Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation



Abstract
1. Introduction
1.1. NAD+ in Redox Reactions
1.2. Cellular NAD+ Pools
1.3. NAD+ Biogenesis/Synthesis
1.4. NAD+ Signaling
1.5. ADP-Ribosylation and Carbohydrate Metabolism
2. Cytoplasmic Crosstalk of ADP-Ribosylation and the Carbohydrate Metabolism
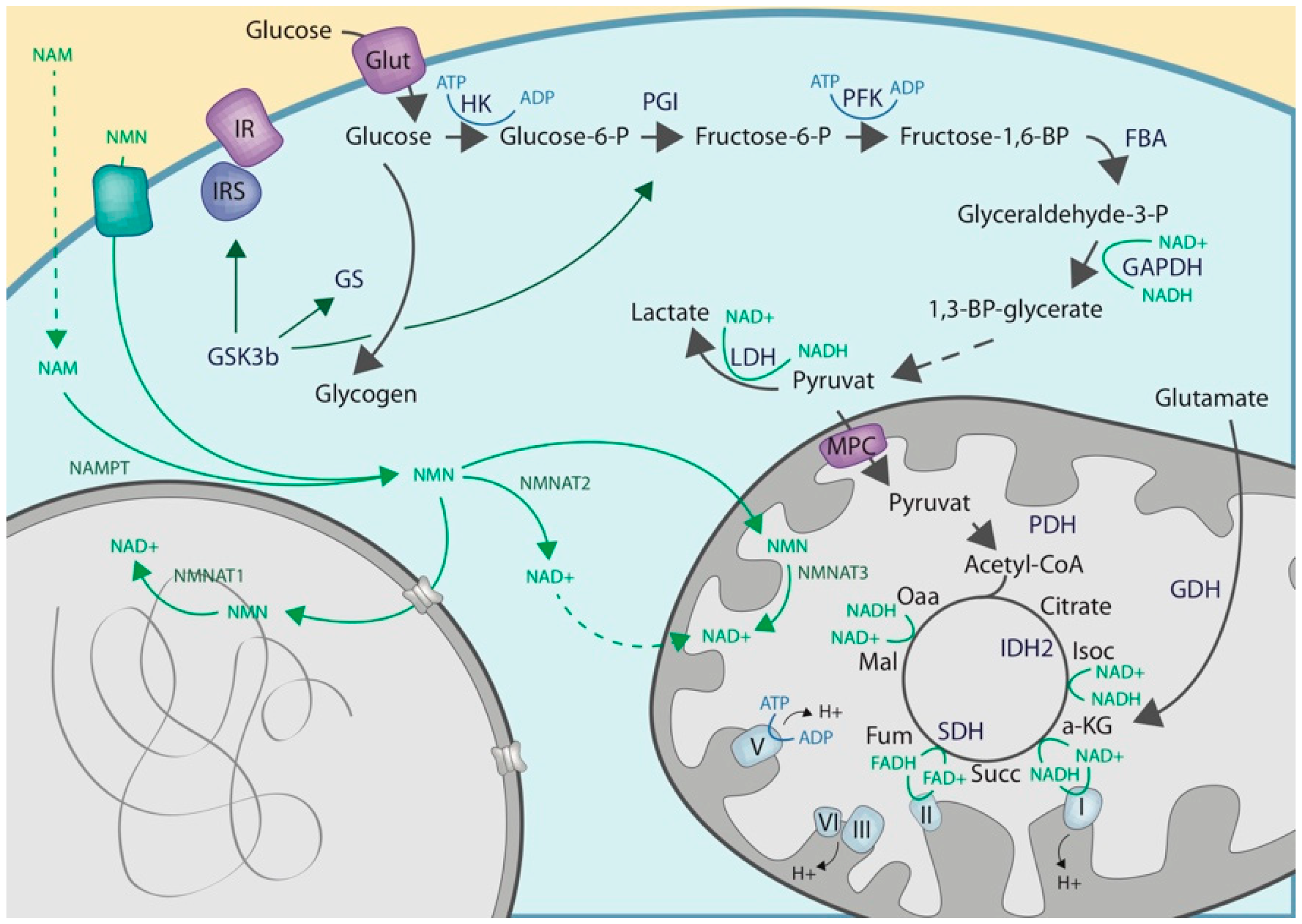
2.1. Cytoplasmic NAD Biosynthesis
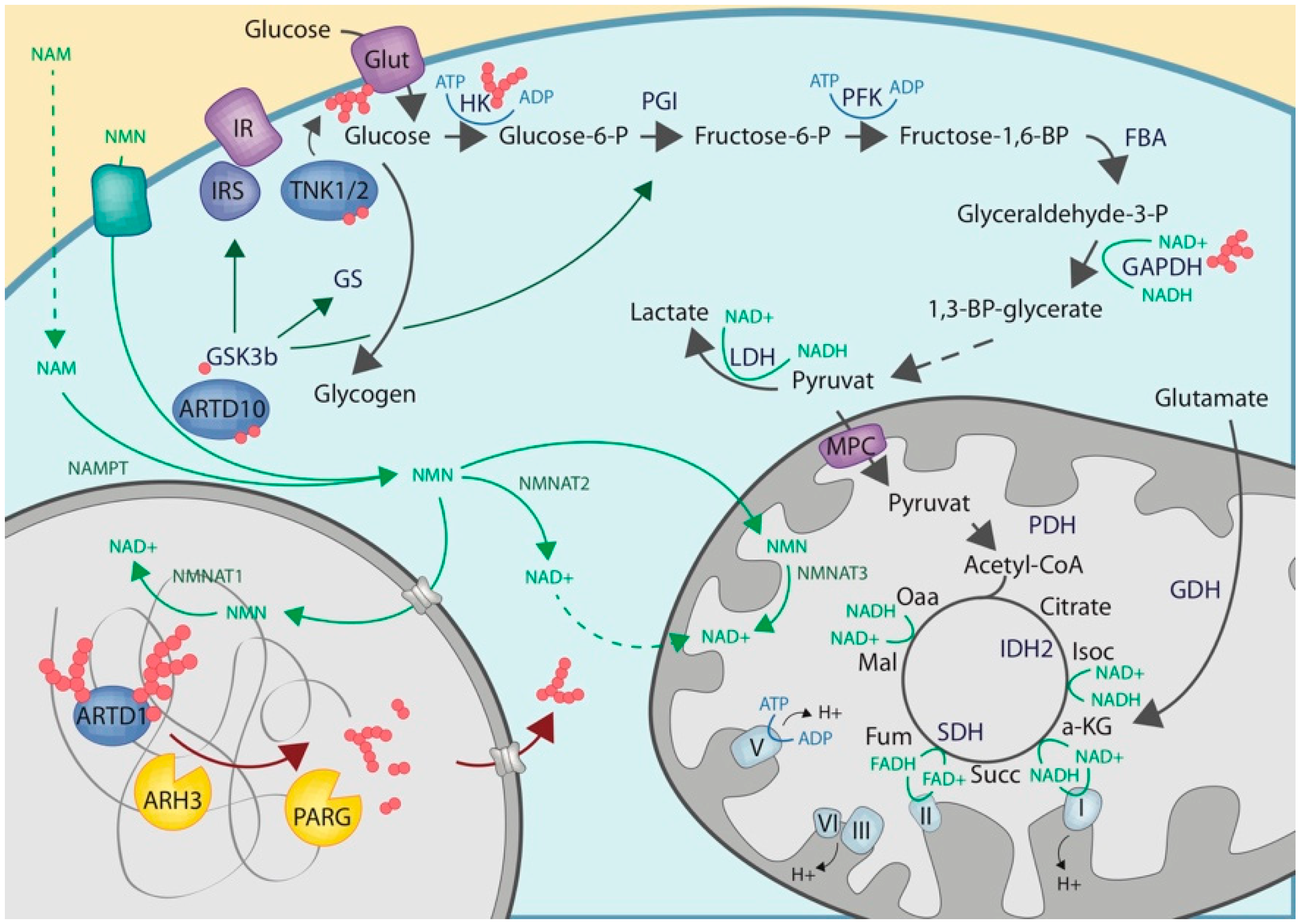
2.2. Cytoplasmic ADP-Ribosylation
2.3. Crosstalk with Enzymes of the Carbohydrate Metabolisms
3. Mitochondrial NAD Biosynthesis, ADP-Ribosylation Crosstalk and Carbohydrate Metabolism
3.1. Mitochondrial NAD Biosynthesis
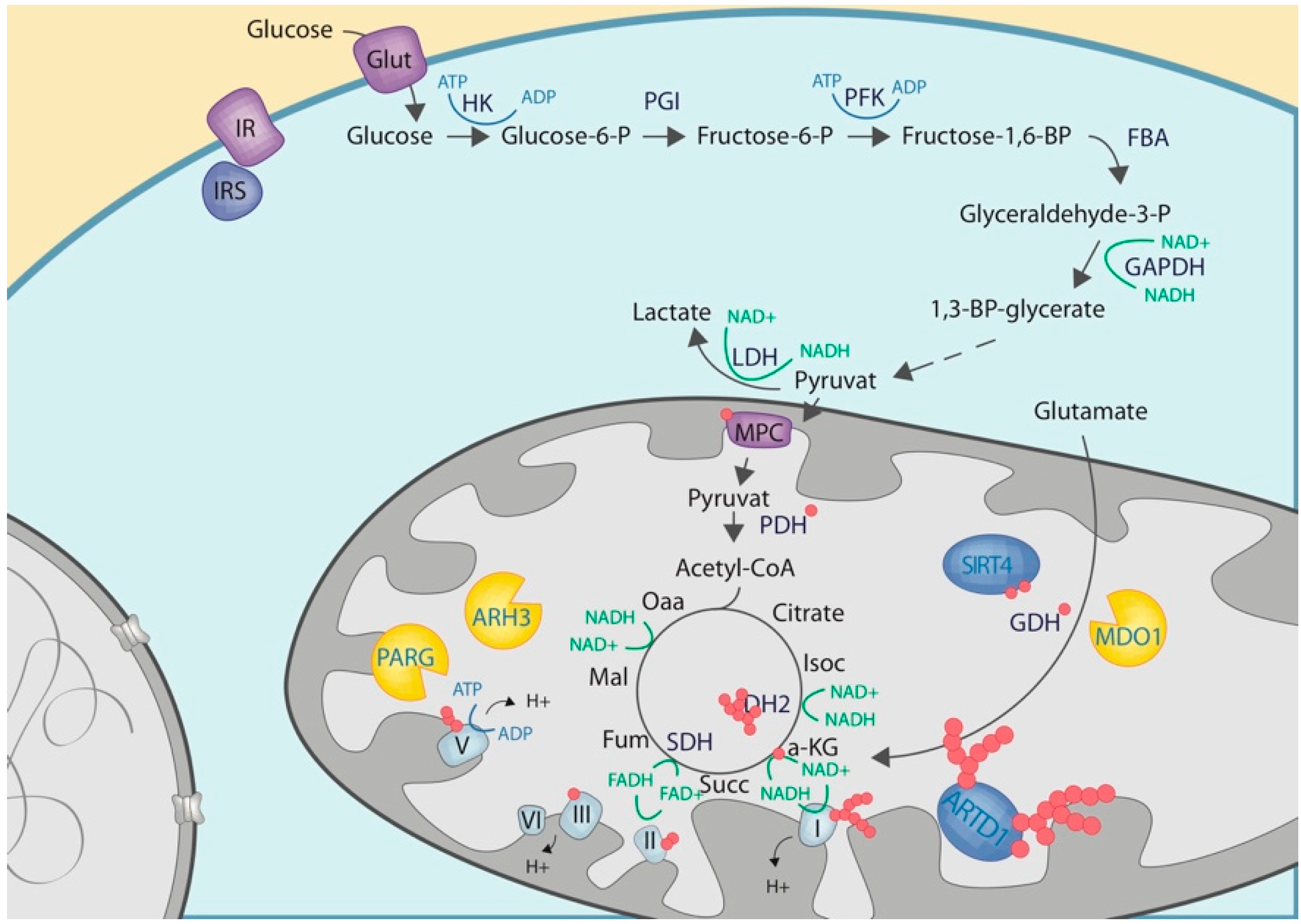
3.2. Mitochondrial ADP-Ribosylation
3.3. Crosstalk with Proteins or Enzymes of the Carbohydrate Metabolisms
4. Crosstalk between Nuclear ADP-Ribosylation and the Carbohydrate Metabolism
4.1. Nuclear NAD Biosynthesis
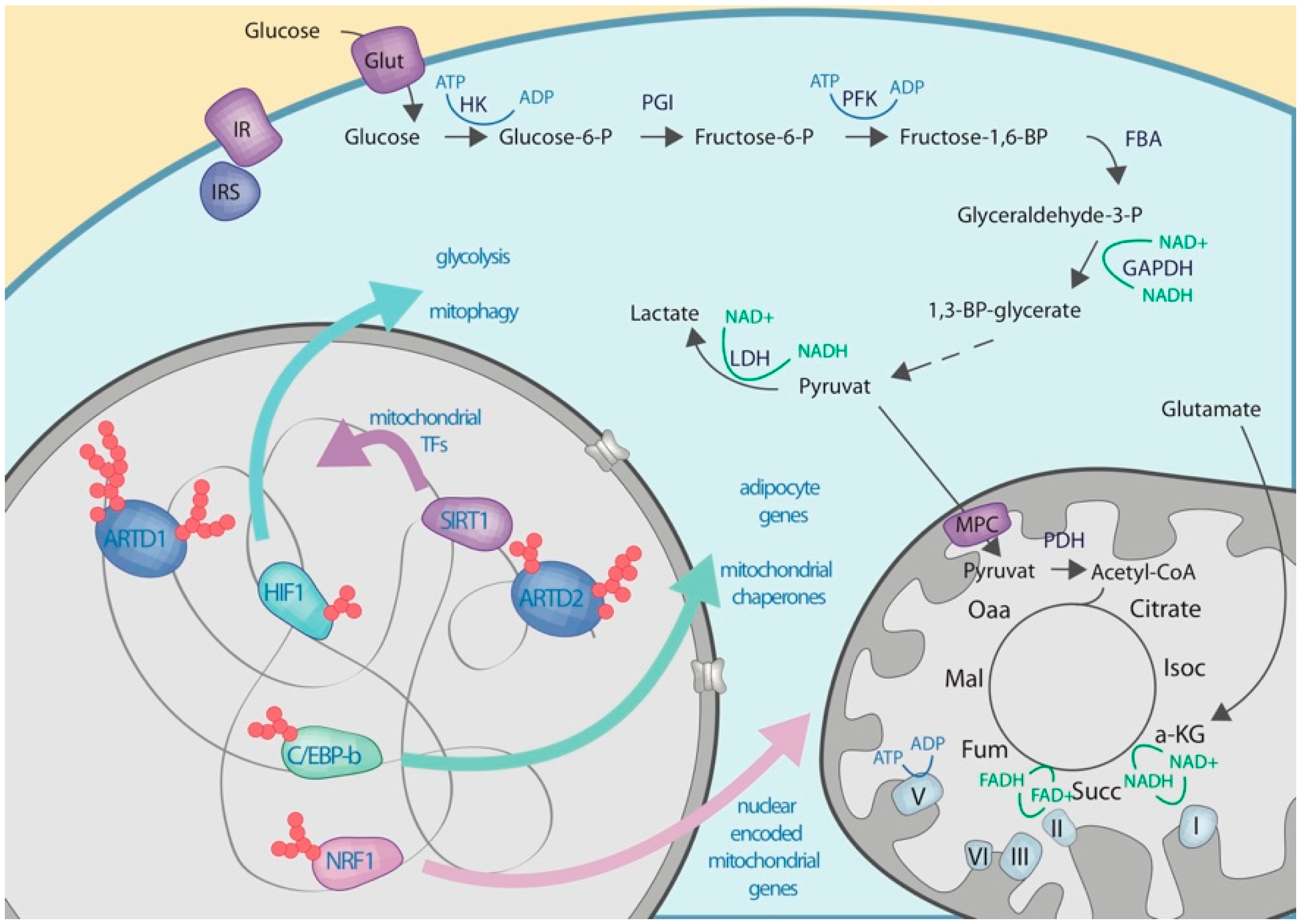
4.2. Nuclear ADP-riboslyation
4.3. Crosstalk with Proteins Regulating the Expression of Enzymes Involved in the Carbohydrate Metabolisms
5. Intercompartmental NAD+ Cross-Talks after Genotoxic Stress
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Verdin, E. NAD+ in aging, metabolism, and neurodegeneration. Science 2015, 350, 1208–1213. [Google Scholar] [CrossRef]
- Katsyuba, E.; Auwerx, J. Modulating NAD+ metabolism, from bench to bedside. EMBO J. 2017, 36, 2670–2683. [Google Scholar] [CrossRef]
- Chiarugi, A.; Dölle, C.; Felici, R.; Ziegler, M. The NAD metabolome—A key determinant of cancer cell biology. Nat. Rev. Cancer 2012, 12, 741–752. [Google Scholar] [CrossRef]
- Pearce, E.L.; Poffenberger, M.C.; Chang, C.H.; Jones, R.G. Fueling immunity: Insights into metabolism and lymphocyte function. Science 2013. [Google Scholar] [CrossRef]
- Van der Heiden, M.G.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef]
- MacDonald, M.J. High content of mitochondrial glycerol-3-phosphate dehydrogenase in pancreatic islets and its inhibition by diazoxide. J. Biol. Chem. 1981, 256, 8287–8290. [Google Scholar]
- MacDonald, M.J. Evidence for the malate aspartate shuttle in pancreatic islets. Arch. Biochem. Biophys. 1982, 213, 643–649. [Google Scholar] [CrossRef]
- Pollak, N.; Dölle, C.; Ziegler, M. The power to reduce: Pyridine nucleotides—Small molecules with a multitude of functions. Biochem. J. 2007. [Google Scholar] [CrossRef]
- Liu, L.; Su, X.; Quinn, W.J.; Hui, S.; Krukenberg, K.; Frederick, D.W.; Redpath, P.; Zhan, L.; Chellappa, K.; White, E.; et al. Quantitative Analysis of NAD Synthesis-Breakdown Fluxes. Cell Metab. 2018. [Google Scholar] [CrossRef]
- Hikosaka, K.; Ikutani, M.; Shito, M.; Kazuma, K.; Gulshan, M.; Nagai, Y.; Takatsu, K.; Konno, K.; Tobe, K.; Kanno, H.; et al. Deficiency of nicotinamide mononucleotide adenylyltransferase 3 (Nmnat3) causes hemolytic anemia by altering the glycolytic flow in mature erythrocytes. J. Biol. Chem. 2014. [Google Scholar] [CrossRef]
- Dölle, C.; Niere, M.; Lohndal, E.; Ziegler, M. Visualization of subcellular NAD pools and intra-organellar protein localization by poly-ADP-ribose formation. Cell. Mol. Life Sci. 2010, 67, 433–443. [Google Scholar] [CrossRef]
- VanLinden, M.R.; Niere, M.; Nikiforov, A.A.; Ziegler, M.; Dölle, C. Compartment-specific poly-ADP-ribose formation as a biosensor for subcellular NAD pools. Method. Mol. Biol. 2017, 1608, 45–56. [Google Scholar]
- Cambronne, X.A.; Stewart, M.L.; Kim, D.; Jones-Brunette, A.M.; Morgan, R.K.; Farrens, D.L.; Cohen, M.S.; Goodman, R.H. Biosensor reveals multiple sources for mitochondrial NAD+. Science 2016. [Google Scholar] [CrossRef]
- Cameron, W.D.; Bui, C.V.; Hutchinson, A.; Loppnau, P.; Gräslund, S.; Rocheleau, J.V. Apollo-NADP+: A spectrally tunable family of genetically encoded sensors for NADP+. Nat. Methods 2016, 13, 352–358. [Google Scholar] [CrossRef]
- Sallin, O.; Reymond, L.; Gondrand, C.; Raith, F.; Koch, B.; Johnsson, K. Semisynthetic biosensors for mapping cellular concentrations of nicotinamide adenine dinucleotides. Elife 2018. [Google Scholar] [CrossRef]
- Koch-Nolte, F.; Fischer, S.; Haag, F.; Ziegler, M. Compartmentation of NAD+-dependent signalling. FEBS Lett. 2011. [Google Scholar] [CrossRef]
- Alano, C.C.; Tran, A.; Tao, R.; Ying, W.; Karliner, J.S.; Swanson, R.A. Differences among cell types in NAD+ compartmentalization: A comparison of neurons, astrocytes, and cardiac myocytes. J. Neurosci. Res. 2007. [Google Scholar] [CrossRef]
- Di Lisa, F.; Menabò, R.; Canton, M.; Barile, M.; Bernardi, P. Opening of the Mitochondrial Permeability Transition Pore Causes Depletion of Mitochondrial and Cytosolic NAD+ and Is a Causative Event in the Death of Myocytes in Postischemic Reperfusion of the Heart. J. Biol. Chem. 2001. [Google Scholar] [CrossRef]
- Fjeld, C.C.; Birdsong, W.T.; Goodman, R.H. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc. Natl. Acad. Sci. USA 2003. [Google Scholar] [CrossRef]
- Veech, R.L.; Eggleston, L.V.; Krebs, H.A. The redox state of free nicotinamide-adenine dinucleotide phosphate in the cytoplasm of rat liver. Biochem. J. 1969. [Google Scholar] [CrossRef]
- Zhang, Q.; Piston, D.W.; Goodman, R.H. Regulation of corepressor function by nuclear NADH. Science 2002. [Google Scholar] [CrossRef]
- Stein, L.R.; Imai, S.I. The dynamic regulation of NAD metabolism in mitochondria. Trends Endocrinol. Metab. 2012. [Google Scholar] [CrossRef]
- Anderson, R.M.; Bitterman, K.J.; Wood, J.G.; Medvedik, O.; Sinclair, D.A. Nicatinamide and PNC1 govern lifespan extension by calorie restriction in Saccharomyces cerevisiae. Nature 2003, 423, 181–185. [Google Scholar] [CrossRef]
- Nikiforov, A.; Kulikova, V.; Ziegler, M. The human NAD metabolome: Functions, metabolism and compartmentalization. Crit. Rev. Biochem. Mol. Biol. 2015. [Google Scholar] [CrossRef]
- Hassa, P.O.; Haenni, S.S.; Elser, M.; Hottiger, M.O. Nuclear ADP-Ribosylation Reactions in Mammalian Cells: Where Are We Today and Where Are We Going? Microbiol. Mol. Biol. Rev. 2006. [Google Scholar] [CrossRef]
- Bogan, K.L.; Brenner, C. Nicotinic Acid, Nicotinamide, and Nicotinamide Riboside: A Molecular Evaluation of NAD+ Precursor Vitamins in Human Nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef]
- Mori, V.; Amici, A.; Mazzola, F.; Di Stefano, M.; Conforti, L.; Magni, G.; Ruggieri, S.; Raffaelli, N.; Orsomando, G. Metabolic profiling of alternative NAD biosynthetic routes in mouse tissues. PLoS ONE 2014. [Google Scholar] [CrossRef]
- Strømland, Ø.; Niere, M.; Nikiforov, A.A.; VanLinden, M.R.; Heiland, I.; Ziegler, M. Keeping the balance in NAD metabolism. Biochem. Soc. Trans. 2019. [Google Scholar] [CrossRef]
- Cantó, C.; Menzies, K.J.; Auwerx, J. NAD+ Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015. [Google Scholar] [CrossRef]
- Dölle, C.; Skoge, R.; VanLinden, M.; Ziegler, M. NAD Biosynthesis in Humans - Enzymes, Metabolites and Therapeutic Aspects. Curr. Top. Med. Chem. 2013, 13, 2907–2917. [Google Scholar] [CrossRef]
- Revollo, J.R.; Grimm, A.A.; Imai, S.I. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004. [Google Scholar] [CrossRef]
- Yang, H.; Yang, T.; Baur, J.A.; Perez, E.; Matsui, T.; Carmona, J.J.; Lamming, D.W.W.; Souza-Pinto, N.C.; Bohr, V.A.; Rosenzweig, A.; et al. Nutrient-Sensitive Mitochondrial NAD+ Levels Dictate Cell Survival. Cell 2007. [Google Scholar] [CrossRef]
- Revollo, J.R.; Körner, A.; Mills, K.F.; Satoh, A.; Wang, T.; Garten, A.; Dasgupta, B.; Sasaki, Y.; Wolberger, C.; Townsend, R.R.; et al. Nampt/PBEF/Visfatin Regulates Insulin Secretion in β Cells as a Systemic NAD Biosynthetic Enzyme. Cell Metab. 2007. [Google Scholar] [CrossRef]
- Brazill, J.M.; Li, C.; Zhu, Y.; Zhai, R.G. NMNAT: It’s an NAD+ synthase… It’s a chaperone… It’s a neuroprotector. Curr. Opin. Genet. Dev. 2017. [Google Scholar] [CrossRef]
- Berger, F.; Lau, C.; Dahlmann, M.; Ziegler, M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005. [Google Scholar] [CrossRef]
- Felici, R.; Lapucci, A.; Ramazzotti, M.; Chiarugi, A. Insight into Molecular and Functional Properties of NMNAT3 Reveals New Hints of NAD Homeostasis within Human Mitochondria. PLoS ONE 2013. [Google Scholar] [CrossRef][Green Version]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 2018. [Google Scholar] [CrossRef]
- Johnson, S.; Imai, S. NAD+ biosynthesis, aging, and disease. F1000Research 2018. [Google Scholar] [CrossRef]
- Haigis, M.C.; Sinclair, D.A. Mammalian Sirtuins: Biological Insights and Disease Relevance. Annu. Rev. Pathol. Mech. Dis. 2010. [Google Scholar] [CrossRef]
- Ansari, H.R.; Raghava, G.P.S. Identification of NAD interacting residues in proteins. BMC Bioinformat. 2010. [Google Scholar] [CrossRef]
- Berger, F.; Ramírez-Hernández, M.H.; Ziegler, M. The new life of a centenarian: Signalling functions of NAD(P). Trends Biochem. Sci. 2004. [Google Scholar] [CrossRef]
- Houtkooper, R.H.; Williams, R.W.; Auwerx, J. Metabolic Networks of Longevity. Cell 2010. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarragó, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016. [Google Scholar] [CrossRef]
- Chini, E.N.; Chini, C.C.S.; Espindola Netto, J.M.; de Oliveira, G.C.; van Schooten, W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol. Sci. 2018. [Google Scholar] [CrossRef]
- Malavasi, F.; Deaglio, S.; Funaro, A.; Ferrero, E.; Horenstein, A.L.; Ortolan, E.; Vaisitti, T.; Aydin, S. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol. Rev. 2008. [Google Scholar] [CrossRef]
- Opitz, C.A.; Heiland, I. Dynamics of NAD-metabolism: Everything but constant. Biochem. Soc. Trans. 2015. [Google Scholar] [CrossRef]
- Kirchberger, T.; Guse, A.H. Measuring CD38 (ADP-ribosyl Cyclase/Cyclic ADP-ribose hydrolase) activity by reverse-phase HPLC. Cold Spring Harb. Protoc. 2013. [Google Scholar] [CrossRef]
- Horenstein, A.L.; Sizzano, F.; Lusso, R.; Besso, F.G.; Ferrero, E.; Deaglio, S.; Corno, F.; Malavasi, F. CD38 and CD157 Ectoenzymes Mark Cell Subsets in the Human Corneal Limbus. Mol. Med. 2009. [Google Scholar] [CrossRef]
- Hottiger, M.O. Nuclear ADP-Ribosylation and Its Role in Chromatin Plasticity, Cell Differentiation, and Epigenetics. Annu. Rev. Biochem. 2015. [Google Scholar] [CrossRef]
- Gupte, R.; Liu, Z.; Kraus, W.L. Parps and adp-ribosylation: Recent advances linking molecular functions to biological outcomes. Genes Dev. 2017. [Google Scholar] [CrossRef]
- Grube, K.; Bürkle, A. Poly(ADP-ribose) polymerase activity in mononuclear leukocytes of 13 mammalian species correlates with species-specific life span. Proc. Natl. Acad. Sci. USA 1992. [Google Scholar] [CrossRef]
- Guarente, L. Sirtuins, aging, and metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011. [Google Scholar] [CrossRef]
- Chambon, P.; Weill, J.D.; Mandel, P. Nicotinamide mononucleotide activation of a new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem. Biophys. Res. Commun. 1963. [Google Scholar] [CrossRef]
- Leung, A.K.L. PARPs. Curr. Biol. 2017, 27, R1249–R1267. [Google Scholar] [CrossRef]
- Haag, F.; Adriouch, S.; Braß, A.; Jung, C.; Möller, S.; Scheuplein, F.; Bannas, P.; Seman, M.; Koch-Nolte, F. Extracellular NAD and ATP: Partners in immune cell modulation. Purinergic Signal. 2007. [Google Scholar] [CrossRef]
- Di Girolamo, M.; Fabrizio, G. Overview of the Mammalian ADP-Ribosyl-Transferases Clostridia Toxin-Like (ARTCs) Family. Biochem. Pharmacol. 2019. [Google Scholar] [CrossRef]
- Imai, S.I.; Guarente, L. It takes two to tango: Nad+ and sirtuins in aging/longevity control. npj Aging Mech. Dis. 2016. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Amé, J.C.; De Murcia, G. Poly(ADP-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell Biol. 2006. [Google Scholar] [CrossRef]
- Imai, S.I.; Armstrong, C.M.; Kaeberlein, M.; Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000. [Google Scholar] [CrossRef]
- Rajman, L.; Chwalek, K.; Sinclair, D.A. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, J.; Park, J.; Rai, P.; Zhai, R.G. Subcellular compartmentalization of NAD + and its role in cancer: A sereNADe of metabolic melodies. Pharmacol. Ther. 2019. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Lüscher, B.; Schüler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian ADP-ribosyltransferases. Trends Biochem. Sci. 2010. [Google Scholar] [CrossRef]
- Dölle, C.; Rack, J.G.M.; Ziegler, M. NAD and ADP-ribose metabolism in mitochondria. FEBS J. 2013. [Google Scholar] [CrossRef]
- McLennan, A.G. The Nudix hydrolase superfamily. Cell. Mol. Life Sci. 2006. [Google Scholar] [CrossRef]
- Lin, S.; Gasmi, L.; Xie, Y.; Ying, K.; Gu, S.; Wang, Z.; Jin, H.; Chao, Y.; Wu, C.; Zhou, Z.; et al. Cloning, expression and characterisation of a human Nudix hydrolase specific for adenosine 5′-diphosphoribose (ADP-ribose). Biochim. Biophys. Acta 2002. [Google Scholar] [CrossRef]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. ADP-ribose gating of the calcium-permeable LTRPC2 channel revealed by Nudix motif homology. Nature 2001. [Google Scholar] [CrossRef]
- Perraud, A.L.; Shen, B.; Dunn, C.A.; Rippe, K.; Smith, M.K.; Bessman, M.J.; Stoddard, B.L.; Scharenberg, A.M. NUDT9, a member of the Nudix hydrolase family, is an evolutionarily conserved mitochondrial ADP-ribose pyrophosphatase. J. Biol. Chem. 2003. [Google Scholar] [CrossRef]
- Ying, W.; Alano, C.C.; Garnier, P.; Swanson, R.A. NAD+ as a metabolic link between DNA damage and cell death. J. Neurosci. Res. 2005. [Google Scholar] [CrossRef]
- Ryu, K.W.; Nandu, T.; Kim, J.; Challa, S.; DeBerardinis, R.J.; Lee Kraus, W. Metabolic regulation of transcription through compartmentalized NAD+ biosynthesis. Science 2018. [Google Scholar] [CrossRef]
- Cameron, A.M.; Castoldi, A.; Sanin, D.E.; Flachsmann, L.J.; Field, C.S.; Puleston, D.J.; Kyle, R.L.; Patterson, A.E.; Hässler, F.; Buescher, J.M.; et al. Inflammatory macrophage dependence on NAD+ salvage is a consequence of reactive oxygen species–mediated DNA damage. Nat. Immunol. 2019. [Google Scholar] [CrossRef]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019. [Google Scholar] [CrossRef]
- Alano, C.C.; Ying, W.; Swanson, R.A. Poly(ADP-ribose) Polymerase-1-mediated Cell Death in Astrocytes Requires NAD+ Depletion and Mitochondrial Permeability Transition. J. Biol. Chem. 2004. [Google Scholar] [CrossRef]
- Erener, S.; Hesse, M.; Kostadinova, R.; Hottiger, M.O. Poly(ADP-Ribose)Polymerase-1 (PARP1) Controls Adipogenic Gene Expression and Adipocyte Function. Mol. Endocrinol. 2011. [Google Scholar] [CrossRef]
- Bai, P. Biology of Poly(ADP-Ribose) Polymerases: The Factotums of Cell Maintenance. Mol. Cell 2015. [Google Scholar] [CrossRef]
- Leutert, M.; Menzel, S.; Braren, R.; Rissiek, B.; Hopp, A.K.; Nowak, K.; Bisceglie, L.; Gehrig, P.; Li, H.; Zolkiewska, A.; et al. Proteomic Characterization of the Heart and Skeletal Muscle Reveals Widespread Arginine ADP-Ribosylation by the ARTC1 Ectoenzyme. Cell Rep. 2018. [Google Scholar] [CrossRef]
- Larsen, S.C.; Hendriks, I.A.; Lyon, D.; Jensen, L.J.; Nielsen, M.L. Systems-wide Analysis of Serine ADP-Ribosylation Reveals Widespread Occurrence and Site-Specific Overlap with Phosphorylation. Cell Rep. 2018. [Google Scholar] [CrossRef]
- Luo, X.; Ryu, K.W.; Kim, D.S.; Nandu, T.; Medina, C.J.; Gupte, R.; Gibson, B.A.; Soccio, R.E.; Yu, Y.; Gupta, R.K.; et al. PARP-1 Controls the Adipogenic Transcriptional Program by PARylating C/EBPβ and Modulating Its Transcriptional Activity. Mol. Cell 2017. [Google Scholar] [CrossRef]
- Akram, M. Citric Acid Cycle and Role of its Intermediates in Metabolism. Cell Biochem. Biophys. 2014. [Google Scholar] [CrossRef]
- Vyas, S.; Matic, I.; Uchima, L.; Rood, J.; Zaja, R.; Hay, R.T.; Ahel, I.; Chang, P. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat. Commun. 2014. [Google Scholar] [CrossRef]
- Vyas, S.; Chesarone-Cataldo, M.; Todorova, T.; Huang, Y.H.; Chang, P. A systematic analysis of the PARP protein family identifies new functions critical for cell physiology. Nat. Commun. 2013. [Google Scholar] [CrossRef]
- Verheugd, P.; Bütepage, M.; Eckei, L.; Lüscher, B. Players in ADP-ribosylation: Readers and Erasers. Curr. Protein Pept. Sci. 2016, 17, 654–667. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.S.; Jividen, K.; Spencer, A.; Dworak, N.; Ni, L.; Oostdyk, L.T.; Chatterjee, M.; Kuśmider, B.; Reon, B.; Parlak, M.; et al. Ubiquitin Modification by the E3 Ligase/ADP-Ribosyltransferase Dtx3L/Parp9. Mol. Cell 2017. [Google Scholar] [CrossRef] [PubMed]
- Slade, D.; Dunstan, M.S.; Barkauskaite, E.; Weston, R.; Lafite, P.; Dixon, N.; Ahel, M.; Leys, D.; Ahel, I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature 2011. [Google Scholar] [CrossRef] [PubMed]
- Ohashi, S.; Kanai, M.; Hanai, S.; Uchiumi, F.; Maruta, H.; Tanuma, S.I.; Miwa, M. Subcellular localization of poly(ADP-ribose) glycohydrolase in mammalian cells. Biochem. Biophys. Res. Commun. 2003. [Google Scholar] [CrossRef]
- Mashimo, M.; Kato, J.; Moss, J. Structure and function of the ARH family of ADP-ribosyl-acceptor hydrolases. DNA Repair (Amst). 2014. [Google Scholar] [CrossRef]
- Abplanalp, J.; Leutert, M.; Frugier, E.; Nowak, K.; Feurer, R.; Kato, J.; Kistemaker, H.V.A.; Filippov, D. V.; Moss, J.; Caflisch, A.; et al. Proteomic analyses identify ARH3 as a serine mono-ADP-ribosylhydrolase. Nat. Commun. 2017. [Google Scholar] [CrossRef]
- Fontana, P.; Bonfiglio, J.J.; Palazzo, L.; Bartlett, E.; Matic, I.; Ahel, I. Serine ADP-ribosylation reversal by the hydrolase ARH3. Elife 2017. [Google Scholar] [CrossRef]
- Rosenthal, F.; Feijs, K.L.H.; Frugier, E.; Bonalli, M.; Forst, A.H.; Imhof, R.; Winkler, H.C.; Fischer, D.; Caflisch, A.; Hassa, P.O.; et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat. Struct. Mol. Biol. 2013. [Google Scholar] [CrossRef]
- Jankevicius, G.; Hassler, M.; Golia, B.; Rybin, V.; Zacharias, M.; Timinszky, G.; Ladurner, A.G. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat. Struct. Mol. Biol. 2013. [Google Scholar] [CrossRef]
- Feijs, K.L.H.; Forst, A.H.; Verheugd, P.; Lüscher, B. Macrodomain-containing proteins: Regulating new intracellular functions of mono(ADP-ribosyl)ation. Nat. Rev. Mol. Cell Biol. 2013. [Google Scholar] [CrossRef]
- Liszt, G.; Ford, E.; Kurtev, M.; Guarente, L. Mouse Sir2 homolog SIRT6 is a nuclear ADP-ribosyltransferase. J. Biol. Chem. 2005. [Google Scholar] [CrossRef] [PubMed]
- Niere, M.; Kernstock, S.; Koch-Nolte, F.; Ziegler, M. Functional Localization of Two Poly(ADP-Ribose)-Degrading Enzymes to the Mitochondrial Matrix. Mol. Cell. Biol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Agnew, T.; Munnur, D.; Crawford, K.; Palazzo, L.; Mikoc, A.; Ahel, I. MacroD1 is a promiscuous ADP-ribosyl hydrolase localized to mitochondria. Front. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Haigis, M.C.; Mostoslavsky, R.; Haigis, K.M.; Fahie, K.; Christodoulou, D.C.; Murphy, A.J.J.; Valenzuela, D.M.; Yancopoulos, G.D.; Karow, M.; Blander, G.; et al. SIRT4 Inhibits Glutamate Dehydrogenase and Opposes the Effects of Calorie Restriction in Pancreatic β Cells. Cell 2006. [Google Scholar] [CrossRef] [PubMed]
- Grimaldi, G.; Corda, D. ADP-ribosylation and intracellular traffic: an emerging role for PARP enzymes. Biochem. Soc. Trans. 2019. [Google Scholar] [CrossRef] [PubMed]
- Catara, G.; Grimaldi, G.; Schembri, L.; Spano, D.; Turacchio, G.; Lo Monte, M.; Beccari, A.R.; Valente, C.; Corda, D. PARP1-produced poly-ADP-ribose causes the PARP12 translocation to stress granules and impairment of Golgi complex functions. Sci. Rep. 2017. [Google Scholar] [CrossRef]
- Jwa, M.; Chang, P. PARP16 is a tail-anchored endoplasmic reticulum protein required for the PERK-and IRE1α-mediated unfolded protein response. Nat. Cell Biol. 2012. [Google Scholar] [CrossRef]
- Di Paola, S.; Micaroni, M.; Di Tullio, G.; Buccione, R.; Di Girolamo, M. PARP16/ARTD15 is a novel endoplasmic-reticulum-associated mono-ADP-ribosyltransferase that interacts with, and modifies karyopherin-ß1. PLoS ONE 2012. [Google Scholar] [CrossRef]
- Yeh, T.Y.J.; Beiswenger, K.K.; Li, P.; Bolin, K.E.; Lee, R.M.; Tsao, T.S.; Murphy, A.N.; Hevener, A.L.; Chi, N.W. Hypermetabolism, hyperphagia, and reduced adiposity in tankyrase-deficient mice. Diabetes 2009. [Google Scholar] [CrossRef]
- Feijs, K.L.; Kleine, H.; Braczynski, A.; Forst, A.H.; Herzog, N.; Verheugd, P.; Linzen, U.; Kremmer, E.; Lüscher, B. ARTD10 substrate identification on protein microarrays: Regulation of GSK3β by mono-ADP-ribosylation. Cell Commun. Signal. 2013. [Google Scholar] [CrossRef]
- Lüscher, B.; Bütepage, M.; Eckei, L.; Krieg, S.; Verheugd, P.; Shilton, B.H. ADP-Ribosylation, a Multifaceted Posttranslational Modification Involved in the Control of Cell Physiology in Health and Disease. Chem. Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Mueckler, M.; Thorens, B. The SLC2 (GLUT) family of membrane transporters. Mol. Aspects Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Czech, M.P. The GLUT4 Glucose Transporter. Cell Metab. 2007. [Google Scholar] [CrossRef] [PubMed]
- Su, Z.; Deshpande, V.; James, D.E.; Stöckli, J. Tankyrase modulates insulin sensitivity in skeletal muscle cells by regulating the stability of GLUT4 vesicle proteins. J. Biol. Chem. 2018. [Google Scholar] [CrossRef]
- Robey, R.B.; Hay, N. Mitochondrial hexokinases, novel mediators of the antiapoptotic effects of growth factors and Akt. Oncogene 2006. [Google Scholar] [CrossRef]
- Fouquerel, E.; Goellner, E.M.; Yu, Z.; Gagné, J.P.; de Moura, M.B.; Feinstein, T.; Wheeler, D.; Redpath, P.; Li, J.; Romero, G.; et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD + depletion. Cell Rep. 2014. [Google Scholar] [CrossRef]
- Lochhead, P.A.; Coghlan, M.; Rice, S.Q.J.; Sutherland, C. Inhibition of GSK-3 selectively reduces glucose-6-phosphatase and phosphoenolpyruvate carboxykinase gene expression. Diabetes 2001. [Google Scholar] [CrossRef]
- Liberman, Z.; Eldar-Finkelman, H. Serine 332 phosphorylation of insulin receptor substrate-1 by glycogen synthase kinase-3 attenuates insulin signaling. J. Biol. Chem. 2005. [Google Scholar] [CrossRef]
- Márton, J.; Fodor, T.; Nagy, L.; Vida, A.; Kis, G.; Brunyánszki, A.; Antal, M.; Lüscher, B.; Bai, P. PARP10 (ARTD10) modulates mitochondrial function. PLoS ONE 2018. [Google Scholar] [CrossRef]
- Nicholls, C.; Li, H.; Liu, J.P. GAPDH: A common enzyme with uncommon functions. Clin. Exp. Pharmacol. Physiol. 2012. [Google Scholar] [CrossRef]
- Sirover, M.A. Pleiotropic effects of moonlighting glyceraldehyde-3-phosphate dehydrogenase (GAPDH) in cancer progression, invasiveness, and metastases. Cancer Metastasis Rev. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kots, A.Y.; Sergienko, E.A.; Bulargina, T. V.; Severin, E.S. Glyceraldehyde-3-phosphate activates auto-ADP-ribosylation of glyceraldehyde-3-phosphate dehydrogenase. FEBS Lett. 1993. [Google Scholar] [CrossRef]
- Du, X.; Matsumura, T.; Edelstein, D.; Rossetti, L.; Zsengellér, Z.; Szabó, C.; Brownlee, M. Inhibition of GAPDH activity by poly(ADP-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. J. Clin. Invest. 2003. [Google Scholar] [CrossRef] [PubMed]
- Mayo, E.; Fabrizio, G.; Scarpa, E.; Stilla, A.; Dani, N.; Chiacchiera, F.; Kleine, H.; Attanasio, F.; Lüscher, B.; Di Girolamo, M. ARTD10/PARP10 Induces ADP-Ribosylation of GAPDH and Recruits GAPDH into Cytosolic Membrane-Free Cell Bodies When Overexpressed in Mammalian Cells. Challenges 2018. [Google Scholar] [CrossRef]
- Cantó, C.; Houtkooper, R.H.; Pirinen, E.; Youn, D.Y.; Oosterveer, M.H.; Cen, Y.; Fernandez-Marcos, P.J.; Yamamoto, H.; Andreux, P.A.; Cettour-Rose, P.; et al. The NAD+ precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012. [Google Scholar] [CrossRef]
- Pittelli, M.; Felici, R.; Pitozzi, V.; Giovannelli, L.; Bigagli, E.; Cialdai, F.; Romano, G.; Moroni, F.; Chiarugi, A. Pharmacological Effects of Exogenous NAD on Mitochondrial Bioenergetics, DNA Repair, and Apoptosis. Mol. Pharmacol. 2011. [Google Scholar] [CrossRef]
- Kun, E.; Zimber, P.H.; Chang, A.C.; Puschendorf, B.; Grunicke, H. Macromolecular enzymatic product of NAD+ in liver mitochondria. Proc. Natl. Acad. Sci. USA 1975. [Google Scholar] [CrossRef]
- Nikiforov, A.; Dölle, C.; Niere, M.; Ziegler, M. Pathways and subcellular compartmentation of NAD biosynthesis in human cells: From entry of extracellular precursors to mitochondrial NAD generation. J. Biol. Chem. 2011. [Google Scholar] [CrossRef]
- Son, M.J.; Kwon, Y.; Son, T.; Cho, Y.S. Restoration of Mitochondrial NAD+ Levels Delays Stem Cell Senescence and Facilitates Reprogramming of Aged Somatic Cells. Stem Cells 2016. [Google Scholar] [CrossRef]
- Yamamoto, M.; Hikosaka, K.; Mahmood, A.; Tobe, K.; Shojaku, H.; Inohara, H.; Nakagawa, T. Nmnat3 is dispensable in mitochondrial NAD level maintenance in vivo. PLoS ONE 2016. [Google Scholar] [CrossRef]
- Peek, C.B.; Affinati, A.H.; Ramsey, K.M.; Kuo, H.Y.; Yu, W.; Sena, L.A.; Ilkayeva, O.; Marcheva, B.; Kobayashi, Y.; Omura, C.; et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013. [Google Scholar] [CrossRef] [PubMed]
- Ying, W. NAD + /NADH and NADP + /NADPH in Cellular Functions and Cell Death: Regulation and Biological Consequences. Antioxid. Redox Signal. 2008. [Google Scholar] [CrossRef] [PubMed]
- Davila, A.; Liu, L.; Chellappa, K.; Redpath, P.; Nakamaru-Ogiso, E.; Paolella, L.M.; Zhang, Z.; Migaud, M.E.; Rabinowitz, J.D.; Baur, J.A. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife 2018. [Google Scholar] [CrossRef] [PubMed]
- Roberts, J.H.; Stark, P.; Giri, C.P.; Smulson, M. Cytoplasmic poly(ADP-ribose) polymerase during the HeLa cell cycle. Arch. Biochem. Biophys. 1975. [Google Scholar] [CrossRef]
- Burzio, L.O.; Sáez, L.; Cornejo, R. Poly (ADP-ribose) synthetase activity in rat testis mitochondria. Biochem. Biophys. Res. Commun. 1981. [Google Scholar] [CrossRef]
- Williams, E.G.; Wu, Y.; Wolski, W.; Kim, J.Y.; Lan, J.; Hasan, M.; Halter, C.; Jha, P.; Ryu, D.; Auwerx, J.; et al. Quantifying and Localizing the Mitochondrial Proteome Across Five Tissues in A Mouse Population. Mol. Cell. Proteom. 2018. [Google Scholar] [CrossRef]
- Ahuja, N.; Schwer, B.; Carobbio, S.; Waltregny, D.; North, B.J.; Castronovo, V.; Maechler, P.; Verdin, E. Regulation of insulin secretion by SIRT4, a mitochondrial ADP-ribosyltransferase. J. Biol. Chem. 2007. [Google Scholar] [CrossRef]
- Niere, M.; Mashimo, M.; Agledal, L.; Dölle, C.; Kasamatsu, A.; Kato, J.; Moss, J.; Ziegler, M. ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose). J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- Neuvonen, M.; Ahola, T. Differential Activities of Cellular and Viral Macro Domain Proteins in Binding of ADP-Ribose Metabolites. J. Mol. Biol. 2009. [Google Scholar] [CrossRef]
- Richter, C.; Winterhalter, K.H.; Baumhuter, S.; Lotscher, H.R.; Moser, B. ADP-ribosylation in inner membrane of rat liver mitochondria. Proc. Natl. Acad. Sci. USA 2006. [Google Scholar] [CrossRef]
- Schwer, B.; North, B.J.; Frye, R.A.; Ott, M.; Verdin, E. The human silent information regulator (Sir)2 homologue hSIRT3 is a mitochondrial nicotinamide adenine dinucleotide-dependent deacetylase. J. Cell Biol. 2002. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 2014. [Google Scholar] [CrossRef] [PubMed]
- Onyango, P.; Celic, I.; McCaffery, J.M.; Boeke, J.D.; Feinberg, A.P. SIRT3, a human SIR2 homologue, is an NAD- dependent deacetylase localized to mitochondria. Proc. Natl. Acad. Sci. USA 2002. [Google Scholar] [CrossRef] [PubMed]
- Martello, R.; Leutert, M.; Jungmichel, S.; Bilan, V.; Larsen, S.C.; Young, C.; Hottiger, M.O.; Nielsen, M.L. Proteome-wide identification of the endogenous ADP-ribosylome of mammalian cells and tissue. Nat. Commun. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.N.; Carbone, M.; Mostocotto, C.; Mancone, C.; Tripodi, M.; Malone, R.; Amati, P. Mitochondrial localization of PARP-1 requires interaction with mitofilin and is involved in the maintenance of mitochondrial DNA integrity. J. Biol. Chem. 2009. [Google Scholar] [CrossRef]
- Lai, Y.; Chen, Y.; Watkins, S.C.; Nathaniel, P.D.; Guo, F.; Kochanek, P.M.; Jenkins, L.W.; Szabó, C.; Clark, R.S.B. Identification of poly-ADP-ribosylated mitochondrial proteins after traumatic brain injury. J. Neurochem. 2008. [Google Scholar] [CrossRef]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Carcillo, J.A.; Szabó, C.; Clark, R.S.B. Intra-mitochondrial poly(ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003. [Google Scholar] [CrossRef]
- Pankotai, E.; Lacza, Z.; Murányi, M.; Szabó, C. Intra-mitochondrial poly(ADP-ribosyl)ation: Potential role for alpha-ketoglutarate dehydrogenase. Mitochondrion 2009. [Google Scholar] [CrossRef]
- Módis, K.; Gerö, D.; Erdélyi, K.; Szoleczky, P.; Dewitt, D.; Szabo, C. Cellular bioenergetics is regulated by PARP1 under resting conditions and during oxidative stress. Biochem. Pharmacol. 2012. [Google Scholar] [CrossRef]
- Brunyanszki, A.; Olah, G.; Coletta, C.; Szczesny, B.; Szabo, C. Regulation of Mitochondrial Poly(ADP-Ribose) Polymerase Activation by the -Adrenoceptor/cAMP/Protein Kinase A Axis during Oxidative Stress. Mol. Pharmacol. 2014. [Google Scholar] [CrossRef]
- Yu, S.W.; Wang, H.; Poitras, M.F.; Coombs, C.; Bowers, W.J.; Federoff, H.J.; Poirier, G.G.; Dawson, T.M.; Dawson, V.L. Mediation of poty(ADP-ribose) polymerase-1 - Dependent cell death by apoptosis-inducing factor. Science 2002. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, G.; Rapizzi, E.; Vannacci, A.; Rizzuto, R.; Moroni, F.; Chiarugi, A. Nuclear poly(ADP-ribose) polymerase-1 rapidly triggers mitochondrial dysfunction. J. Biol. Chem. 2005. [Google Scholar] [CrossRef] [PubMed]
- Poitras, M.F.; Koh, D.W.; Yu, S.W.; Andrabi, S.A.; Mandir, A.S.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Spatial and functional relationship between poly(ADP-ribose) polymerase-1 and poly(ADP-ribose) glycohydrolase in the brain. Neuroscience 2007. [Google Scholar] [CrossRef] [PubMed]
- Lapucci, A.; Pittelli, M.; Rapizzi, E.; Felici, R.; Moroni, F.; Chiarugi, A. Poly(ADP-ribose) Polymerase-1 Is a Nuclear Epigenetic Regulator of Mitochondrial DNA Repair and Transcription. Mol. Pharmacol. 2011. [Google Scholar] [CrossRef] [PubMed]
- Druzhyna, N.; Smulson, M.E.; LeDoux, S.P.; Wilson, G.L. Poly(ADP-ribose) polymerase facilitates the repair of N-methylpurines in mitochondrial DNA. Diabetes 2000. [Google Scholar] [CrossRef]
- Ueda, K.; Oka, J.; Narumiya, S.; Miyakawa, N.; Hayaishi, O. Poly ADP-ribose glycohydrolase from rat liver nuclei, a novel enzyme degrading the polymer. Biochem. Biophys. Res. Commun. 1972. [Google Scholar] [CrossRef]
- Mashimo, M.; Bu, X.; Aoyama, K.; Kato, J.; Ishiwata-Endo, H.; Stevens, L.A.; Kasamatsu, A.; Wolfe, L.A.; Toro, C.; Adams, D.; et al. PARP1 inhibition alleviates injury in ARH3-deficient mice and human cells. JCI Insight 2019. [Google Scholar] [CrossRef]
- Danhauser, K.; Alhaddad, B.; Makowski, C.; Piekutowska-Abramczuk, D.; Syrbe, S.; Gomez-Ospina, N.; Manning, M.A.; Kostera-Pruszczyk, A.; Krahn-Peper, C.; Berutti, R.; et al. Bi-allelic ADPRHL2 Mutations Cause Neurodegeneration with Developmental Delay, Ataxia, and Axonal Neuropathy. Am. J. Hum. Genet. 2018. [Google Scholar] [CrossRef]
- Lattin, J.E.; Schroder, K.; Su, A.I.; Walker, J.R.; Zhang, J.; Wiltshire, T.; Saijo, K.; Glass, C.K.; Hume, D.A.; Kellie, S.; et al. Expression analysis of G Protein-Coupled Receptors in mouse macrophages. Immunome Res. 2008. [Google Scholar] [CrossRef]
- Zang, L.; Xue, B.; Lu, Z.; Li, X.; Yang, G.; Guo, Q.; Ba, J.; Zou, X.; Dou, J.; Lu, J.; et al. Identification of LRP16 as a negative regulator of insulin action and adipogenesis in 3T3-L1 adipocytes. Horm. Metab. Res. 2013. [Google Scholar] [CrossRef]
- Li, X.; Xue, B.; Wang, X.; Sun, L.; Zhang, T.; Qu, L.; Zou, X.; Mu, Y. Reduced expression of the LRP16 gene in mouse insulinoma (MIN6) cells exerts multiple effects on insulin content, proliferation and apoptosis. J. Huazhong Univ. Sci. Technol. - Med. Sci. 2012. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Guo, Q.H.; Wang, X.; Xue, B.; Sun, L.Q.; Meng, Q.T.; Lu, J.M.; Mu, Y.M. LRP16 gene protects mouse insulinoma MIN6 cells against fatty acid-induced apoptosis through Akt/FoxO1 signaling. Chin. Med. J. (Engl). 2012. [Google Scholar] [CrossRef]
- Li, Y.Z.; Zhao, P.; Han, W.D. Clinicopathological significance of LRP16 protein in 336 gastric carcinoma patients. World J. Gastroenterol. 2009. [Google Scholar] [CrossRef] [PubMed]
- Brunyanszki, A.; Szczesny, B.; Virág, L.; Szabo, C. Mitochondrial poly(ADP-ribose) polymerase: The Wizard of Oz at work. Free Radic. Biol. Med. 2016. [CrossRef]
- Zhou, H.-Z.; Swanson, R.A.; Simonis, U.; Ma, X.; Cecchini, G.; Gray, M.O. Poly(ADP-ribose) polymerase-1 hyperactivation and impairment of mitochondrial respiratory chain complex I function in reperfused mouse hearts. Am. J. Physiol. Heart Circ. Physiol. 2006. [Google Scholar] [CrossRef]
- Mayer, P.R.; Huang, N.; Dewey, C.M.; Dries, D.R.; Zhang, H.; Yu, G. Expression, localization, and biochemical characterization of nicotinamide mononucleotide adenylyltransferase 2. J. Biol. Chem. 2010. [Google Scholar] [CrossRef]
- Lau, C. The NMN/NaMN adenylyltransferase (NMNAT) protein family. Front. Biosci. 2009, 14, 410–431. [Google Scholar] [CrossRef]
- Zhang, T.; Berrocal, J.G.; Yao, J.; DuMond, M.E.; Krishnakumar, R.; Ruhl, D.D.; Ryu, K.W.; Gamble, M.J.; Kraus, W.L. Regulation of poly(ADP-ribose) polymerase-1-dependent gene expression through promoter-directed recruitment of a nuclear NAD + synthase. J. Biol. Chem. 2012. [Google Scholar] [CrossRef]
- Hottiger, M.O. SnapShot: ADP-Ribosylation Signaling. Mol. Cell 2015. [Google Scholar] [CrossRef]
- Fouquerel, E.; Sobol, R.W. ARTD1 (PARP1) activation and NAD+ in DNA repair and cell death. DNA Repair (Amst). 2014. [Google Scholar] [CrossRef]
- Kraus, W.L.; Hottiger, M.O. PARP-1 and gene regulation: Progress and puzzles. Mol. Aspects Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Posavec Marjanović, M.; Crawford, K.; Ahel, I. PARP, transcription and chromatin modeling. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Jubin, T.; Kadam, A.; Gani, A.R.; Singh, M.; Dwivedi, M.; Begum, R. Poly ADP-ribose polymerase-1: Beyond transcription and towards differentiation. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Knudsen, K.E. Transcriptional Roles of PARP1 in Cancer. Mol. Cancer Res. 2014. [Google Scholar] [CrossRef]
- Abplanalp, J.; Hottiger, M.O. Cell fate regulation by chromatin ADP-ribosylation. Semin. Cell Dev. Biol. 2017. [Google Scholar] [CrossRef]
- Chalkiadaki, A.; Guarente, L. The multifaceted functions of sirtuins in cancer. Nat. Rev. Cancer 2015. [Google Scholar] [CrossRef]
- Mao, Z.; Hine, C.; Tian, X.; Van Meter, M.; Au, M.; Vaidya, A.; Seluanov, A.; Gorbunova, V. SIRT6 promotes DNA repair under stress by activating PARP1. Science 2011. [Google Scholar] [CrossRef]
- Rezazadeh, S.; Yang, D.; Tombline, G.; Simon, M.; Regan, S.P.; Seluanov, A.; Gorbunova, V. SIRT6 promotes transcription of a subset of NRF2 targets by mono-ADP-ribosylating BAF170. Nucleic Acids Res. 2019. [Google Scholar] [CrossRef]
- Darlington, G.J.; Ross, S.E.; MacDougald, O.A. The role of C/EBP genes in adipocyte differentiation. J. Biol. Chem. 1998. [Google Scholar] [CrossRef]
- Poli, V. The role of C/EBP isoforms in the control of inflammatory and native immunity functions. J. Biol. Chem. 1998. [Google Scholar] [CrossRef]
- Westmacott, A.; Burke, Z.D.; Oliver, G.; Slack, J.M.W.; Tosh, D. C/EBPα and C/EBPβ are markers of early liver development. Int. J. Dev. Biol. 2006. [Google Scholar] [CrossRef] [PubMed]
- Tiranti, V.; Rossi, E.; Rocchi, M.; DiDonato, S.; Zuffardi, O.; Zeviani, M. The gene (nfe2l1) for human nrf-1, an activator involved in nuclear- mitochondrial interactions, maps to 7q32. Genomics 1995. [Google Scholar] [CrossRef] [PubMed]
- Hossain, M.B.; Ji, P.; Anish, R.; Jacobson, R.H.; Takada, S. Poly(ADP-ribose) polymerase 1 interacts with nuclear respiratory factor 1 (NRF-1) and plays a role in NRF-1 transcriptional regulation. J. Biol. Chem. 2009. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.G.; Robbins, P.A.; Ratcliffe, P.J. The human side of hypoxia-inducible factor Br. J. Haematol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Hulse, M.; Caruso, L.B.; Madzo, J.; Tan, Y.; Johnson, S.; Tempera, I. Poly(ADP-ribose) polymerase 1 is necessary for coactivating hypoxia-inducible factor-1-dependent gene expression by Epstein-Barr virus latent membrane protein 1. PLoS Pathog. 2018. [Google Scholar] [CrossRef]
- Martínez-Romero, R.; Martínez-Lara, E.; Aguilar-Quesada, R.; Peralta, A.; Oliver, F.J.; Siles, E. PARP-1 modulates deferoxamine-induced HIF-1α accumulation through the regulation of nitric oxide and oxidative stress. J. Cell. Biochem. 2008. [Google Scholar] [CrossRef]
- Rahman, S.; Islam, R. Mammalian Sirt1: Insights on its biological functions. Cell Commun. Signal. 2011. [Google Scholar] [CrossRef]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S.I. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 Homeobox 1 in the DMH and LH. Cell Metab. 2013. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Brunyánszki, A.; Huber, A.; Szántó, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011. [Google Scholar] [CrossRef]
- Mohamed, J.S.; Hajira, A.; Pardo, P.S.; Boriek, A.M. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1α network in skeletal muscle. Diabetes 2014. [Google Scholar] [CrossRef]
- Szántó, M.; Rutkai, I.; Hegedus, C.; Czikora, Á.; Rózsahegyi, M.; Kiss, B.; Virág, L.; Gergely, P.; Tóth, A.; Bai, P. Poly(ADP-ribose) polymerase-2 depletion reduces doxorubicin-induced damage through SIRT1 induction. Cardiovasc. Res. 2011. [Google Scholar] [CrossRef] [PubMed]
- Geng, B.; Cai, Y.; Gao, S.; Lu, J.; Zhang, L.; Zou, J.; Liu, M.; Yu, S.; Ye, J.; Liu, P. PARP-2 knockdown protects cardiomyocytes from hypertrophy via activation of SIRT1. Biochem. Biophys. Res. Commun. 2013. [Google Scholar] [CrossRef] [PubMed]
- Iyengar, S.; Farnham, P.J. KAP1 protein: An enigmatic master regulator of the genome. J. Biol. Chem. 2011. [Google Scholar] [CrossRef] [PubMed]
- Van Meter, M.; Kashyap, M.; Rezazadeh, S.; Geneva, A.J.; Morello, T.D.; Seluanov, A.; Gorbunova, V. SIRT6 represses LINE1 retrotransposons by ribosylating KAP1 but this repression fails with stress and age. Nat. Commun. 2014. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Sauve, A.A.; Bai, P. Crosstalk between poly(ADP-ribose) polymerase and sirtuin enzymes. Mol. Aspects Med. 2013. [Google Scholar] [CrossRef] [PubMed]
- Zong, W.X.; Ditsworth, D.; Bauer, D.E.; Wang, Z.Q.; Thompson, C.B. Alkylating DNA damage stimulates a regulated form of necrotic cell death. Genes Dev. 2004. [Google Scholar] [CrossRef]
- Bai, P.; Cantó, C.; Oudart, H.; Brunyánszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011. [Google Scholar] [CrossRef]
- Pirinen, E.; Cantó, C.; Jo, Y.S.; Morato, L.; Zhang, H.; Menzies, K.J.; Williams, E.G.; Mouchiroud, L.; Moullan, N.; Hagberg, C.; et al. Pharmacological inhibition of poly(ADP-ribose) polymerases improves fitness and mitochondrial function in skeletal muscle. Cell Metab. 2014. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Modification Side | Function | Function Affected by ADPR | Localization | References |
|---|---|---|---|---|---|
| ATP5A1 | E508, K506 (m) S184, S513 (h) | ATP synthesis | N/A | m | [75,76] |
| ATP5B | E98, R121 (m) | ATP synthesis | N/A | m | [75] |
| ATP5F1 | H164, S226 (h) | ATP synthesis | N/A | m | [76] |
| ATP5F1B | S415, R458, H477 (h) | ATP synthesis | N/A | m | [76] |
| ATP5F1C | S195 (h) | ATP synthesis | N/A | m | [76] |
| ATP50 | R29, S126 (m) S163, S166 (h) | ATP synthesis | N/A | m | [75,76] |
| C/EBP-b | E135, K133, E139 (m) S141, S257 (h) | Transcription factor, gene expression | Yes (activity dampened) | n | [76,77] |
| COX4I1 | H51 (h) | Electron transport chain | N/A | m | [76] |
| CYCS | S34 (h) | Electron transport chain | N/A | m | [76] |
| GAPDH | R198, R232 (m) R80, S122, S192, R197, S210 (h) | Glycolysis | N/A | c, n | [75,76] |
| MDH1 | S241 (h) | Carbohydrate metabolism | N/A | m | [76] |
| MDH2 | S261, S246 (m) S317 (h) | Carbohydrate metabolism | N/A | m | [75,76] |
| NDUFB1 | R49 (m) | Electron transport | N/A | m | [75] |
| NDUFB5 | R93 (m) | Electron transport | N/A | m | [75] |
| NDUFAF7 | R415 (m) | Electron transport | N/A | m | [75] |
| NDUFC2 | R120 (m) | Electron transport | N/A | m | [75] |
| NDUFAB1 | S99 (h) | Electron transport | N/A | m | [76] |
| NDUFV1 | R449 (m) | Electron transport | N/A | m | [75] |
| PDPR | R866 (m) | Carbohydrate metabolism | N/A | m | [75] |
| PDHA1 | R304 (m) | Carbohydrate metabolism | N/A | m | [75] |
| PDHX | S131 (m) | Carbohydrate metabolism | N/A | m | [75] |
| SDHA | S505, H522 (h) | Electron transport | N/A | m | [76] |
| UQCRC1 | S221 (h) | Electron transport | N/A | m | [76] |
| UQCRC2 | R241 (m) S221 (h) | Electron transport | N/A | m | [75,76] |
| UQCRFS1 | E95 (m) | Electron transport | N/A | m | [75] |
| Nucleus | ||||
| Family | Name | NAD+ Metabolism | ART/ARH Activity | References |
| ARTs | ARTD1 | Consumption | Poly (branching) | [79,80,81] |
| ARTD2 | Consumption | Poly (branching) | [79,80,81] | |
| ARTD3 | Consumption | Mono | [79,80,81] | |
| ARTD4 | Consumption | Mono | [79,80,81] | |
| ARTD5 | Consumption | Poly/Oligo | [79,80,81] | |
| ARTD6 | Consumption | Poly/Oligo | [79,80,81] | |
| ARTD8 | Consumption | Mono | [79,80,81] | |
| ARTD9 | Consumption | Inactive/Mono | [79,80,82] | |
| ARTD10 | Consumption | Mono | [79,80,81] | |
| ARTD11 | Consumption | Mono | [79,80,81] | |
| ARTD14 | Consumption | Mono | [79,80,81] | |
| ARHs | PARG | Poly | [83,84] | |
| ARH3 | Poly/Mono | [85,86,87] | ||
| TARG | Mono | [88,89,90] | ||
| SIRTs | SIRT1 | Consumption | N/A | [39] |
| SIRT2 | Consumption | N/A | [39] | |
| SIRT6 | Consumption | Mono | [39,91] | |
| SIRT7 | Consumption | N/A | [39] | |
| NAMPT | NAMPT | Synthesis | N/A | [2] |
| NMNAT | NMNAT1 | Synthesis | N/A | [2] |
| Cytoplasm | ||||
| Family | Name | NAD+ Metabolism | ART/ARH Activity | References |
| ARTs | ARTD2 | Consumption | Poly (branching) | [79,80,81] |
| ARTD3 | Consumption | Mono | [79,80,81] | |
| ARTD4 | Consumption | Mono | [79,80,81] | |
| ARTD5 | Consumption | Poly/Oligo | [79,80,81] | |
| ARTD6 | Consumption | Poly/Oligo | [79,80,81] | |
| ARTD7 | Consumption | Mono | [79,80,81] | |
| ARTD8 | Consumption | Mono | [79,80,81] | |
| ARTD9 | Consumption | Inactive/Mono | [79,80,82] | |
| ARTD10 | Consumption | Mono | [79,80,81] | |
| ARTD11 | Consumption | Mono | [79,80,81] | |
| ARTD12 | Consumption | Mono | [79,80,81] | |
| ARTD13 | Consumption | Inactive | [79,80,81] | |
| ARTD14 | Consumption | Mono | [79,80,81] | |
| ARTD15 | Consumption | Mono | [79,80,81] | |
| ARTD16 | Consumption | Mono | [79,80,81] | |
| ARTD17 | Consumption | Mono | [79,80,81] | |
| ARTD18 | Consumption | Mono | [79,80,81] | |
| ARHs | PARG | Poly | [83,84] | |
| ARH1 | Mono | [85] | ||
| ARH2 | Inactive | [85] | ||
| MacroD2 | Mono | [88,89,90] | ||
| SIRTs | SIRT1 | Consumption | N/A | [39] |
| SIRT2 | Consumption | N/A | [39] | |
| NAMPT | NAMPT | Synthesis | N/A | [2] |
| NMNAT | NMNAT2 | Synthesis | N/A | [2] |
| Mitochondria | ||||
| Family | Name | NAD+ Metabolism | ART/ARH Activity | References |
| ARTs | ARTD1 | Consumption | Poly (branching) | [79,80] |
| ARHs | PARG | Poly | [83,84,92] | |
| ARH3 | Poly/Mono | [85,86,87] | ||
| MacroD1 | Mono | [88,89,90,93] | ||
| SIRTs | SIRT3 | Consumption | N/A | [39] |
| SIRT4 | Consumption | Mono | [39,94] | |
| SIRT5 | Consumption | N/A | [39] | |
| NAMPT | NAMPT | Synthesis | N/A | [2] |
| NMNAT | NMNAT3 | Synthesis | N/A | [2] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hopp, A.-K.; Grüter, P.; Hottiger, M.O. Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells 2019, 8, 890. https://doi.org/10.3390/cells8080890
Hopp A-K, Grüter P, Hottiger MO. Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells. 2019; 8(8):890. https://doi.org/10.3390/cells8080890
Chicago/Turabian StyleHopp, Ann-Katrin, Patrick Grüter, and Michael O. Hottiger. 2019. "Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation" Cells 8, no. 8: 890. https://doi.org/10.3390/cells8080890
APA StyleHopp, A.-K., Grüter, P., & Hottiger, M. O. (2019). Regulation of Glucose Metabolism by NAD+ and ADP-Ribosylation. Cells, 8(8), 890. https://doi.org/10.3390/cells8080890