Genome-Wide Identification and Characterization of Long Noncoding RNAs of Brown to White Adipose Tissue Transformation in Goats

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Ethics Statement
2.2. Animal and Sample Collection
2.3. Histology Analysis
2.4. Immunohistochemistry
2.5. RNA Extraction, Library Construction, and Sequencing
2.6. Quality Control and Transcriptome Assembly
2.7. Identification and Expression Analysis of Potential lncRNA
2.8. Target Gene Prediction and Enrichment Analysis
2.9. Quantitative Real-Time PCR
3. Results
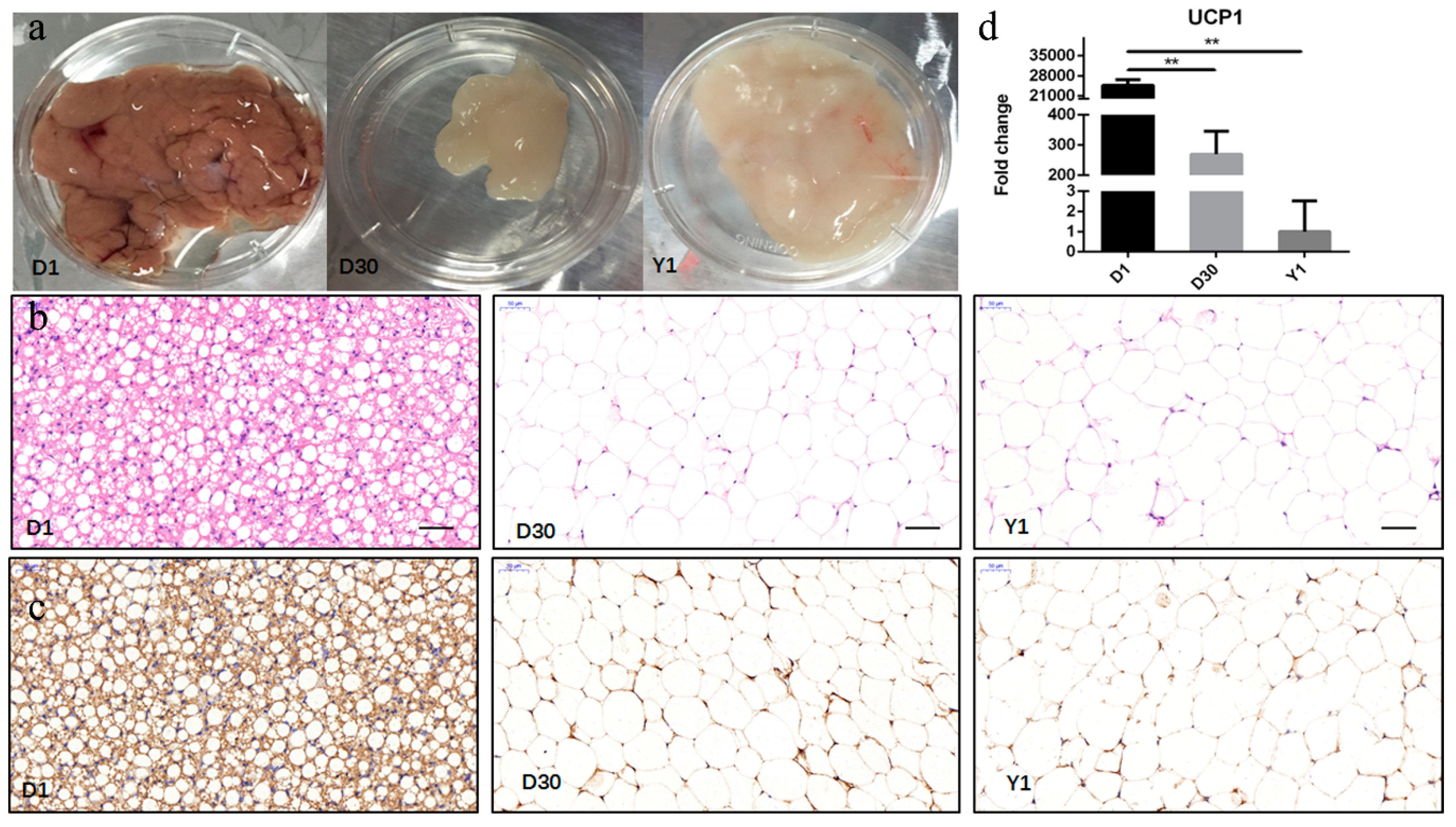
3.1. Characterization of the Transformation from BAT to WAT during Perirenal Fat Development
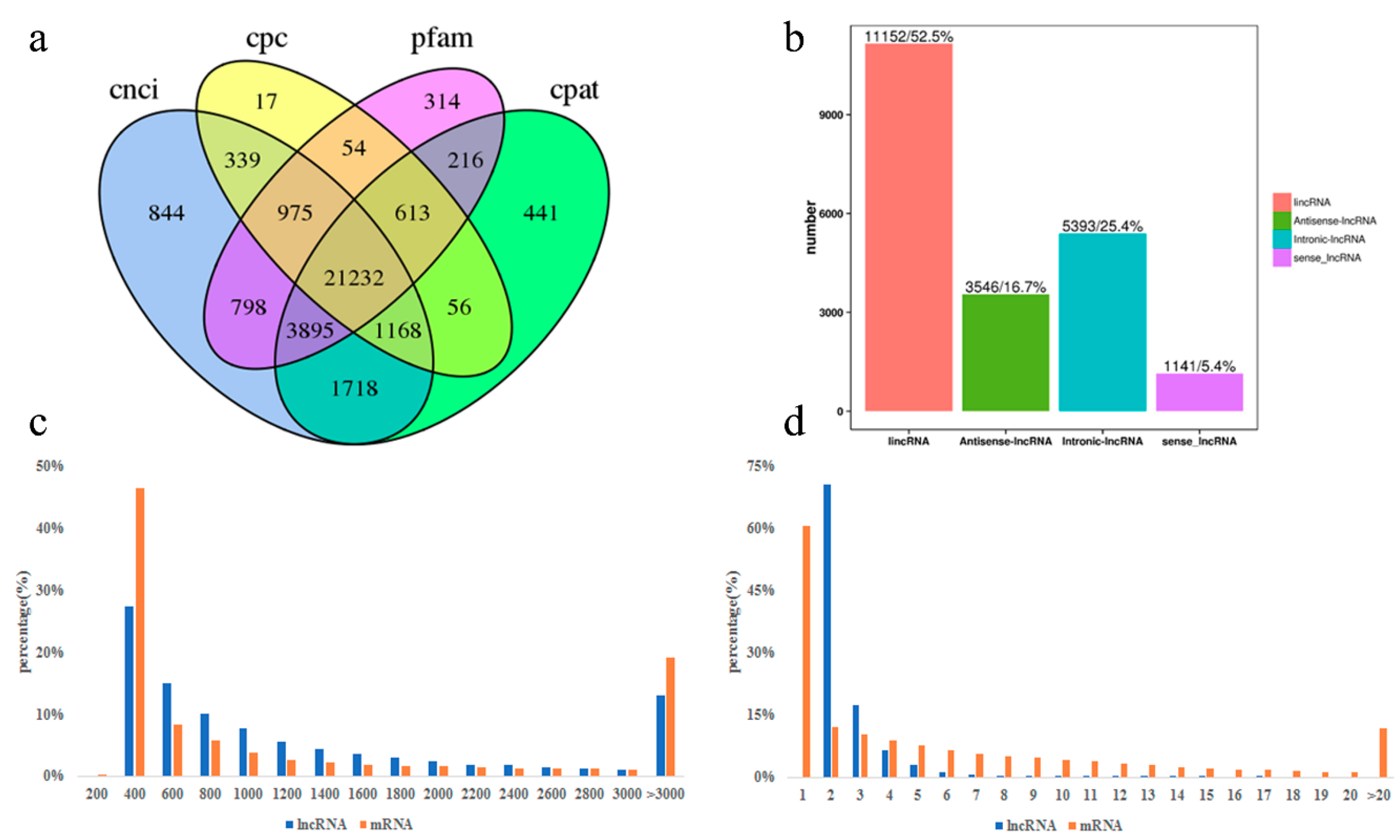
3.2. Identification and Characterization of lncRNA in Perirenal Fat at Three Stages
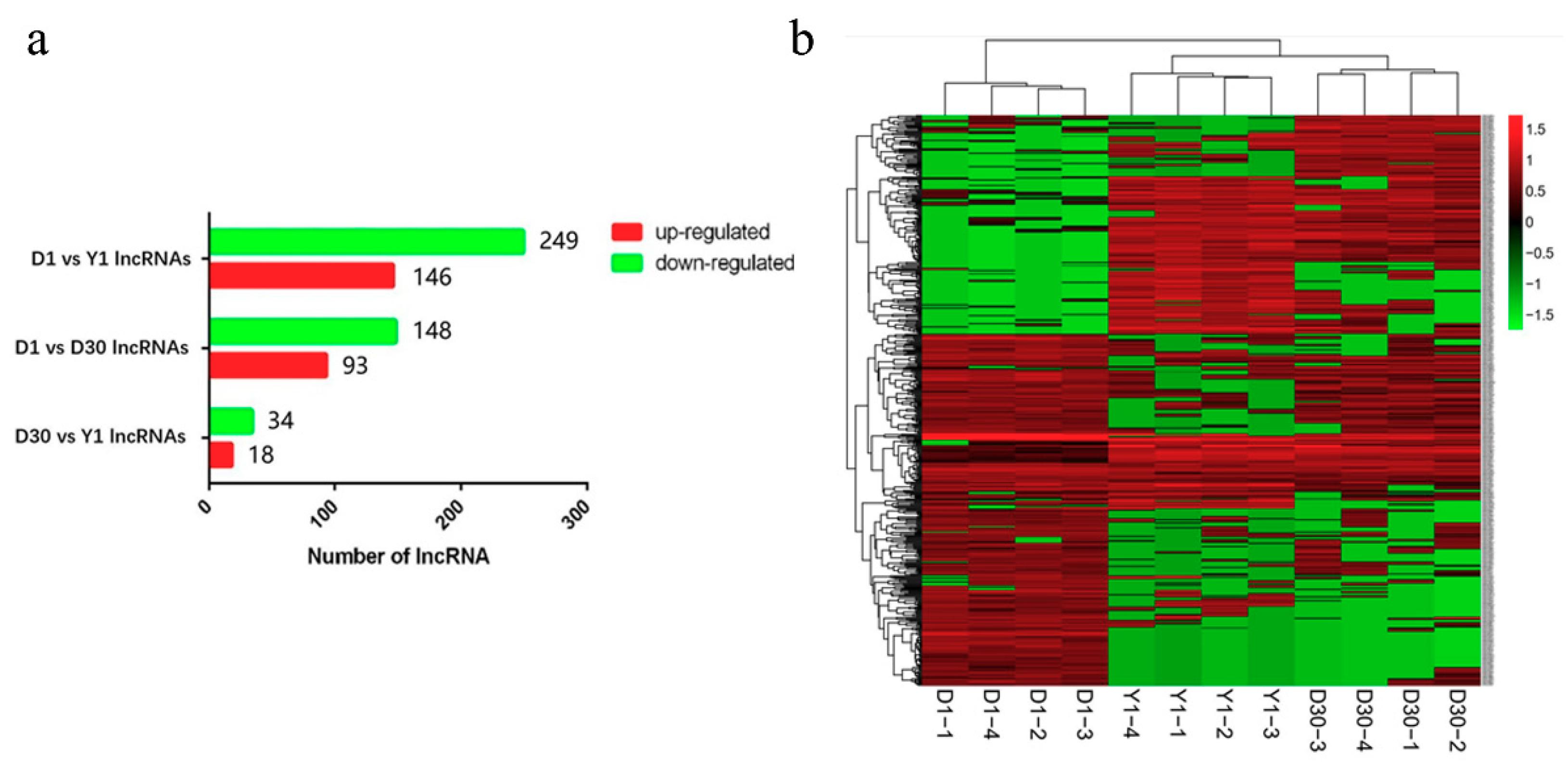
3.3. Identification of Differentially Expressed lncRNAs during the Transformation from Brown to White Adipose Tissue
3.4. The Cis-Regulated Target Genes of lncRNAs
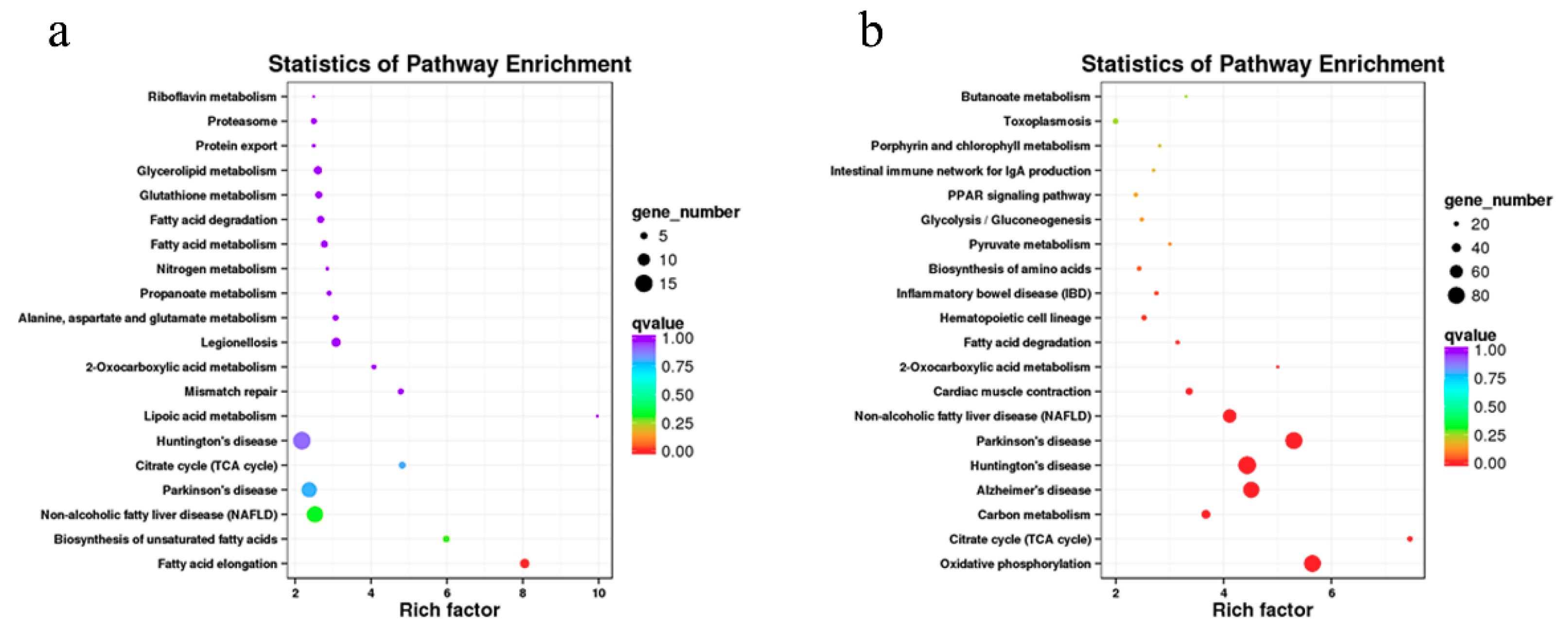
3.5. Enrichment Analysis of Coexpressed Genes of lncRNAs
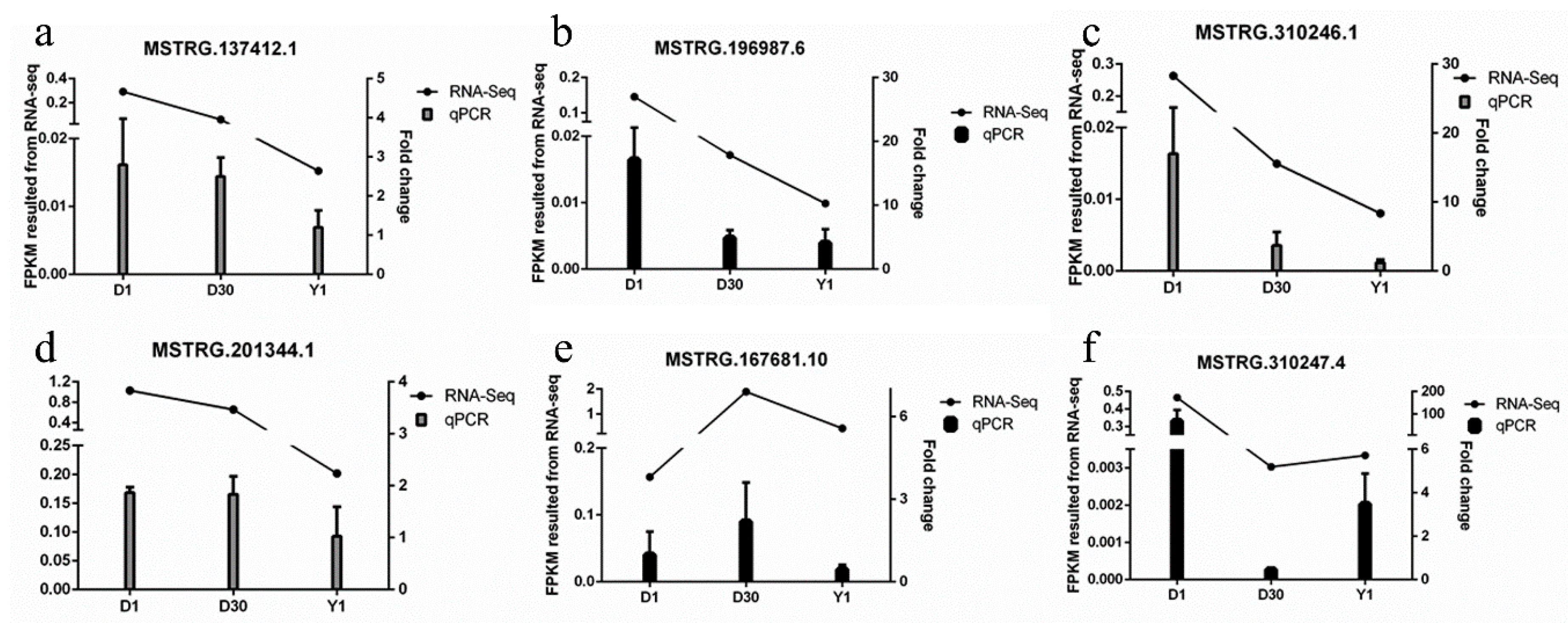
3.6. Validation of lncRNAs by qPCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Peirce, V.; Carobbio, S.; Vidal-Puig, A. The different shades of fat. Nature 2014, 510, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.E.; Pope, M.; Budge, H. The ontogeny of brown adipose tissue. Annu. Rev. Nutr. 2015, 35, 295–320. [Google Scholar] [CrossRef]
- Nedergaard, J.; Golozoubova, V.; Matthias, A.; Asadi, A.; Jacobsson, A.; Cannon, B. Ucp1: The only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochim. Biophys. Acta 2001, 1504, 82–106. [Google Scholar] [CrossRef]
- Ishibashi, J.; Seale, P. Medicine. Beige can be slimming. Science 2010, 328, 1113–1114. [Google Scholar] [CrossRef] [PubMed]
- Kiefer, F.W. Browning and thermogenic programing of adipose tissue. Best Pract. Res. Clin. Endocrinol. Metab. 2016, 30, 479–485. [Google Scholar] [CrossRef] [PubMed]
- Barbatelli, G.; Murano, I.; Madsen, L.; Hao, Q.; Jimenez, M.; Kristiansen, K.; Giacobino, J.P.; De Matteis, R.; Cinti, S. The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E1244–E1253. [Google Scholar] [CrossRef] [Green Version]
- Cao, L.; Choi, E.Y.; Liu, X.; Martin, A.; Wang, C.; Xu, X.; During, M.J. White to brown fat phenotypic switch induced by genetic and environmental activation of a hypothalamic-adipocyte axis. Cell Metab. 2011, 14, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Harms, M.; Seale, P. Brown and beige fat: Development, function and therapeutic potential. Nat. Med. 2013, 19, 1252–1263. [Google Scholar] [CrossRef]
- Cypess, A.M.; White, A.P.; Vernochet, C.; Schulz, T.J.; Xue, R.; Sass, C.A.; Huang, T.L.; Roberts-Toler, C.; Weiner, L.S.; Sze, C.; et al. Anatomical localization, gene expression profiling and functional characterization of adult human neck brown fat. Nat. Med. 2013, 19, 635–639. [Google Scholar] [CrossRef]
- Wu, J.; Bostrom, P.; Sparks, L.M.; Ye, L.; Choi, J.H.; Giang, A.H.; Khandekar, M.; Virtanen, K.A.; Nuutila, P.; Schaart, G.; et al. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 2012, 150, 366–376. [Google Scholar] [CrossRef]
- van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar] [CrossRef] [PubMed]
- Townsend, K.L.; Tseng, Y.H. Of mice and men: Novel insights regarding constitutive and recruitable brown adipocytes. Int. J. Obes. Suppl. 2015, 5, S15–S20. [Google Scholar] [CrossRef] [PubMed]
- Basse, A.L.; Dixen, K.; Yadav, R.; Tygesen, M.P.; Qvortrup, K.; Kristiansen, K.; Quistorff, B.; Gupta, R.; Wang, J.; Hansen, J.B. Global gene expression profiling of brown to white adipose tissue transformation in sheep reveals novel transcriptional components linked to adipose remodeling. BMC Genom. 2015, 16, 215. [Google Scholar] [CrossRef] [PubMed]
- Henry, B.A.; Pope, M.; Birtwistle, M.; Loughnan, R.; Alagal, R.; Fuller-Jackson, J.P.; Perry, V.; Budge, H.; Clarke, I.J.; Symonds, M.E. Ontogeny and thermogenic role for sternal fat in female sheep. Endocrinology 2017, 158, 2212–2225. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.Y.; Lin, J.D. Long noncoding rnas: A new regulatory code in metabolic control. Trends Biochem. Sci. 2015, 40, 586–596. [Google Scholar] [CrossRef] [PubMed]
- Knoll, M.; Lodish, H.F.; Sun, L. Long non-coding rnas as regulators of the endocrine system. Nat. Rev. Endocrinol. 2015, 11, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Kornfeld, J.W.; Bruning, J.C. Regulation of metabolism by long, non-coding rnas. Front. Genet. 2014, 5, 57. [Google Scholar] [CrossRef]
- Huang, Y.; Zheng, Y.; Jin, C.; Li, X.; Jia, L.; Li, W. Long non-coding rna h19 inhibits adipocyte differentiation of bone marrow mesenchymal stem cells through epigenetic modulation of histone deacetylases. Sci. Rep. 2016, 6, 28897. [Google Scholar] [CrossRef]
- Cui, X.; You, L.; Li, Y.; Zhu, L.; Zhang, F.; Xie, K.; Cao, Y.; Ji, C.; Guo, X. A transcribed ultraconserved noncoding rna, uc.417, serves as a negative regulator of brown adipose tissue thermogenesis. FASEB J. 2016, 30, 4301–4312. [Google Scholar] [CrossRef]
- Zhao, X.Y.; Li, S.M.; Wang, G.X.; Yu, Q.; Lin, J.D. A long noncoding rna transcriptional regulatory circuit drives thermogenic adipocyte differentiation. Mol. Cell 2014, 55, 372–382. [Google Scholar] [CrossRef]
- Xiong, Y.; Yue, F.; Jia, Z.H.; Gao, Y.; Jin, W.; Hu, K.P.; Zhang, Y.; Zhu, D.H.; Yang, G.S.; Kuang, S.H. A novel brown adipocyte-enriched long non-coding rna that is required for brown adipocyte differentiation and sufficient to drive thermogenic gene program in white adipocytes. Biochim. Biophys. Acta-Mol. Cell. Biol. Lipids 2018, 1863, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Bai, Z.; Chai, X.R.; Yoon, M.J.; Kim, H.J.; Lo, K.A.; Zhang, Z.C.; Xu, D.; Siang, D.T.C.; Walet, A.C.E.; Xu, S.H.; et al. Dynamic transcriptome changes during adipose tissue energy expenditure reveal critical roles for long noncoding rna regulators. PLoS Biol. 2017, 15, e2002176. [Google Scholar] [CrossRef] [PubMed]
- Alvarez-Dominguez, J.R.; Bai, Z.Q.; Xu, D.; Yuan, B.B.; Lo, K.A.; Yoon, M.J.; Lim, Y.C.; Knoll, M.; Slavov, N.; Chen, S.; et al. De novo reconstruction of adipose tissue transcriptomes reveals long non-coding rna regulators of brown adipocyte development. Cell Metab. 2015, 21, 764–776. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Landmead, B.; Salzberg, S.L. Hisat: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Xie, M.; Jiang, Y.; Xiao, N.Q.; Du, X.Y.; Zhang, W.G.; Tosser-Klopp, G.; Wang, J.H.; Yang, S.; Liang, J.; et al. Sequencing and automated whole-genome optical mapping of the genome of a domestic goat (capra hircus). Nat. Biotechnol. 2013, 31, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Pertea, M.; Pertea, G.M.; Antonescu, C.M.; Chang, T.C.; Mendell, J.T.; Salzberg, S.L. Stringtie enables improved reconstruction of a transcriptome from rna-seq reads. Nat. Biotechnol. 2015, 33, 290–295. [Google Scholar] [CrossRef]
- Kelley, D.; Rinn, J. Transposable elements reveal a stem cell-specific class of long noncoding rnas. Genome Biol. 2012, 13, R107. [Google Scholar] [CrossRef]
- Kong, L.; Zhang, Y.; Ye, Z.Q.; Liu, X.Q.; Zhao, S.Q.; Wei, L.; Gao, G. Cpc: Assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007, 35, W345–W349. [Google Scholar] [CrossRef]
- Wang, L.; Park, H.J.; Dasari, S.; Wang, S.; Kocher, J.P.; Li, W. Cpat: Coding-potential assessment tool using an alignment-free logistic regression model. Nucleic Acids Res. 2013, 41, e74. [Google Scholar] [CrossRef]
- Sun, L.; Luo, H.; Bu, D.; Zhao, G.; Yu, K.; Zhang, C.; Liu, Y.; Chen, R.; Zhao, Y. Utilizing sequence intrinsic composition to classify protein-coding and long non-coding transcripts. Nucleic Acids Res. 2013, 41, e166. [Google Scholar] [CrossRef]
- El-Gebali, S.; Mistry, J.; Bateman, A.; Eddy, S.R.; Luciani, A.; Potter, S.C.; Qureshi, M.; Richardson, L.J.; Salazar, G.A.; Smart, A.; et al. The pfam protein families database in 2019. Nucleic Acids Res. 2019, 47, D427–D432. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by rna-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Anders, S.; Huber, W. Differential expression analysis for sequence count data. Genome Biol. 2010, 11, R106. [Google Scholar] [CrossRef] [PubMed]
- Li, J.W.; Ma, W.; Zeng, P.; Wang, J.Y.; Geng, B.; Yang, J.C.; Cui, Q.H. Lnctar: A tool for predicting the rna targets of long noncoding rnas. Brief. Bioinform. 2015, 16, 806–812. [Google Scholar] [CrossRef] [PubMed]
- Young, M.D.; Wakefield, M.J.; Smyth, G.K.; Oshlack, A. Gene ontology analysis for rna-seq: Accounting for selection bias. Genome Biol. 2010, 11, R14. [Google Scholar] [CrossRef] [PubMed]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. Kobas 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef] [PubMed]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, F.; Hao, G.; Shao, M.; Nham, K.; An, Y.; Wang, Q.; Zhu, Y.; Kusminski, C.M.; Hassan, G.; Gupta, R.K.; et al. An adipose tissue atlas: An image-guided identification of human-like bat and beige depots in rodents. Cell Metab. 2018, 27, 252–262.e3. [Google Scholar] [CrossRef]
- Lidell, M.E. Brown adipose tissue in human infants. Handb. Exp. Pharmacol. 2019, 251, 107–123. [Google Scholar]
- Sun, L.; Goff, L.A.; Trapnell, C.; Alexander, R.; Lo, K.A.; Hacisuleyman, E.; Sauvageau, M.; Tazon-Vega, B.; Kelley, D.R.; Hendrickson, D.G.; et al. Long noncoding rnas regulate adipogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 3387–3392. [Google Scholar] [CrossRef]
- Ding, C.; Lim, Y.C.; Chia, S.Y.; Walet, A.C.E.; Xu, S.; Lo, K.A.; Zhao, Y.; Zhu, D.; Shan, Z.; Chen, Q.; et al. De novo reconstruction of human adipose transcriptome reveals conserved lncrnas as regulators of brown adipogenesis. Nat. Commun. 2018, 9, 1329. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, M.; Jin, Y.Y.; Erdenee, S.; Hu, L.Y.; Chen, H.; Cai, Y.; Lan, X.Y. Comparative transcriptome profiling of mrna and lncrna related to tail adipose tissues of sheep. Front. Genet. 2018, 9, 365. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.H.; Ge, W.; Luo, Z.X.; Guo, Y.; Jiao, B.L.; Qu, L.; Zhang, Z.Y.; Wang, X. Integrated analysis of coding genes and non-coding rnas during hair follicle cycle of cashmere goat (capra hircus). BMC Genom. 2017, 18, 767. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.X.; Ye, J.; Yang, C.; Zhang, K.F.; Li, X.M.; Luo, L.; Ding, J.P.; Li, Y.S.; Cao, H.G.; Ling, Y.H.; et al. Screening and evaluating of long noncoding rnas in the puberty of goats. BMC Genom. 2017, 18, 164. [Google Scholar] [CrossRef] [PubMed]
- Zhan, S.Y.; Dong, Y.; Zhao, W.; Guo, J.Z.; Zhong, T.; Wang, L.J.; Li, L.; Zhang, H.P. Genome-wide identification and characterization of long non-coding rnas in developmental skeletal muscle of fetal goat. BMC Genom. 2016, 17, 666. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Trapnell, C.; Goff, L.; Koziol, M.; Tazon-Vega, B.; Regev, A.; Rinn, J.L. Integrative annotation of human large intergenic noncoding rnas reveals global properties and specific subclasses. Genes Dev. 2011, 25, 1915–1927. [Google Scholar] [CrossRef] [PubMed]
- Derrien, T.; Johnson, R.; Bussotti, G.; Tanzer, A.; Djebali, S.; Tilgner, H.; Guernec, G.; Martin, D.; Merkel, A.; Knowles, D.G.; et al. The gencode v7 catalog of human long noncoding rnas: Analysis of their gene structure, evolution, and expression. Genome Res. 2012, 22, 1775–1789. [Google Scholar] [CrossRef] [PubMed]
- Weng, B.; Ran, M.; Chen, B.; He, C.; Dong, L.; Peng, F. Genome-wide analysis of long non-coding rnas and their role in postnatal porcine testis development. Genomics 2017, 109, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mendell, J.T. Functional classification and experimental dissection of long noncoding rnas. Cell 2018, 172, 393–407. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.B.; Du, Y.; Zhang, C.H.; Sun, C.; He, Y.L.; Wu, Y.H.; Liu, J.X.; Luo, J.; Loor, J.J. Fatty acid elongase 5 (elovl5) alters the synthesis of long-chain unsaturated fatty acids in goat mammary epithelial cells. J. Dairy Sci. 2018, 101, 4586–4594. [Google Scholar] [CrossRef] [PubMed]
- Boerman, J.P.; Lock, A.L. Effect of unsaturated fatty acids and triglycerides from soybeans on milk fat synthesis and biohydrogenation intermediates in dairy cattle. J. Dairy Sci. 2014, 97, 7031–7042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sain, J.; Gonzalez, M.A.; Lavandera, J.V.; Scalerandi, M.V.; Bernal, C.A. The effects of trans-fatty acids on tag regulation in mice depend on dietary unsaturated fatty acids. Br. J. Nutr. 2016, 116, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Hodson, L.; Rosqvist, F.; Parry, S.A. The influence of dietary fatty acids on liver fat content and metabolism. Proc. Nutr. Soc. 2019, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gnoni, A.; Giudetti, A.M. Dietary long-chain unsaturated fatty acids acutely and differently reduce the activities of lipogenic enzymes and of citrate carrier in rat liver. J. Physiol. Biochem. 2016, 72, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Chang, J.S.; Chen, C.L.; Yeh, C.L.; Chien, Y.W. Effects of different amounts and types of dietary fatty acids on the body weight, fat accumulation, and lipid metabolism in hamsters. Nutrition 2016, 32, 601–608. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, M.T.; Yudell, B.E.; Loor, J.J. Regulation of energy metabolism by long-chain fatty acids. Prog. Lipid Res. 2014, 53, 124–144. [Google Scholar] [CrossRef] [PubMed]
- Shabalina, I.G.; Kalinovich, A.V.; Cannon, B.; Nedergaard, J. Metabolically inert perfluorinated fatty acids directly activate uncoupling protein 1 in brown-fat mitochondria. Arch. Toxicol. 2016, 90, 1117–1128. [Google Scholar] [CrossRef]
- Maeyashiki, C.; Oshima, S.; Otsubo, K.; Kobayashi, M.; Nibe, Y.; Matsuzawa, Y.; Onizawa, M.; Nemoto, Y.; Nagaishi, T.; Okamoto, R.; et al. Hadha, the alpha subunit of the mitochondrial trifunctional protein, is involved in long-chain fatty acid-induced autophagy in intestinal epithelial cells. Biochem. Biophys. Res. Commun. 2017, 484, 636–641. [Google Scholar] [CrossRef]
- Zhou, Z.; Zhou, J.; Du, Y. Estrogen receptor alpha interacts with mitochondrial protein hadhb and affects beta-oxidation activity. Mol. Cell. Proteom. 2012, 11, M111.011056. [Google Scholar] [CrossRef]
- Briant, L.J.B.; Dodd, M.S.; Chibalina, M.V.; Rorsman, N.J.G.; Johnson, P.R.V.; Carmeliet, P.; Rorsman, P.; Knudsen, J.G. Cpt1a-dependent long-chain fatty acid oxidation contributes to maintaining glucagon secretion from pancreatic islets. Cell Rep. 2018, 23, 3300–3311. [Google Scholar] [CrossRef]
- Dai, J.; Liang, K.; Zhao, S.; Jia, W.; Liu, Y.; Wu, H.; Lv, J.; Cao, C.; Chen, T.; Zhuang, S.; et al. Chemoproteomics reveals baicalin activates hepatic cpt1 to ameliorate diet-induced obesity and hepatic steatosis. Proc. Natl. Acad. Sci. USA 2018, 115, E5896–E5905. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Cheng, K.K. Hypothalamic ampk as a mediator of hormonal regulation of energy balance. Int. J. Mol. Sci. 2018, 19, 3552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Ai, W.; Hu, X.; Meng, Y.; Yuan, C.; Su, H.; Wang, L.; Zhu, X.; Gao, P.; Shu, G.; et al. Phytol stimulates the browning of white adipocytes through the activation of amp-activated protein kinase (ampk) alpha in mice fed high-fat diet. Food Funct. 2018, 9, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.Y.; Zhang, L.N.; Li, B.H.; Jiang, H.W.; Duan, Y.N.; Xie, Z.F.; Shuai, L.; Li, J.; Li, J.Y. Amp-activated protein kinase (ampk) regulates energy metabolism through modulating thermogenesis in adipose tissue. Front. Physiol. 2018, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.X.; Yang, Q.Y.; Zhang, L.P.; Liang, X.W.; Sun, X.F.; Wang, B.; Chen, Y.T.; Zhu, M.J.; Du, M. Ampk alpha 1 deficiency suppresses brown adipogenesis in favor of fibrogenesis during brown adipose tissue development. Biochem. Biophys. Res. Commun. 2017, 491, 508–514. [Google Scholar] [CrossRef]
- Mottillo, E.P.; Desjardins, E.M.; Crane, J.D.; Smith, B.K.; Green, A.E.; Ducommun, S.; Henriksen, T.I.; Rebalka, I.A.; Razi, A.; Sakamoto, K.; et al. Lack of adipocyte ampk exacerbates insulin resistance and hepatic steatosis through brown and beige adipose tissue function. Cell Metab. 2016, 24, 118–129. [Google Scholar] [CrossRef] [PubMed]
- Neupane, P.; Bhuju, S.; Thapa, N.; Bhattarai, H.K. Atp synthase: Structure, function and inhibition. Biomol. Concepts 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Dabritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470 e413. [Google Scholar] [CrossRef]
- Gowthami, N.; Sunitha, B.; Kumar, M.; Keshava Prasad, T.S.; Gayathri, N.; Padmanabhan, B.; Srinivas Bharath, M.M. Mapping the protein phosphorylation sites in human mitochondrial complex i (nadh: Ubiquinone oxidoreductase): A bioinformatics study with implications for brain aging and neurodegeneration. J. Chem. Neuroanat. 2019, 95, 13–28. [Google Scholar] [CrossRef]
- Govatati, S.; Deenadayal, M.; Shivaji, S.; Bhanoori, M. Mitochondrial nadh:Ubiquinone oxidoreductase alterations are associated with endometriosis. Mitochondrion 2013, 13, 782–790. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Yang, X.; Zhu, Y.; Zhan, S.; Chao, Z.; Zhong, T.; Guo, J.; Wang, Y.; Li, L.; Zhang, H. Genome-Wide Identification and Characterization of Long Noncoding RNAs of Brown to White Adipose Tissue Transformation in Goats. Cells 2019, 8, 904. https://doi.org/10.3390/cells8080904
Wang L, Yang X, Zhu Y, Zhan S, Chao Z, Zhong T, Guo J, Wang Y, Li L, Zhang H. Genome-Wide Identification and Characterization of Long Noncoding RNAs of Brown to White Adipose Tissue Transformation in Goats. Cells. 2019; 8(8):904. https://doi.org/10.3390/cells8080904
Chicago/Turabian StyleWang, Linjie, Xin Yang, Yuehua Zhu, Siyuan Zhan, Zhe Chao, Tao Zhong, Jiazhong Guo, Yan Wang, Li Li, and Hongping Zhang. 2019. "Genome-Wide Identification and Characterization of Long Noncoding RNAs of Brown to White Adipose Tissue Transformation in Goats" Cells 8, no. 8: 904. https://doi.org/10.3390/cells8080904