astrocytes, or microglia [] and from the vasculature [,] and muscle fibers they innervate, all of which might provide either toxic or protective factors.
This evidence supports the notion that NMJ dismantlement can occur independently from motor neuron degeneration and may represent an early pathogenic signature of muscle-nerve communication defects.
2.1.1. PKCθ as a Potential Signaling Involved in NMJ Dysfunction in ALS Disease
It has been demonstrated that Protein Kinase C and A (PKC and PKA) activities have antagonistic effects on NMJ stability and Acetylcholine Receptors (AChRs) recycling []. Stimulation of PKC or inactivation of PKA significantly accelerates the removal of postsynaptic AChRs and depresses AChR recycling. A recent work disclosed the PKC isoform, namely PKCθ, which triggers NMJ dismantlement in a mouse model expressing SOD1G93A mutant gene. PKCθ is a kinase that triggers in physiological condition the dismantlement of supernumerary NMJs during the first postnatal days [,]. It has been demonstrated that perturbation in redox signaling cascades, induced by muscle-specific accumulation of mutant SOD1G93A gene, promotes the activation of PKCθ that in turn acts as mediator of endplates destabilization []. In contrast, PKCθ selective pharmacological inhibition reduces oxidative damage and guarantees the stabilization of AChR turnover and the rescue of the NMJ morphological complexity [].
PKCθ also modulates the activation of two sensors of nerve activity, Calcineurin (CN) and Nuclear Factor of Activated T cells (NFATc1), which promote slow muscle phenotype through the activation of their target Myocyte Enhancer Factor 2D (MEF2D), involved in muscle glucose homeostasis. Furthermore, it has been demonstrated that PKCθ-lacking mice display an impaired glucose homeostasis in fast muscles [,], suggesting that PKCθ can participate to muscle metabolic changes associated to muscle denervation and ALS disease progression.
2.1.2. Metabolic Changes and Mitochondrial Alteration May Influence NMJ Stability
Metabolic homeostasis and energy balance result severely compromised in both ALS patients and mouse models, where the energy expenditure is higher than the intake and it is associated to an increased energy demand and to an abnormal lipid metabolism [,,,,]. In fact, a recent study revealed that ALS patients start losing weight almost 10 years before the clinical onset of the disease and show a higher daily energy intake to compensate the elevated energy consumption []. Likewise, the ALS mouse model that ubiquitously overexpress SOD1G93A mutant gene shows similar energetic alterations, such as an increased energy expenditure and a concomitant skeletal muscle hypermetabolism [].
Further evidence has also pointed out that metabolic alterations in SOD1G93A mouse model can be distinct from denervation [] and that muscle specific accumulation of SOD1G93A is able to induce metabolic changes in glucose and lipid pathways, independently from motor neuron degeneration and preceding muscle denervation []. These data suggest that some of the metabolic changes represent part of the earliest signs of morphological and functional alterations occurring in the motor nerve terminals. Among cell components, dysfunctional mitochondria represent key regulators of metabolic changes [,,,]. Indeed, treatment with mitochondria protective drug can preserve NMJ function and structure in ALS mouse model at late stages of the disease, suggesting a central role of mitochondria in the pathology []. The link between muscle functional mitochondrial alterations and neurodegeneration has been clarified by Dupuis and collaborators. The analysis of a transgenic mouse model overexpressing the potent mitochondrial uncoupler protein (UCP1) exclusively in skeletal muscle revealed that muscle specific expression of UCP1 induces an age-dependent deterioration of the NMJ []. This defect correlated with progressive signs of denervation, supporting the idea of the crucial role of skeletal muscle on nerve homeostasis and revealing the potential molecular signature associated with the dying back phenomenon [].
It has been demonstrated that antioxidant treatment of mice expressing human mutated SOD1 gene (SOD1G93A) exclusively in skeletal muscle was able to rescue mitochondrial functionality and NMJ stability [,]. Moreover, studies on SOD1G93A mice overexpressing the neurotrophic factors GDNF or IGF-1 in muscle tissue revealed an hyperinnervation of muscle fibers [,,,,,,,,], NMJ stability, and motor neuron preservation [,,]. This suggests that a sort of “saving back” approach can be envisaged. Nevertheless, not all treatments guarantee the same efficacy. Neuromuscular Magnetic Stimulation (NMMS) was able to induce a significant increase of muscle strength, without significant change in Compound Muscle Action Potential (CMAP) amplitude, suggesting that the improvement was not related to reinnervation phenomena []. In particular, NMMS activated a molecular mini-circuit to counteract muscle atrophy, to promote muscle robustness and maintenance of fiber type composition, and to induce NMJ stabilization. In addition, NMMS induced an improvement of AChR functionality, characterized by the reduction of the expression level of gamma subunit of AChR [], whose expression increases in denervated muscle or under conditions that alter NMJ functionality [,].
It has been also demonstrated that Musk activation preserves synapses at the NMJ in diaphragm muscles of mutant SOD1 transgenic mice, without any positive effects on motor neuron death and survival []. Cleveland and colleagues demonstrated that the over expression of Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1alpha in the skeletal muscle of ALS transgenic mice is able to increase mitochondria energy production, ameliorating muscle endurance and performance and reducing muscle atrophy, without any effects on muscle innervation and motor neuron survival []. Nevertheless, the authors did not detail NMJ morphology in PGC1α overexpressing ALS mice even if it has been demonstrated that muscle-specific PGC-1α expression induces functional improvement in NMJ []. These data suggest that additional work is necessary to define the best experimental conditions to support a “saving back” therapeutic approach. Moreover, all these data reinforce the evidence that ALS is a complex multi-systemic disease in which alterations in structural, physiological and metabolic parameters in different cell types, including motor neurons, glia, NMJ, vasculature, and muscle, act synergistically to exacerbate the disease. Moreover, all these works suggest that a single treatment or the modulation of a single pathway may be not enough to significantly counteract motor neuron degeneration and increase survival. Combinatorial approaches on different tissue targets might be necessary to achieve satisfactory therapeutic benefits.
2.2. Aging and NMJ Defect
Another physio-pathologic condition where the functional communication between muscle and nerve is compromised is aging. As mammals grow older, many functional and structural changes occur in skeletal muscle and NMJ, leading to gradual loss of mobility [].
It is widely accepted that not all muscles share the same susceptibility to age-related alterations. Several studies indicate diversity in muscle fibers susceptibility to the denervation process, based on motor neuron type. The NMJ associated with faster-contracting motor neurons are more susceptible to age-related structural changes than those associated with slower-contracting motor neurons [,,].
One of the first evidences of the aging process occurring in NMJ is the formation of new branches of terminal axons that form new synaptic sites on muscle fibers []. NMJ undergo remodeling processes with cyclical extension and retraction of motor nerve terminals, leading to an increase in the complexity of the nerve terminal arborization []. In particular, this remodeling process is characterized by the increase in size and complexity of the branches of terminal axons [], associated with fragmentation [,,,,,,,,] and folds reduction of postsynaptic sites [,]. It has been demonstrated that oxidative stress, along with compromised mitochondria and increased intracellular calcium, amplifies the presynaptic decline in NMJ, accompanied with a decreased number of synaptic vesicles, similarly to what observed in ALS mouse models [,,]. This initial NMJ dysfunction is followed by a neurodegeneration promoted by increased production of inflammatory cytokines and loss of trophic support []. Moreover, the evidence that some denervated fibers are not successfully re-innervated during aging raise the prospect that this alteration is at the base of the progressive decline in muscle mass and strength with aging, a condition known as sarcopenia.
The triggers of the NMJ alterations during aging are still debated. It is still unclear whether NMJ changes in aged muscles are caused by alterations in motor neurons or rather in skeletal muscle fiber []. One of the possible causes is the death of a fraction of motor neurons, occurring in humans between the age of 60 and 90 []. It has been suggested that motor neuron death provokes a temporary denervation of downstream muscles and induces a collateral reinnervation process. This process is driven by Perisynaptic Schwann cells that grow from the axon stump to guide surviving motor neurons that sprout their axons to create new synaptic terminals on muscle fibers [,]. Nevertheless, in advanced age, motor neurons show impaired capacity to re-innervated denervated fibers, suggesting that alterations in different components of nerve-muscle circuit impinge the potential compensatory mechanisms.
Other studies suggest that changes in the end-plate morphology and NMJ remodeling that occur with aging precede the loss of fast motor units and suggest the involvement of retrograde signaling. It has been reported that proteolytic cleavage of agrin, a proteoglycan involved in NMJ development, maturation, and AChRs clustering [], induces early onset sarcopenia in young adult mice [], whereas the injection of a neurotrypsin-resistant agrin fragment stabilized NMJ and improved the phenotype of neurotrypsin-overexpressing mice [].
Other studies employed the effect of Muscle Electrical Stimulation (ES) to improve muscle functionality and to counteract NMJ decline during aging []. ES treatment in old sedentary people improved muscle performance, increasing fiber size, stimulating satellite cells and modulating the degeneration of the mitochondrial apparatus [,]. Moreover, it has been reported that ES treatment increases the number of fast twitch fibers [] and counteracts neuromuscular disabilities from age related NMJ degeneration in paraplegic patients [,]. At molecular level ES treatment activates signaling pathways that decode for a specific calcium signaling involved in metabolic and structural adaptation of muscle fibers, such as the Calcineurin-NFAT and CamKII pathways that control the maintenance or switching of muscle fiber type []. Moreover, it has been described that biphasic electrical stimulation significantly increases the number and size of AChRs clusters available for NMJ formation during innervation [] and that electrical stimulation promotes axonal growth and sensorimotor functional recovery after injury [].
Factors that Influence NMJ Stability during Aging
Among the factors that can affect the stability of the NMJ there are metabolic changes associated to mitochondrial dysfunction and lifestyle.
Several recent evidence demonstrated that mitochondrial energy metabolism is altered during muscle aging, which is also accompanied by a reduction in the rate of oxygen consumption [,]. In human skeletal muscle, as well as in animal models, the mitochondrial capability to produce ATP decreases with age [,,,,,,].
It is widely recognized that mitochondrial dysfunctions have a causal link with cellular ROS accumulation and therefore, as organisms grow old, there is an increase of intracellular ROS and consequently an accumulation of damaged cell components in all the tissues []. Since muscle and nerve are highly metabolic active tissues, their mutual communication is affected by mitochondrial dysfunction.
Another factor playing a crucial role in sarcopenia progression is lifestyle. Physical inactivity and impaired nutrition stimulate loss of muscle mass and NMJ defect, exacerbating sarcopenia [,]. It has been demonstrated that exercise induces muscle hypertrophy and NMJ remodeling, and improves recovery of peripheral nervous terminal after nerve injury [,]. In old mice exercise training is able to minimize NMJ expansion that turn back to levels comparable to young mice [].
How skeletal muscle and NMJ send retrograde signals to motor neurons represents an intriguing field of research. Skeletal muscle has been identified as an endocrine organ that produce and secrete growth factors, cytokines, and peptides, collectively indicated as myokines, which make muscle capable to communicate with other tissues and organs, including bone, intestine, adipose tissue, liver, pancreas, and brain []. One of the critical players that integrates muscle fiber function with motor neuron signaling following exercise is Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1alpha. PGC-1α expression activates a broad NMJ gene program and improves postsynaptic NMJ architecture [,]. It has been demonstrated that loss of skeletal muscle PGC-1α hampers acetylcholine receptor (AChR) clustering and the transcription of NMJ genes including AChRs, muscle-specific kinase, and utrophin [].
Among other factors, Insulin-like Growth Factor-1 (IGF-1) plays a pivotal role in muscle growth, differentiation, and regeneration [,]. In mammalian organisms the majority of IGF-1 is produced and released by the liver as systemic growth factor, while a minor fraction is produced by other tissues and participates to autocrine and paracrine signals. In skeletal muscle, the binding of IGF-1 with its receptor activates anabolic, anticatabolic, and antiapoptotic signaling pathways that preserves muscle mass and strength []. It has been demonstrated that the overexpression of IGF-1 isoforms selectively in skeletal muscle maintains muscle mass and activates several pathways that promote the clearance of dysfunctional mitochondria and the maintenance of NMJ stability in senescence mice []. Moreover, it has been demonstrated that IGF-1 overexpression is sufficient to downregulate the levels of two cytokines, namely IL-1β and IL-6, in aged mice, counteracting the chronic inflammation process typical of aging (inflammaging) []. In this context, a crucial role is played by senescent Schwann cells that overexpress IL-6 cytokine and negatively affect the nerve microenvironment during muscle innervation [].
3. Conclusions
Significant advances have been provided in the understanding the critical components involved in aging and ALS. Morphological and functional alterations of the NMJ represent part of the earliest signs of aging and ALS. Nevertheless, no consensus has emerged to the cells, tissues, and pathways that are directly affected. Although several pieces of the aging and ALS pathogenic puzzles have been defined, the picture of these diseases remained to be completed. The field will benefit when the pathologic events, the factors, the cell and tissue components, and the late “restriction point”, which might trigger the final catastrophic event, will be sequentially characterized. This will also help to: (i) verify whether a sort of “saving back” approach can be envisaged and tested and to (ii) design more effective therapeutic approaches, acting in a timely fashion on the different cells and tissues involved in the diseases (Figure 2).
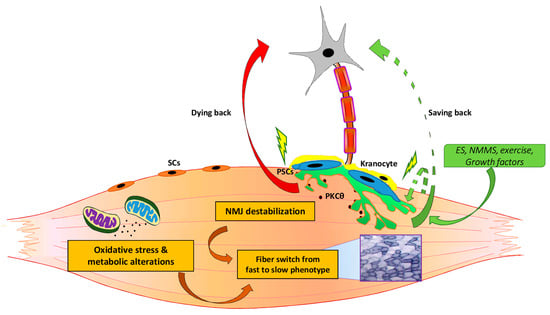
Figure 2.
A summary of the relevant changes, cells, and signaling involved in the pathogenesis of ALS and aging. Morphological and functional alterations of the neuromuscular junction (NMJ) represent part of the earliest signs of aging and ALS. NMJ development, remodelling, and stability is also influenced by different signals and cells, including PSCs and kranocytes. According to the dying back hypothesis, and based on studies regarding how skeletal muscle interventions can influence NMJ, further studies should be encouraged to verify whether combinatorial approaches on different cell and tissue targets induce more satisfactory therapeutic benefits and to define whether a sort of “saving back” approach can be promoted to preserve NMJ integrity, to delay motor neuron impairment, and to counteract muscle-nerve dysfunction.
Author Contributions
A.M. conceptualized the study; E.L. and I.C. wrote the original draft; A.M. and G.D. wrote/reviewed and edited the text and figures.
Funding
This research was funded by ASI, progetti di Ateneo, Progetto Finalizzata, Fondazione Roma.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rudolf, R.; Khan, M.M.; Witzemann, V. Motor Endplate—Anatomical, Functional, and Molecular Concepts in the Historical Perspective. Cells 2019, 8, 387. [Google Scholar] [CrossRef] [PubMed]
- Mège, R.M.; Goudou, D.; Giaume, C.; Nicolet, M.; Rieger, F. Is intercellular communication via gap junctions required for myoblast fusion? Cell Adhes. Commun. 1994, 2, 329–343. [Google Scholar] [CrossRef] [PubMed]
- Schiaffino, S.; Reggiani, C. Molecular diversity of myofibrillar proteins: Gene regulation and functional significance. Physiol. Rev. 1996, 76, 371–423. [Google Scholar] [CrossRef] [PubMed]
- Hughes, S.M. Muscle development: Electrical control of gene expression. Curr. Biol. 1998, 8, R892–R894. [Google Scholar] [CrossRef]
- Olson, E.N.; Williams, R.S. Calcineurin signaling and muscle remodeling. Cell 2000, 101, 689–692. [Google Scholar] [CrossRef]
- Buller, A.J.; Eccles, J.C.; Eccles, R.M. Differentiation of fast and slow muscles in the cat hind limb. J. Physiol. 1960, 150, 399–416. [Google Scholar] [CrossRef] [PubMed]
- Pette, D.; Vrbová, G. Neural control of phenotypic expression in mammalian muscle fibers. Muscle Nerve 1985, 8, 676–689. [Google Scholar] [CrossRef] [PubMed]
- Kablar, B.; Rudnicki, M.A. Development in the Absence of Skeletal Muscle Results in the Sequential Ablation of Motor Neurons from the Spinal Cord to the Brain. Dev. Biol. 1999, 208, 93–109. [Google Scholar] [CrossRef]
- Liu, W.; Wei-LaPierre, L.; Klose, A.; Dirksen, R.T.; Chakkalakal, J.V. Inducible depletion of adult skeletal muscle stem cells impairs the regeneration of neuromuscular junctions. eLife 2015, 4, e09221. [Google Scholar] [CrossRef]
- Feng, Z.; Ko, C.-P. Schwann Cells Promote Synaptogenesis at the Neuromuscular Junction via Transforming Growth Factor- 1. J. Neurosci. 2008, 28, 9599–9609. [Google Scholar] [CrossRef]
- Koirala, S.; Reddy, L.V.; Ko, C.-P. Roles of glial cells in the formation, function, and maintenance of the neuromuscular junction. J. Neurosci. 2003, 32, 987–1002. [Google Scholar] [CrossRef] [PubMed]
- Lin, W.; Sanchez, H.B.; Deerinck, T.; Morris, J.K.; Ellisman, M.; Lee, K.-F. Aberrant development of motor axons and neuromuscular synapses in erbB2-deficient mice. Proc. Natl. Acad. Sci. USA 2000. [Google Scholar] [CrossRef] [PubMed]
- Rousse, I.; St.-Amour, A.; Darabid, H.; Robitaille, R. Synapse-glia interactions are governed by synaptic and intrinsic glial properties. Neuroscience 2010. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Ko, C.-P. The role of glial cells in the formation and maintenance of the neuromuscular junction. Ann. N. Y. Acad. Sci. 2008, 1132, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Court, F.A.; Gillingwater, T.H.; Melrose, S.; Sherman, D.L.; Greenshields, K.N.; Morton, A.J.; Harris, J.B.; Willison, H.J.; Ribchester, R.R. Identity, developmental restriction and reactivity of extralaminar cells capping mammalian neuromuscular junctions. J. Cell Sci. 2008. [Google Scholar] [CrossRef] [PubMed]
- Weis, J.; Fine, S.M.; David, C.; Savarirayan, S.; Sanes, J.R. Integration site-dependent expression of a transgene reveals specialized features of cells associated with neuromuscular junctions. J. Cell Biol. 1991. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, Y.; Lin, W. Neuron-glia interactions: The roles of Schwann cells in neuromuscular synapse formation and function. Biosci. Rep. 2011, 31, 295–302. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A. Understanding ALS: New therapeutic approaches. FEBS J. 2013, 280, 4315–4322. [Google Scholar] [CrossRef]
- Park, K.H.J. Mechanisms of Muscle Denervation in Aging: Insights from a Mouse Model of Amyotrophic Lateral Sclerosis. Aging Dis. 2015, 6, 380–389. [Google Scholar] [CrossRef]
- García, M.L.; Fernández, A.; Solas, M.T. Mitochondria, motor neurons and aging. J. Neurol. Sci. 2013, 330, 18–26. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musarò, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2018, 170, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Martini, M.; Scicchitano, B.M.; Romanello, V.; Boncompagni, S.; Nicoletti, C.; Pietrangelo, L.; De Panfilis, S.; Catizone, A.; Bouchè, M.; et al. Muscle Expression of SOD1G93A Triggers the Dismantlement of Neuromuscular Junction via PKC-Theta. Antioxid. Redox Signal. 2018, 28, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Magrané, J.; Cortez, C.; Gan, W.B.; Manfredi, G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum. Mol. Genet. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, J.; Putman, C.T.; Gordon, T. Time course of preferential motor unit loss in the SOD1 G93A mouse model of amyotrophic lateral sclerosis. Neurobiol. Dis. 2007, 28, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, J.; Putman, C.T.; Tyreman, N.; Gordon, T. Preferential motor unit loss in the SOD1G93A transgenic mouse model of amyotrophic lateral sclerosis. J. Physiol. 2008, 586, 3337–3351. [Google Scholar] [CrossRef] [PubMed]
- De Winter, F.; Vo, T.; Stam, F.J.; Wisman, L.A.B.; Bär, P.R.; Niclou, S.P.; van Muiswinkel, F.L.; Verhaagen, J. The expression of the chemorepellent Semaphorin 3A is selectively induced in terminal Schwann cells of a subset of neuromuscular synapses that display limited anatomical plasticity and enhanced vulnerability in motor neuron disease. Mol. Cell. Neurosci. 2006, 32, 102–117. [Google Scholar] [CrossRef]
- Chance, P.F.; Rabin, B.A.; Ryan, S.G.; Ding, Y.; Scavina, M.; Crain, B.; Griffin, J.W.; Cornblath, D.R. Linkage of the gene for an autosomal dominant form of juvenile amyotrophic lateral sclerosis to chromosome 9q34. Am. J. Hum. Genet. 1998, 62, 633–640. [Google Scholar] [CrossRef]
- Yang, Y.; Hentati, A.; Deng, H.X.; Dabbagh, O.; Sasaki, T.; Hirano, M.; Hung, W.Y.; Ouahchi, K.; Yan, J.; Azim, A.C.; et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat. Genet. 2001, 29, 160–165. [Google Scholar] [CrossRef]
- Hadano, S.; Hand, C.K.; Osuga, H.; Yanagisawa, Y.; Otomo, A.; Devon, R.S.; Miyamoto, N.; Showguchi-Miyata, J.; Okada, Y.; Singaraja, R.; et al. A gene encoding a putative GTPase regulator is mutated in familial amyotrophic lateral sclerosis 2. Nat. Genet. 2001. [Google Scholar] [CrossRef]
- Puls, I.; Jonnakuty, C.; LaMonte, B.H.; Holzbaur, E.L.F.; Tokito, M.; Mann, E.; Floeter, M.K.; Bidus, K.; Drayna, D.; Oh, S.J.; et al. Mutant dynactin in motor neuron disease. Nat. Genet. 2003, 33, 455–456. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, A.L.; Mitne-Neto, M.; Silva, H.C.A.; Richieri-Costa, A.; Middleton, S.; Cascio, D.; Kok, F.; Oliveira, J.R.M.; Gillingwater, T.; Webb, J.; et al. A Mutation in the Vesicle-Trafficking Protein VAPB Causes Late-Onset Spinal Muscular Atrophy and Amyotrophic Lateral Sclerosis. Am. J. Hum. Genet. 2004. [Google Scholar] [CrossRef] [PubMed]
- Vance, C.; Rogelj, B.; Hortobágyi, T.; De Vos, K.J.; Nishimura, A.L.; Sreedharan, J.; Hu, X.; Smith, B.; Ruddy, D.; Wright, P.; et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009. [Google Scholar] [CrossRef] [PubMed]
- Wegorzewska, I.; Bell, S.; Cairns, N.J.; Miller, T.M.; Baloh, R.H. TDP-43 mutant transgenic mice develop features of ALS and frontotemporal lobar degeneration. Proc. Natl. Acad. Sci. USA 2009. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.H.; Fallini, C.; Ticozzi, N.; Keagle, P.J.; Sapp, P.C.; Piotrowska, K.; Lowe, P.; Koppers, M.; McKenna-Yasek, D.; Baron, D.M.; et al. Mutations in the profilin 1 gene cause familial amyotrophic lateral sclerosis. Nature 2012. [Google Scholar] [CrossRef] [PubMed]
- Chia, R.; Chiò, A.; Traynor, B.J. Novel genes associated with amyotrophic lateral sclerosis: Diagnostic and clinical implications. Lancet Neurol. 2018, 17, 94–102. [Google Scholar] [CrossRef]
- Ludolph, A.C.; Bendotti, C.; Blaugrund, E.; Chio, A.; Greensmith, L.; Loeffler, J.P.; Mead, R.; Niessen, H.G.; Petri, S.; Pradat, P.F.; et al. Guidelines for preclinical animal research in ALS/MND: A consensus meeting. Amyotroph. Lateral Scler. 2010. [Google Scholar] [CrossRef] [PubMed]
- De Giorgio, F.; Maduro, C.; Fisher, E.M.C.; Acevedo-Arozena, A. Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model. Mech. 2019, 12. [Google Scholar] [CrossRef]
- Lino, M.M.; Schneider, C.; Caroni, P. Accumulation of SOD1 mutants in postnatal motoneurons does not cause motoneuron pathology or motoneuron disease. J. Neurosci. 2002, 22, 4825–4832. [Google Scholar] [CrossRef]
- Clement, A.M.; Nguyen, M.D.; Roberts, E.A.; Garcia, M.L.; Boillée, S.; Rule, M.; McMahon, A.P.; Doucette, W.; Siwek, D.; Ferrante, R.J.; et al. Wild-type nonneuronal cells extend survival of SOD1 mutant motor neurons in ALS mice. Science 2003. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Aucello, M.; Rizzuto, E.; Beccafico, S.; Mammucari, C.; Bonconpagni, S.; Belia, S.; Wannenes, F.; Nicoletti, C.; Del Prete, Z.; et al. Skeletal Muscle Is a Primary Target of SOD1G93A-Mediated Toxicity. Cell Metab. 2008, 8, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.; Martin, L.J. Skeletal muscle-restricted expression of human SOD1 causes motor neuron degeneration in transgenic mice. Hum. Mol. Genet. 2010, 19, 2284–2302. [Google Scholar] [CrossRef] [PubMed]
- Luo, G.; Yi, J.; Ma, C.; Xiao, Y.; Yi, F.; Yu, T.; Zhou, J. Defective Mitochondrial Dynamics Is an Early Event in Skeletal Muscle of an Amyotrophic Lateral Sclerosis Mouse Model. PLoS ONE 2013, 8, e82112. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Deschenes, M.R.; Sandri, M. Neuromuscular junction degeneration in muscle wasting. Curr. Opin. Clin. Nutr. Metab. Care 2016, 19, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Fischer, L.R.; Culver, D.G.; Tennant, P.; Davis, A.A.; Wang, M.; Castellano-Sanchez, A.; Khan, J.; Polak, M.A.; Glass, J.D. Amyotrophic lateral sclerosis is a distal axonopathy: Evidence in mice and man. Exp. Neurol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, A.M.; Sanes, J.R.; Lichtman, J.W. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. J. Comp. Neurol. 2005, 490, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Oosthuyse, B.; Moons, L.; Storkebaum, E.; Beck, H.; Nuyens, D.; Brusselmans, K.; Van Dorpe, J.; Hellings, P.; Gorselink, M.; Heymans, S.; et al. Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat. Genet. 2001. [Google Scholar] [CrossRef]
- Horwitz, G.; Jordan, H.; Krekelberg, B.; Richert, M.; Reyes, J.; Foundation, T.S.; Azzouz, M.; Ralph, G.S.; Storkebaum, E.; Walmsley, L.E.; et al. VEGF delivery with retrogradely transported lentivector prolongs survival in a mouse ALS model. Nature 2004, 429, 413–417. [Google Scholar]
- Martinez-Pena, Y.; Valenzuela, I.; Pires-Oliveira, M.; Akaaboune, M. PKC and PKA regulate AChR dynamics at the neuromuscular junction of living mice. PLoS ONE 2013. [Google Scholar] [CrossRef]
- Li, M.-X.; Jia, M.; Yang, L.-X.; Jiang, H.; Lanuza, M.A.; Gonzalez, C.M.; Nelson, P.G. The role of the theta isoform of protein kinase C (PKC) in activity-dependent synapse elimination: Evidence from the PKC theta knock-out mouse in vivo and in vitro. J. Neurosci. 2004, 24, 3762–3769. [Google Scholar] [CrossRef]
- Camerino, G.M.; Bouchè, M.; De Bellis, M.; Cannone, M.; Liantonio, A.; Musaraj, K.; Romano, R.; Smeriglio, P.; Madaro, L.; Giustino, A.; et al. Protein kinase C theta (PKCθ) modulates the ClC-1 chloride channel activity and skeletal muscle phenotype: A biophysical and gene expression study in mouse models lacking the PKCθ. Pflügers Arch. Eur. J. Physiol. 2014, 466, 2215–2228. [Google Scholar] [CrossRef] [PubMed]
- Serra, C.; Federici, M.; Buongiorno, A.; Senni, M.I.; Morelli, S.; Segratella, E.; Pascuccio, M.; Tiveron, C.; Mattei, E.; Tatangelo, L.; et al. Transgenic mice with dominant negative PKC-theta in skeletal muscle: A new model of insulin resistance and obesity. J. Cell. Physiol. 2003, 196, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Kasarskis, E.J.; Neville, H.E. Management of ALS: Nutritional care. Neurology 1996, 47, S118–S120. [Google Scholar] [CrossRef] [PubMed]
- Desport, J.-C.; Torny, F.; Lacoste, M.; Preux, P.-M.; Couratier, P. Hypermetabolism in ALS: Correlations with Clinical and Paraclinical Parameters. Neurodegener. Dis. 2005, 2, 202–207. [Google Scholar] [CrossRef] [PubMed]
- Funalot, B.; Desport, J.-C.; Sturtz, F.; Camu, W.; Couratier, P. High metabolic level in patients with familial amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Bouteloup, C.; Desport, J.-C.; Clavelou, P.; Guy, N.; Derumeaux-Burel, H.; Ferrier, A.; Couratier, P. Hypermetabolism in ALS patients: An early and persistent phenomenon. J. Neurol. 2009, 256, 1236–1242. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Corcia, P.; Fergani, A.; Gonzalez De Aguilar, J.-L.; Bonnefont-Rousselot, D.; Bittar, R.; Seilhean, D.; Hauw, J.-J.; Lacomblez, L.; Loeffler, J.-P.; et al. Dyslipidemia is a protective factor in amyotrophic lateral sclerosis. Neurology 2008, 70, 1004–1009. [Google Scholar] [CrossRef] [PubMed]
- Huisman, M.H.B.; Seelen, M.; van Doormaal, P.T.C.; de Jong, S.W.; de Vries, J.H.M.; van der Kooi, A.J.; de Visser, M.; Schelhaas, H.J.; van den Berg, L.H.; Veldink, J.H. Effect of Presymptomatic Body Mass Index and Consumption of Fat and Alcohol on Amyotrophic Lateral Sclerosis. JAMA Neurol. 2015, 72, 1155–1162. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Oudart, H.; de Tapia, M.; Barbeito, L.; Loeffler, J.-P. Mitochondria in Amyotrophic Lateral Sclerosis: A Trigger and a Target. Neurodegener. Dis. 2004, 1, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Palamiuc, L.; Schlagowski, A.; Ngo, S.T.; Vernay, A.; Dirrig-Grosch, S.; Henriques, A.; Boutillier, A.-L.; Zoll, J.; Echaniz-Laguna, A.; Loeffler, J.-P.; et al. A metabolic switch toward lipid use in glycolytic muscle is an early pathologic event in a mouse model of amyotrophic lateral sclerosis. EMBO Mol. Med. 2015, 7, 526–546. [Google Scholar] [CrossRef]
- Dobrowolny, G.; Lepore, E.; Martini, M.; Barberi, L.; Nunn, A.; Scicchitano, B.M.; Musarò, A. Metabolic Changes Associated with Muscle Expression of SOD1G93A. Front. Physiol. 2018, 9, 831. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg. Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Wei, Y.; Zhang, J.; Gal, J.; Zhu, H. Mitochondrial Dysfunction is a Converging Point of Multiple Pathological Pathways in Amyotrophic Lateral Sclerosis. J. Alzheimer’s Dis. 2010, 20, S311–S324. [Google Scholar] [CrossRef] [PubMed]
- Barrett, E.F.; Barrett, J.N.; David, G. Dysfunctional mitochondrial Ca2+ handling in mutant SOD1 mouse models of fALS: Integration of findings from motor neuron somata and motor terminals. Front. Cell. Neurosci. 2014, 8, 184. [Google Scholar] [CrossRef] [PubMed]
- Villegas, R.; Martinez, N.W.; Lillo, J.; Pihan, P.; Hernandez, D.; Twiss, J.L.; Court, F.A. Calcium Release from Intra-Axonal Endoplasmic Reticulum Leads to Axon Degeneration through Mitochondrial Dysfunction. J. Neurosci. 2014, 34, 7179–7189. [Google Scholar] [CrossRef] [PubMed]
- Talbot, J.D.; Barrett, J.N.; Nonner, D.; Zhang, Z.; Wicomb, K.; Barrett, E.F. Preservation of neuromuscular function in symptomatic SOD1-G93A mice by peripheral infusion of methylene blue. Exp. Neurol. 2016, 285, 96–107. [Google Scholar] [CrossRef]
- Dupuis, L.; Gonzalez de Aguilar, J.-L.; Echaniz-Laguna, A.; Eschbach, J.; Rene, F.; Oudart, H.; Halter, B.; Huze, C.; Schaeffer, L.; Bouillaud, F.; et al. Muscle mitochondrial uncoupling dismantles neuromuscular junction and triggers distal degeneration of motor neurons. PLoS ONE 2009, 4, e5390. [Google Scholar] [CrossRef]
- Mohajeri, M.H.; Figlewicz, D.A.; Bohn, M.C. Intramuscular Grafts of Myoblasts Genetically Modified to Secrete Glial Cell Line-Derived Neurotrophic Factor Prevent Motoneuron Loss and Disease Progression in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. Hum. Gene Ther. 1999, 10, 1853–1866. [Google Scholar] [CrossRef]
- Acsadi, G.; Anguelov, R.A.; Yang, H.; Toth, G.; Thomas, R.; Jani, A.; Wang, Y.; Ianakova, E.; Mohammad, S.; Lewis, R.A.; et al. Increased survival and function of SOD1 mice after glial cell-derived neurotrophic factor gene therapy. Hum. Gene Ther. 2002, 13, 1047–1059. [Google Scholar] [CrossRef]
- Kaspar, B.K. Retrograde Viral Delivery of IGF-1 Prolongs Survival in a Mouse ALS Model. Science 2003, 301, 839–842. [Google Scholar] [CrossRef]
- Rabinovsky, E.D.; Gelir, E.; Gelir, S.; Lui, H.; Kattash, M.; Demayo, F.J.; Shenaq, S.M.; Schwartz, R.J. Targeted expression of IGF-1 transgene to skeletal muscle accelerates muscle and motor neuron regeneration. FASEB J. 2003, 17, 53–55. [Google Scholar] [CrossRef] [PubMed]
- Dobrowolny, G.; Giacinti, C.; Pelosi, L.; Nicoletti, C.; Winn, N.; Barberi, L.; Molinaro, M.; Rosenthal, N.; Musarò, A. Muscle expression of a local Igf-1 isoform protects motor neurons in an ALS mouse model. J. Cell Biol. 2005, 168, 193–199. [Google Scholar] [CrossRef] [PubMed]
- Ozdinler, P.H.; Macklis, J.D. IGF-I specifically enhances axon outgrowth of corticospinal motor neurons. Nat. Neurosci. 2006, 9, 1371–1381. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Li, Y.; Yu, S.; Du, W.; Zhang, L.; Dai, Y.; Liu, Y.; Li, N. Expression of insulin-like growth factors systems in cloned cattle dead within hours after birth. Mol. Reprod. Dev. 2007, 74, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Musarò, A.; McCullagh, K.; Paul, A.; Houghton, L.; Dobrowolny, G.; Molinaro, M.; Barton, E.R.; L Sweeney, H.; Rosenthal, N. Localized Igf-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 2001, 27, 195–200. [Google Scholar] [CrossRef]
- Sakowski, S.A.; Schuyler, A.D.; Feldman, E.L. Insulin-like growth factor-I for the treatment of amyotrophic lateral sclerosis. Amyotroph. Lateral Scler. 2009, 10, 63–73. [Google Scholar] [CrossRef][Green Version]
- Musarò, A.; Dobrowolny, G.; Cambieri, C.; Onesti, E.; Ceccanti, M.; Frasca, V.; Pisano, A.; Cerbelli, B.; Lepore, E.; Ruffolo, G.; et al. Neuromuscular magnetic stimulation counteracts muscle decline in ALS patients: Results of a randomized, double-blind, controlled study. Sci. Rep. 2019, 9, 2837. [Google Scholar] [CrossRef]
- Witzemann, V.; Brenner, H.R.; Sakmann, B. Neural factors regulate AChR subunit mRNAs at rat neuromuscular synapses. J. Cell Biol. 1991, 114, 125–141. [Google Scholar] [CrossRef]
- Sengupta-Ghosh, A.; Dominguez, S.L.; Xie, L.; Barck, K.H.; Jiang, Z.; Earr, T.; Imperio, J.; Phu, L.; Budayeva, H.G.; Kirkpatrick, D.S.; et al. Muscle specific kinase (MuSK) activation preserves neuromuscular junctions in the diaphragm but is not sufficient to provide a functional benefit in the SOD1G93A mouse model of ALS. Neurobiol. Dis. 2019, 124, 340–352. [Google Scholar] [CrossRef]
- Da Cruz, S.; Parone, P.A.; Lopes, V.S.; Lillo, C.; McAlonis-Downes, M.; Lee, S.K.; Vetto, A.P.; Petrosyan, S.; Marsala, M.; Murphy, A.N.; et al. Elevated PGC-1α Activity Sustains Mitochondrial Biogenesis and Muscle Function without Extending Survival in a Mouse Model of Inherited ALS. Cell Metab. 2012, 15, 778–786. [Google Scholar] [CrossRef]
- Arnold, A.-S.; Gill, J.; Christe, M.; Ruiz, R.; McGuirk, S.; St-Pierre, J.; Tabares, L.; Handschin, C. Morphological and functional remodelling of the neuromuscular junction by skeletal muscle PGC-1α. Nat. Commun. 2014, 5, 3569. [Google Scholar] [CrossRef] [PubMed]
- Willadt, S.; Nash, M.; Slater, C. Age-related changes in the structure and function of mammalian neuromuscular junctions. Ann. N. Y. Acad. Sci. 2018, 1412, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Rosenheimer, J.L. Ultraterminal sprouting in innervated and partially denervated adult and aged rat muscle. Neuroscience 1990, 38, 763–770. [Google Scholar] [CrossRef]
- Prakash, Y.S.; Sieck, G.C. Age-related remodeling of neuromuscular junctions on type-identified diaphragm fibers. Muscle Nerve 1998, 21, 887–895. [Google Scholar] [CrossRef]
- Suzuki, T.; Maruyama, A.; Sugiura, T.; Machida, S.; Miyata, H. Age-related changes in two- and three-dimensional morphology of type-identified endplates in the rat diaphragm. J. Physiol. Sci. JPS 2009, 59, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Rudolf, R.; Khan, M.M.; Labeit, S.; Deschenes, M.R. Degeneration of Neuromuscular Junction in Age and Dystrophy. Front. Aging Neurosci. 2014, 6. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Lee, Y.L.; Thompson, W.J. Changes in aging mouse neuromuscular junctions are explained by degeneration and regeneration of muscle fiber segments at the synapse. J. Neurosci. 2011, 31, 14910–14919. [Google Scholar] [CrossRef]
- Courtney, J.; Steinbach, J.H. Age changes in neuromuscular junction morphology and acetylcholine receptor distribution on rat skeletal muscle fibres. J. Physiol. 1981, 320, 435–447. [Google Scholar] [CrossRef]
- Pestronk, A.; Drachman, D.B.; Griffin, J.W. Effects of aging on nerve sprouting and regeneration. Exp. Neurol. 1980, 70, 65–82. [Google Scholar] [CrossRef]
- Fahim, M.A.; Robbins, N. Ultrastructural studies of young and old mouse neuromuscular junctions. J. Neurocytol. 1982, 11, 641–656. [Google Scholar] [CrossRef]
- Banker, B.Q.; Kelly, S.S.; Robbins, N. Neuromuscular transmission and correlative morphology in young and old mice. J. Physiol. 1983, 339, 355–377. [Google Scholar] [CrossRef] [PubMed]
- Fahim, M.A.; Holley, J.A.; Robbins, N. Scanning and light microscopic study of age changes at a neuromuscular junction in the mouse. J. Neurocytol. 1983, 12, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Andonian, M.H.; Fahim, M.A. Nerve terminal morphology in C57BL/6NNia mice at different ages. J. Gerontol. 1989, 44, B43–B51. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Thompson, W.J. Nerve terminal growth remodels neuromuscular synapses in mice following regeneration of the postsynaptic muscle fiber. J. Neurosci. 2011, 31, 13191–13203. [Google Scholar] [CrossRef]
- Andonian, M.H.; Fahim, M.A. Effects of endurance exercise on the morphology of mouse neuromuscular junctions during ageing. J. Neurocytol. 1987, 16, 589–599. [Google Scholar] [CrossRef]
- Wokke, J.H.; Jennekens, F.G.; van den Oord, C.J.; Veldman, H.; Smit, L.M.; Leppink, G.J. Morphological changes in the human end plate with age. J. Neurol. Sci. 1990, 95, 291–310. [Google Scholar] [CrossRef]
- Rocha, M.C.; Pousinha, P.A.; Correia, A.M.; Sebastião, A.M.; Ribeiro, J.A. Early Changes of Neuromuscular Transmission in the SOD1(G93A) Mice Model of ALS Start Long before Motor Symptoms Onset. PLoS ONE 2013, 8, e73846. [Google Scholar] [CrossRef]
- Tintignac, L.A.; Brenner, H.-R.; Rüegg, M.A. Mechanisms Regulating Neuromuscular Junction Development and Function and Causes of Muscle Wasting. Physiol. Rev. 2015, 95, 809–852. [Google Scholar] [CrossRef]
- Tomlinson, B.E.; Irving, D.; Rebeiz, J.J. Total numbers of limb motor neurones in the human lumbosacral cord and an analysis of the accuracy of various sampling procedures. J. Neurol. Sci. 1973, 20, 313–327. [Google Scholar] [CrossRef]
- Brown, M.C.; Holland, R.L.; Hopkins, W.G. Motor Nerve Sprouting. Annu. Rev. Neurosci. 1981, 4, 17–42. [Google Scholar] [CrossRef]
- Son, Y.J.; Trachtenberg, J.T.; Thompson, W.J. Schwann cells induce and guide sprouting and reinnervation of neuromuscular junctions. Trends Neurosci. 1996, 19, 280–285. [Google Scholar] [CrossRef]
- Bolliger, M.F.; Zurlinden, A.; Luscher, D.; Butikofer, L.; Shakhova, O.; Francolini, M.; Kozlov, S.V.; Cinelli, P.; Stephan, A.; Kistler, A.D.; et al. Specific proteolytic cleavage of agrin regulates maturation of the neuromuscular junction. J. Cell Sci. 2010. [Google Scholar] [CrossRef] [PubMed]
- Bütikofer, L.; Zurlinden, A.; Bolliger, M.F.; Kunz, B.; Sonderegger, P. Destabilization of the neuromuscular junction by proteolytic cleavage of agrin results in precocious sarcopenia. FASEB J. 2011. [Google Scholar] [CrossRef] [PubMed]
- Hettwer, S.; Lin, S.; Kucsera, S.; Haubitz, M.; Oliveri, F.; Fariello, R.G.; Ruegg, M.A.; Vrijbloed, J.W. Injection of a soluble fragment of neural agrin (NT-1654) considerably improves the muscle pathology caused by the disassembly of the neuromuscular junction. PLoS ONE 2014, 9, e88739. [Google Scholar] [CrossRef] [PubMed]
- Lozano, R.; Gilmore, K.J.; Thompson, B.C.; Stewart, E.M.; Waters, A.M.; Romero-Ortega, M.; Wallace, G.G. Electrical stimulation enhances the acetylcholine receptors available for neuromuscular junction formation. Acta Biomater. 2016. [Google Scholar] [CrossRef] [PubMed]
- Senger, J.L.; Chan, K.M.; Macandili, H.; Chan, A.W.M.; Verge, V.M.K.; Jones, K.E.; Webber, C.A. Conditioning electrical stimulation promotes functional nerve regeneration. Exp. Neurol. 2019. [Google Scholar] [CrossRef]
- Potes, Y.; Pérez-Martinez, Z.; Bermejo-Millo, J.C.; Rubio-Gonzalez, A.; Fernandez-Fernández, M.; Bermudez, M.; Arche, J.M.; Solano, J.J.; Boga, J.A.; Oliván, M.; et al. Overweight in the Elderly Induces a Switch in Energy Metabolism that Undermines Muscle Integrity. Aging Dis. 2019, 10, 217. [Google Scholar] [CrossRef] [PubMed]
- Mosole, S.; Zampieri, S.; Furlan, S.; Carraro, U.; Löefler, S.; Kern, H.; Volpe, P.; Nori, A. Effects of Electrical Stimulation on Skeletal Muscle of Old Sedentary People. Gerontol. Geriatr. Med. 2018, 4, 2333721418768998. [Google Scholar] [CrossRef]
- Kern, H.; Carraro, U.; Adami, N.; Biral, D.; Hofer, C.; Forstner, C.; Mödlin, M.; Vogelauer, M.; Pond, A.; Boncompagni, S.; et al. Home-Based Functional Electrical Stimulation Rescues Permanently Denervated Muscles in Paraplegic Patients with Complete Lower Motor Neuron Lesion. Neurorehabilit. Neural Repair 2010, 24, 709–721. [Google Scholar] [CrossRef]
- Maddocks, M.; Halliday, V.; Chauhan, A.; Taylor, V.; Nelson, A.; Sampson, C.; Byrne, A.; Griffiths, G.; Wilcock, A. Neuromuscular Electrical Stimulation of the Quadriceps in Patients with Non-Small Cell Lung Cancer Receiving Palliative Chemotherapy: A Randomized Phase II Study. PLoS ONE 2013, 8, e86059. [Google Scholar] [CrossRef]
- Gonzalez-Freire, M.; Adelnia, F.; Moaddel, R.; Ferrucci, L. Searching for a mitochondrial root to the decline in muscle function with ageing. J. Cachexia Sarcopenia Muscle 2018, 9, 435–440. [Google Scholar] [CrossRef] [PubMed]
- Zangarelli, A.; Chanseaume, E.; Morio, B.; Brugère, C.; Mosoni, L.; Rousset, P.; Giraudet, C.; Patrac, V.; Gachon, P.; Boirie, Y.; et al. Synergistic effects of caloric restriction with maintained protein intake on skeletal muscle performance in 21-month-old rats: A mitochondria-mediated pathway. FASEB J. 2006, 20, 2439–2450. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Bourdel-Marchasson, I.; Rouland, R.; Calmettes, G.; Biran, M.; Deschodt-Arsac, V.; Miraux, S.; Thiaudiere, E.; Pasdois, P.; Detaille, D.; et al. Mitochondrial energetics is impaired in vivo in aged skeletal muscle. Aging Cell 2014, 13, 39–48. [Google Scholar] [CrossRef] [PubMed]
- Gouspillou, G.; Bourdel-Marchasson, I.; Rouland, R.; Calmettes, G.; Franconi, J.-M.; Deschodt-Arsac, V.; Diolez, P. Alteration of mitochondrial oxidative phosphorylation in aged skeletal muscle involves modification of adenine nucleotide translocator. BBA Bioenerg. 2010, 1797, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Short, K.R.; Bigelow, M.L.; Kahl, J.; Singh, R.; Coenen-Schimke, J.; Raghavakaimal, S.; Nair, K.S. Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. USA 2005, 102, 5618–5623. [Google Scholar] [CrossRef]
- Drew, B.; Phaneuf, S.; Dirks, A.; Selman, C.; Gredilla, R.; Lezza, A.; Barja, G.; Leeuwenburgh, C. Effects of aging and caloric restriction on mitochondrial energy production in gastrocnemius muscle and heart. Am. J. Physiol. Integr. Comp. Physiol. 2003, 284, R474–R480. [Google Scholar] [CrossRef] [PubMed]
- Chen Scarabelli, C.; McCauley, R.B.; Yuan, Z.; Di Rezze, J.; Patel, D.; Putt, J.; Raddino, R.; Allebban, Z.; Abboud, J.; Scarabelli, G.M.; et al. Oral Administration of Amino Acidic Supplements Improves Protein and Energy Profiles in Skeletal Muscle of Aged Rats: Elongation of Functional Performance and Acceleration of Mitochondrial Recovery in Adenosine Triphosphate After Exhaustive Exertion. Am. J. Cardiol. 2008, 101, S42–S48. [Google Scholar] [CrossRef]
- Mansouri, A.; Muller, F.L.; Liu, Y.; Ng, R.; Faulkner, J.; Hamilton, M.; Richardson, A.; Huang, T.-T.; Epstein, C.J.; Van Remmen, H. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech. Ageing Dev. 2006, 127, 298–306. [Google Scholar] [CrossRef]
- Jackson, M.J.; McArdle, A. Age-related changes in skeletal muscle reactive oxygen species generation and adaptive responses to reactive oxygen species. J. Physiol. 2011, 589, 2139–2145. [Google Scholar] [CrossRef]
- Chapman, I.M. The Anorexia of Aging. Clin. Geriatr. Med. 2007, 23, 735–756. [Google Scholar] [CrossRef]
- Thalacker-Mercer, A.E.; Fleet, J.C.; Craig, B.A.; Carnell, N.S.; Campbell, W.W. Inadequate protein intake affects skeletal muscle transcript profiles in older humans. Am. J. Clin. Nutr. 2007, 85, 1344–1352. [Google Scholar] [CrossRef] [PubMed]
- Mosole, S.; Carraro, U.; Kern, H.; Loefler, S.; Fruhmann, H.; Vogelauer, M.; Burggraf, S.; Mayr, W.; Krenn, M.; Paternostro-Sluga, T.; et al. Long-Term High-Level Exercise Promotes Muscle Reinnervation with Age. J. Neuropathol. Exp. Neurol. 2014, 73, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Zampieri, S.; Mammucari, C.; Romanello, V.; Barberi, L.; Pietrangelo, L.; Fusella, A.; Mosole, S.; Gherardi, G.; Höfer, C.; Löfler, S.; et al. Physical exercise in aging human skeletal muscle increases mitochondrial calcium uniporter expression levels and affects mitochondria dynamics. Physiol. Rep. 2016, 4, e13005. [Google Scholar] [CrossRef] [PubMed]
- Fahim, M.A. Endurance exercise modulates neuromuscular junction of C57BL/6NNia aging mice. J. Appl. Physiol. 1997, 83, 59–66. [Google Scholar] [CrossRef] [PubMed]
- Giudice, J.; Taylor, J.M. Muscle as a paracrine and endocrine organ. Curr. Opin. Pharmacol. 2017, 34, 49–55. [Google Scholar] [CrossRef] [PubMed]
- Handschin, C.; Kobayashi, Y.M.; Chin, S.; Seale, P.; Campbell, K.P.; Spiegelman, B.M. PGC-1alpha regulates the neuromuscular junction program and ameliorates Duchenne muscular dystrophy. Genes Dev. 2007, 21, 770–783. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Dobrowolny, G.; Sica, G.; Musarò, A. Molecular Insights into Muscle Homeostasis, Atrophy and Wasting. Curr. Genom. 2018, 19, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Rizzuto, E.; Musarò, A. Counteracting muscle wasting in aging and neuromuscular diseases: The critical role of IGF-1. Aging 2009, 1, 451–457. [Google Scholar] [CrossRef]
- Sandri, M.; Sandri, C.; Gilbert, A.; Skurk, C.; Calabria, E.; Picard, A.; Walsh, K.; Schiaffino, S.; Lecker, S.H.; Goldberg, A.L. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004, 117, 399–412. [Google Scholar] [CrossRef]
- Ascenzi, F.; Barberi, L.; Dobrowolny, G.; Villa Nova Bacurau, A.; Nicoletti, C.; Rizzuto, E.; Rosenthal, N.; Scicchitano, B.M.; Musarò, A. Effects of IGF-1 isoforms on muscle growth and sarcopenia. Aging Cell 2019, 18, e12954. [Google Scholar] [CrossRef] [PubMed]
- Saheb-Al-Zamani, M.; Yan, Y.; Farber, S.J.; Hunter, D.A.; Newton, P.; Wood, M.D.; Stewart, S.A.; Johnson, P.J.; Mackinnon, S.E. Limited regeneration in long acellular nerve allografts is associated with increased Schwann cell senescence. Exp. Neurol. 2013, 247, 165–177. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).