Thymic Stromal Lymphopoietin Interferes with the Apoptosis of Human Skin Mast Cells by a Dual Strategy Involving STAT5/Mcl-1 and JNK/Bcl-xL




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Purification and Culture of Human Skin Mast Cells and Other Skin Cells
2.2. Cell Treatment
2.3. Analysis of Survival
2.4. Staining of Intracellular and Extracellular Proteins
2.5. siRNA Transfection
2.6. RT-quantitative PCR
| SCF | Forward | GCGTGGACTATCTGCCGCCG |
| Reverse | AGCGCTGCGATCCAGCACAAA | |
| IL-33 | Forward | TGTCAACAGCAGTCTACTGTGG |
| Reverse | TGGACCCCTGATATACCAAAGG |
2.7. Immunoblotting
2.8. Statistical Analysis
3. Results
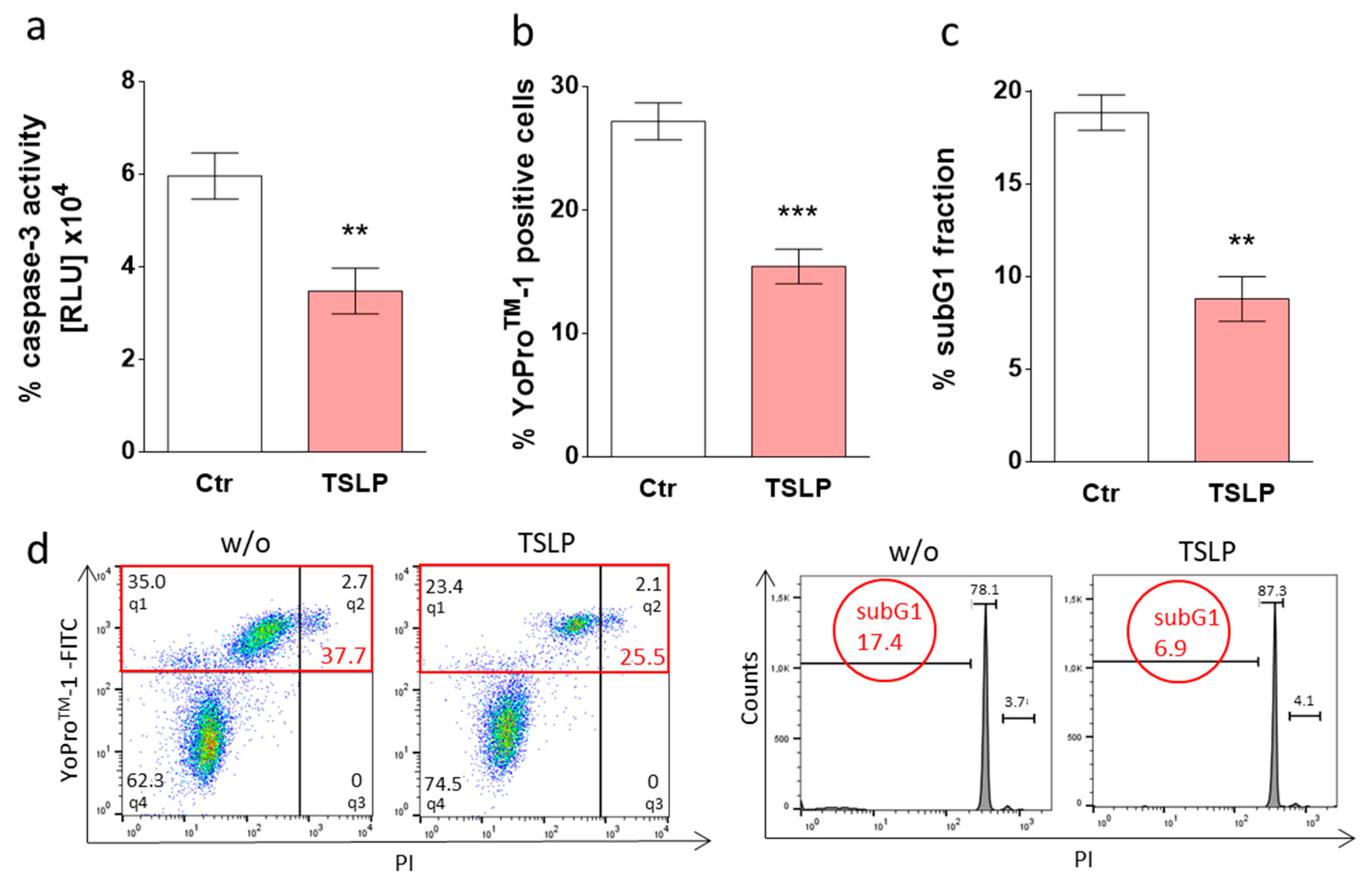
3.1. TSLP Counters Apoptosis of Skin MCs upon Growth Factor Withdrawal
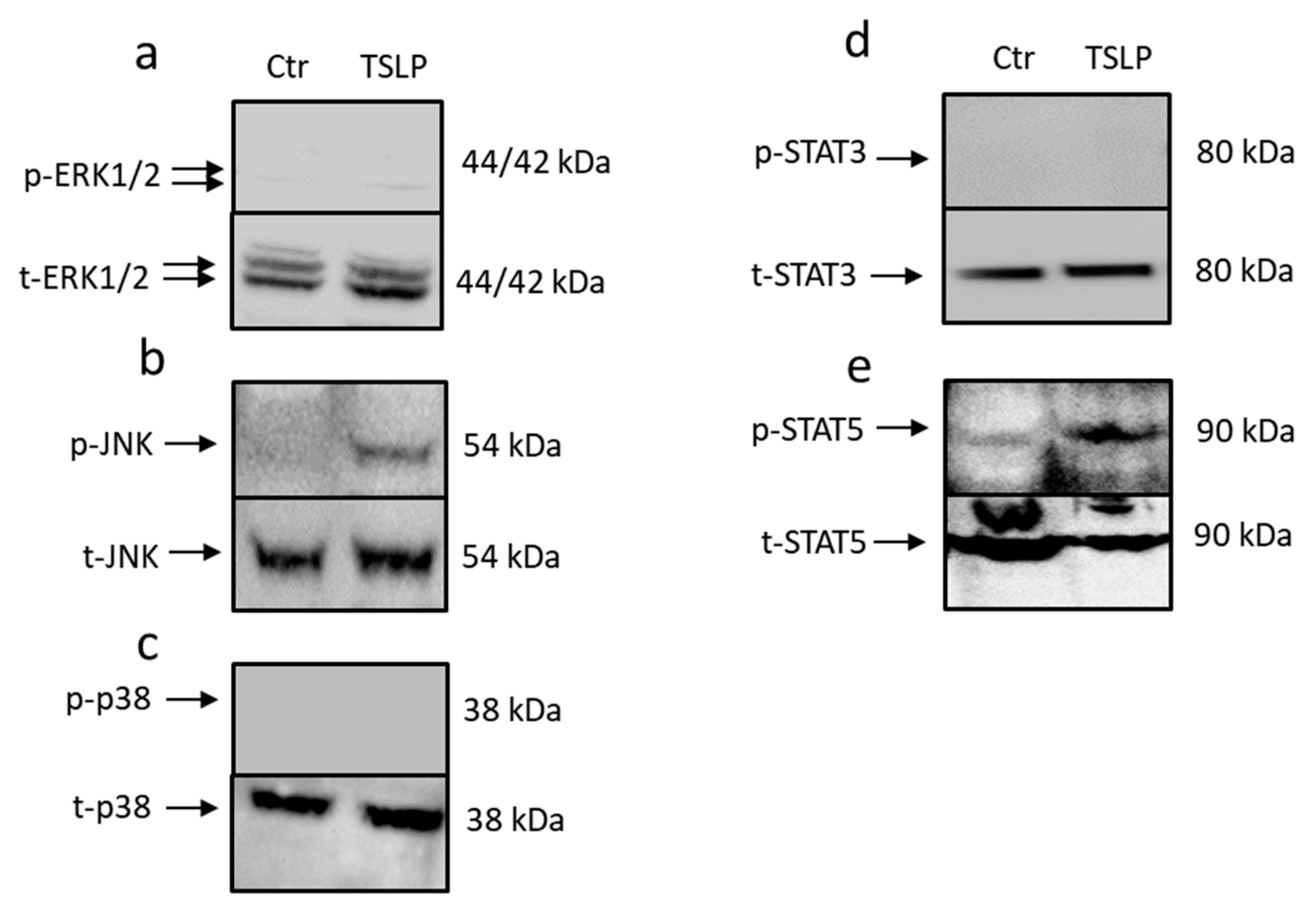
3.2. TSLP Triggers Activation of STAT5 and JNK
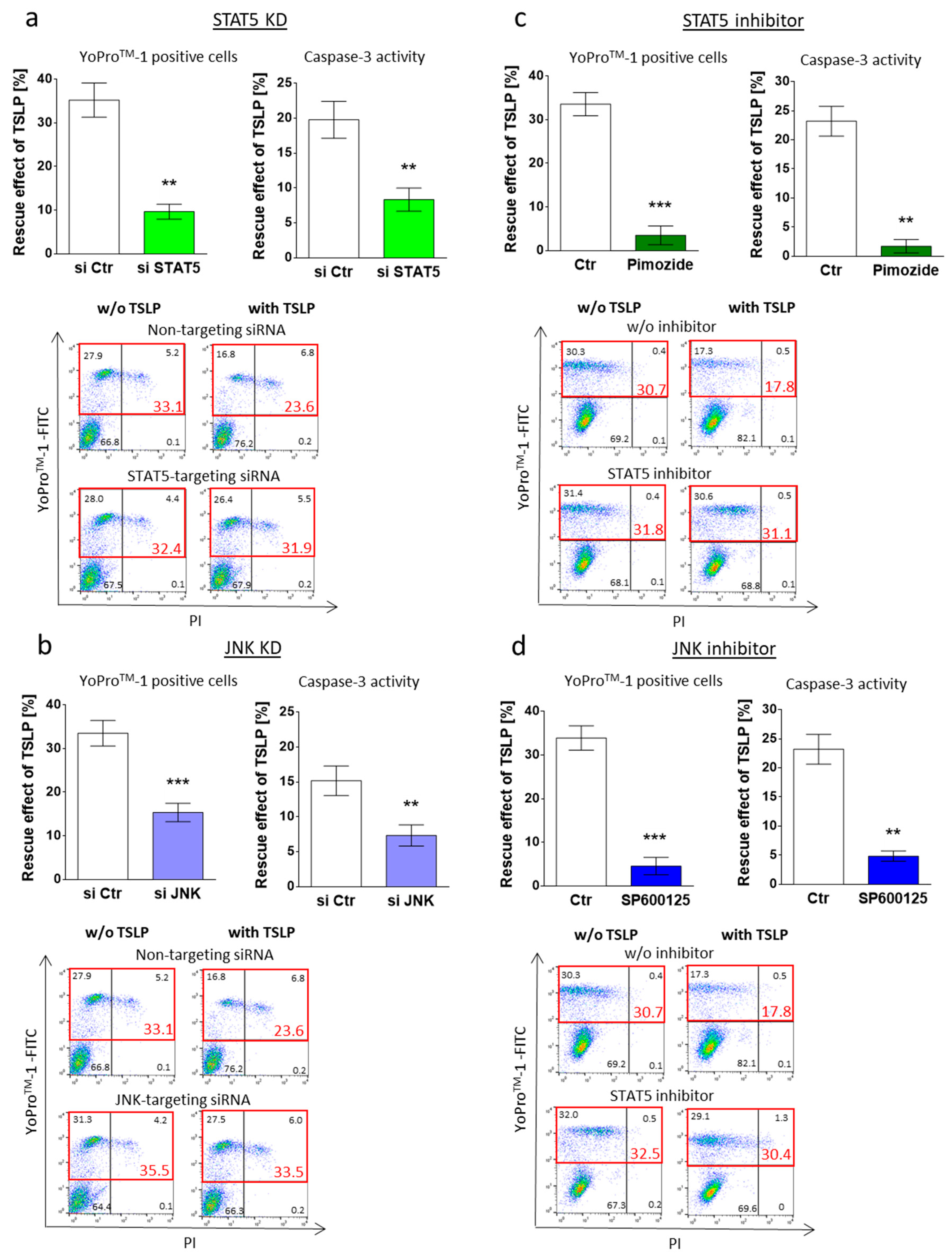
3.3. MC Maintenance by TSLP Critically Depends on JNK and STAT5
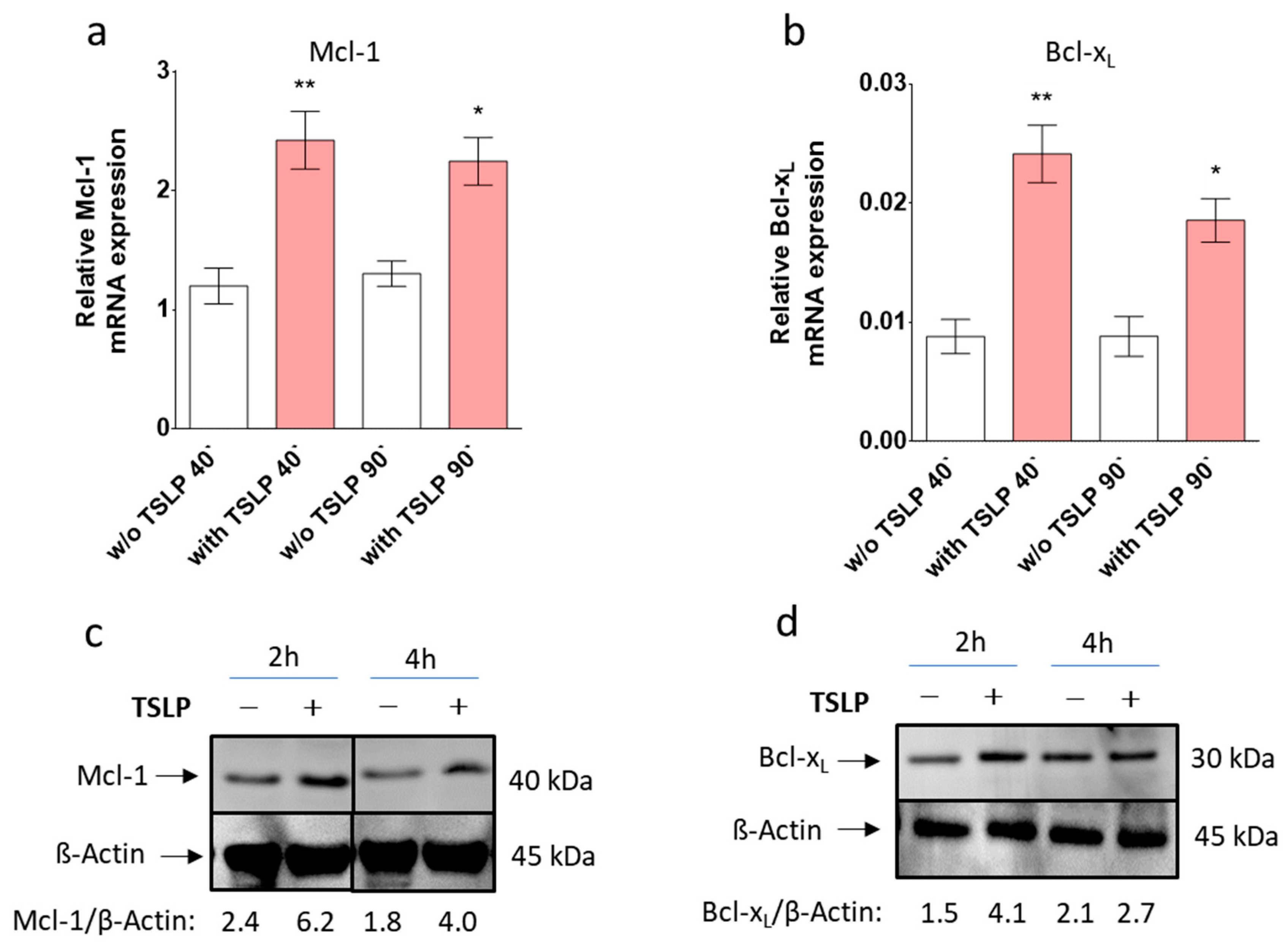
3.4. TSLP up-Regulates Mcl-1 and Bcl-xL
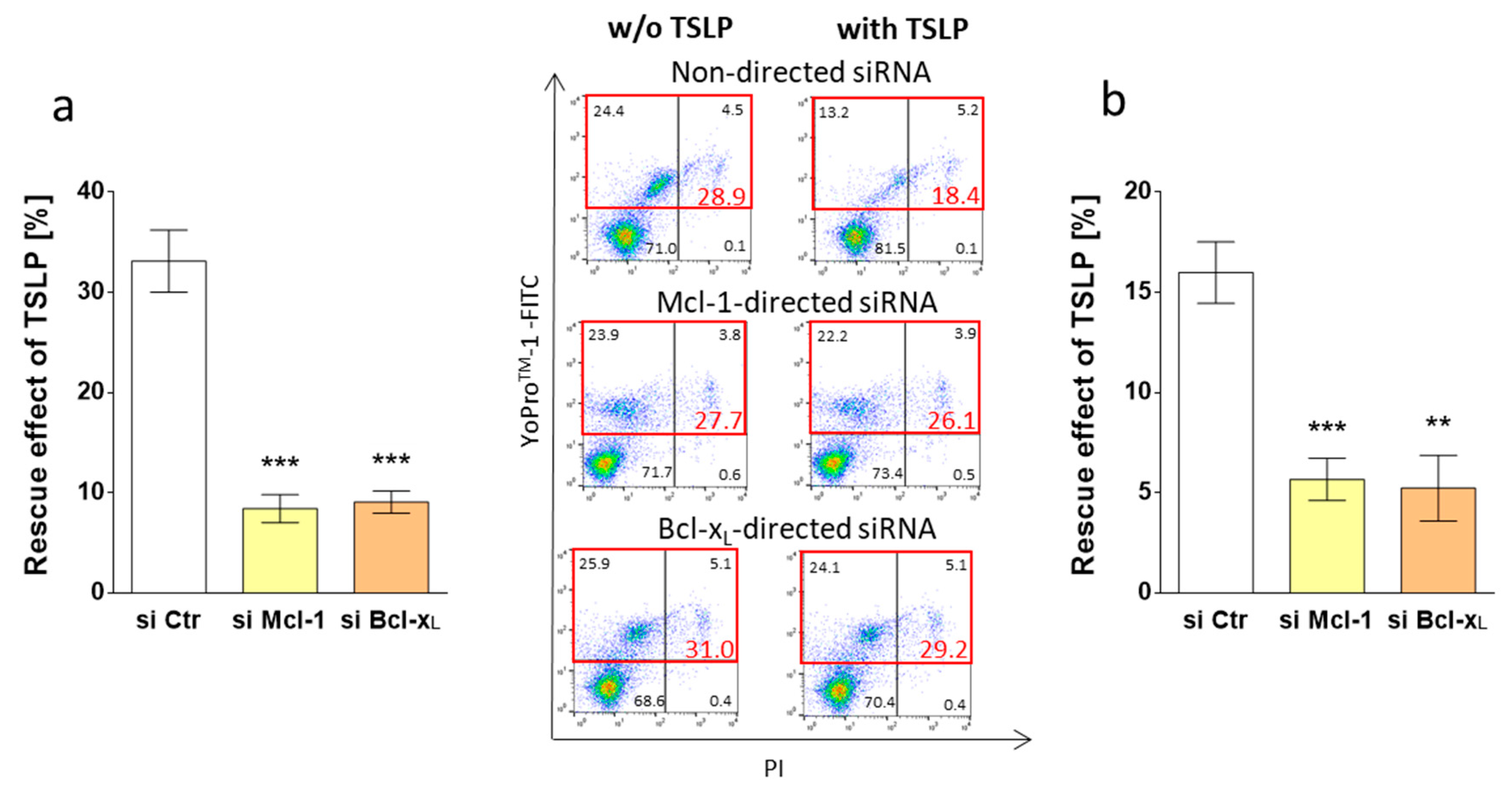
3.5. Survival by TSLP Depends on Mcl-1 and Bcl-xL
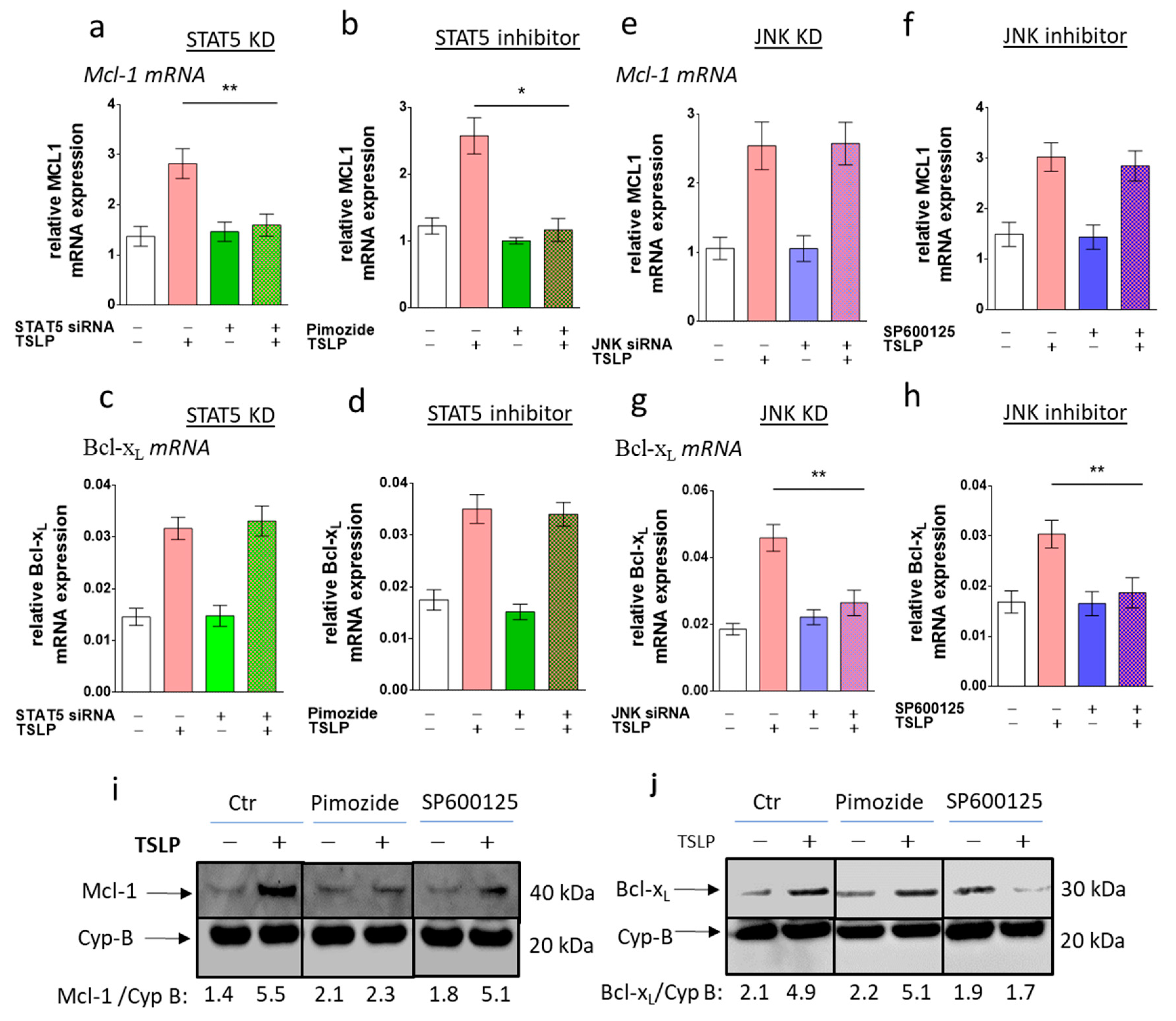
3.6. STAT5 Perturbation Leads to Mcl-1 Downregulation, While Interference with JNK Attenuates Bcl-xL Expression
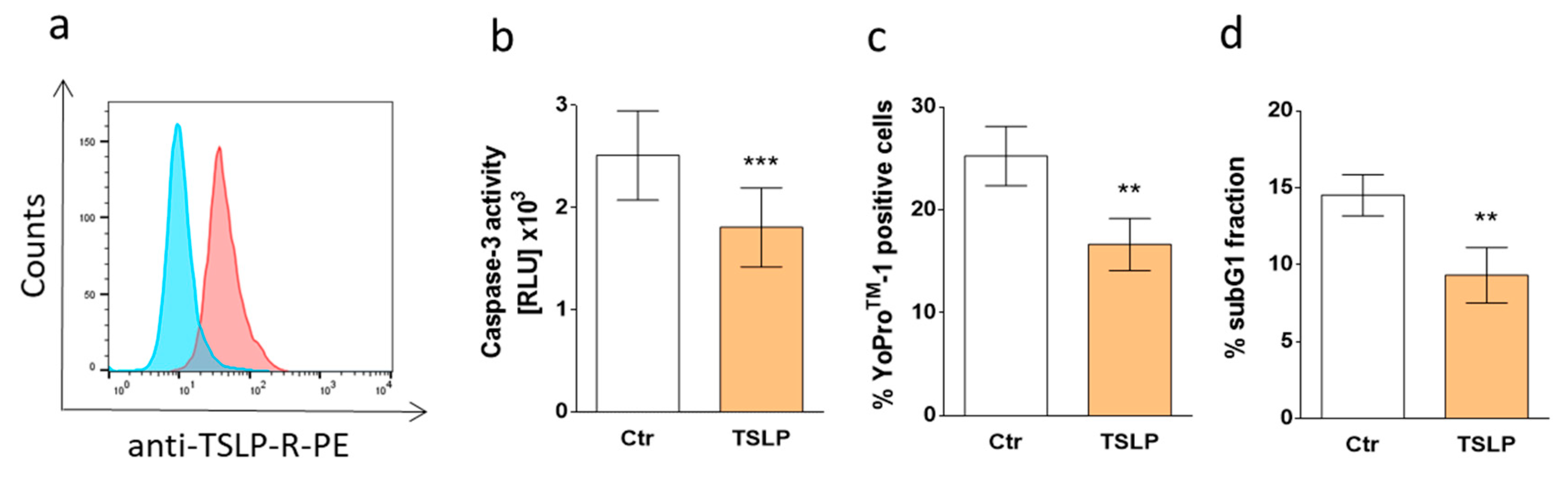
3.7. TSLP Protects Skin MCs Ex Vivo from Cell Death
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Galli, S.J.; Tsai, M.; Piliponsky, A.M. The development of allergic inflammation. Nature 2008, 454, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, T.; Ando, T.; Kimura, M.; Wilson, B.S.; Kawakami, Y. Mast cells in atopic dermatitis. Curr. Opin. Immunol. 2009, 21, 666–678. [Google Scholar] [CrossRef] [PubMed]
- Gilfillan, A.M.; Austin, S.J.; Metcalfe, D.D. Mast cell biology: Introduction and overview. Adv. Exp. Med. Biol. 2011, 716, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Theoharides, T.C.; Alysandratos, K.D.; Angelidou, A.; Delivanis, D.A.; Sismanopoulos, N.; Zhang, B.; Asadi, S.; Vasiadi, M.; Weng, Z.; Miniati, A.; et al. Mast cells and inflammation. Biochim. Biophys. Acta Mol. Basis Dis. 2012, 1822, 21–33. [Google Scholar] [CrossRef] [PubMed]
- Galli, S.J.; Tsai, M. IgE and mast cells in allergic disease. Nat. Med. 2012, 18, 693–704. [Google Scholar] [CrossRef] [PubMed]
- Olivera, A.; Beaven, M.A.; Metcalfe, D.D. Mast cells signal their importance in health and disease. J. Allergy Clin. Immunol. 2018, 142, 381–393. [Google Scholar] [CrossRef]
- Dwyer, D.F.; Barrett, N.A.; Austen, K.F.; Consortium, I.G.P. Expression profiling of constitutive mast cells reveals a unique identity within the immune system. Nat. Immunol. 2016, 17, 878–887. [Google Scholar] [CrossRef] [PubMed]
- Kneilling, M.; Röcken, M. Mast cells: Novel clinical perspectives from recent insights. Exp. Dermatol. 2009, 18, 488–496. [Google Scholar] [CrossRef] [PubMed]
- Ekoff, M.; Nilsson, G. Mast cell apoptosis and survival. Adv. Exp. Med. Biol. 2011, 716, 47–60. [Google Scholar] [CrossRef]
- Gurish, M.F.; Austen, K.F. Developmental origin and functional specialization of mast cell subsets. Immunity 2012, 37, 25–33. [Google Scholar] [CrossRef]
- Bulfone-Paus, S.; Nilsson, G.; Draber, P.; Blank, U.; Levi-Schaffer, F. Positive and Negative Signals in Mast Cell Activation. Trends Immunol. 2017, 38, 657–667. [Google Scholar] [CrossRef]
- Kitamura, Y.; Shimada, M.; Hatanaka, K.; Miyano, Y. Development of mast cells from grafted bone marrow cells in irradiated mice. Nature 1977, 268, 442–443. [Google Scholar] [CrossRef]
- Matsuda, H.; Kitamura, Y.; Sonoda, T.; Imori, T. Precursor of mast cells fixed in the skin of mice. J. Cell. Physiol. 1981, 108, 409–415. [Google Scholar] [CrossRef]
- Hazzan, T.; Eberle, J.; Worm, M.; Babina, M. Apoptotic resistance of human skin mast cells is mediated by Mcl-1. Cell Death Discov. 2017, 3, 17048. [Google Scholar] [CrossRef]
- Okayama, Y.; Kawakami, T. Development, migration, and survival of mast cells. Immunol. Res. 2006, 34, 97–115. [Google Scholar] [CrossRef]
- Wang, J.X.; Kaieda, S.; Ameri, S.; Fishgal, N.; Dwyer, D.; Dellinger, A.; Kepley, C.L.; Gurish, M.F.; Nigrovic, P.A. IL-33/ST2 axis promotes mast cell survival via BCLXL. Proc. Natl. Acad. Sci. USA 2014, 111, 10281–10286. [Google Scholar] [CrossRef]
- Sellge, G.; Lorentz, A.; Gebhardt, T.; Levi-Schaffer, F.; Bektas, H.; Manns, M.P.; Schuppan, D.; Bischoff, S.C. Human intestinal fibroblasts prevent apoptosis in human intestinal mast cells by a mechanism independent of stem cell factor, IL-3, IL-4, and nerve growth factor. J. Immunol. 2004, 172, 260–267. [Google Scholar] [CrossRef]
- Soumelis, V.; Reche, P.A.; Kanzler, H.; Yuan, W.; Edward, G.; Homey, B.; Gilliet, M.; Ho, S.; Antonenko, S.; Lauerma, A.; et al. Human epithelial cells trigger dendritic cell mediated allergic inflammation by producing TSLP. Nat. Immunol. 2002, 3, 673–680. [Google Scholar] [CrossRef]
- Cianferoni, A.; Spergel, J. The importance of TSLP in allergic disease and its role as a potential therapeutic target. Expert Rev. Clin. Immunol. 2014, 10, 1463–1474. [Google Scholar] [CrossRef]
- Ziegler, S.F. Thymic stromal lymphopoietin and allergic disease. J. Allergy Clin. Immunol. 2012, 130, 845–852. [Google Scholar] [CrossRef]
- Volpe, E.; Pattarini, L.; Martinez-Cingolani, C.; Meller, S.; Donnadieu, M.H.; Bogiatzi, S.I.; Fernandez, M.I.; Touzot, M.; Bichet, J.C.; Reyal, F.; et al. Thymic stromal lymphopoietin links keratinocytes and dendritic cell-derived IL-23 in patients with psoriasis. J. Allergy Clin. Immunol. 2014, 134, 373–381. [Google Scholar] [CrossRef]
- Corren, J.; Parnes, J.R.; Wang, L.; Mo, M.; Roseti, S.L.; Griffiths, J.M.; van der Merwe, R. Tezepelumab in Adults with Uncontrolled Asthma. N. Engl. J. Med. 2017, 377, 936–946. [Google Scholar] [CrossRef]
- Simpson, E.L.; Parnes, J.R.; She, D.; Crouch, S.; Rees, W.; Mo, M.; van der Merwe, R. Tezepelumab, an anti-thymic stromal lymphopoietin monoclonal antibody, in the treatment of moderate to severe atopic dermatitis: A randomized phase 2a clinical trial. J. Am. Acad. Dermatol. 2018. [Google Scholar] [CrossRef]
- Nagarkar, D.R.; Poposki, J.A.; Comeau, M.R.; Biyasheva, A.; Avila, P.C.; Schleimer, R.P.; Kato, A. Airway epithelial cells activate TH2 cytokine production in mast cells through IL-1 and thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2012, 130, 225–232. [Google Scholar] [CrossRef]
- Kaur, D.; Doe, C.; Woodman, L.; Heidi Wan, W.Y.; Sutcliffe, A.; Hollins, F.; Brightling, C. Mast cell-airway smooth muscle crosstalk: The role of thymic stromal lymphopoietin. Chest 2012, 142, 76–85. [Google Scholar] [CrossRef]
- Allakhverdi, Z.; Comeau, M.R.; Armant, M.; Agrawal, R.; Woodfolk, J.A.; Sehmi, R.; Howie, K.J.; Gauvreau, G.M.; Delespesse, G. Mast Cell-Activated Bone Marrow Mesenchymal Stromal Cells Regulate Proliferation and Lineage Commitment of CD34(+) Progenitor Cells. Front. Immunol. 2013, 4, 461. [Google Scholar] [CrossRef]
- Han, N.R.; Oh, H.A.; Nam, S.Y.; Moon, P.D.; Kim, D.W.; Kim, H.M.; Jeong, H.J. TSLP induces mast cell development and aggravates allergic reactions through the activation of MDM2 and STAT6. J. Investig. Dermatol. 2014, 134, 2521–2530. [Google Scholar] [CrossRef]
- Rönnberg, E.; Ghaib, A.; Ceriol, C.; Enoksson, M.; Arock, M.; Säfholm, J.; Ekoff, M.; Nilsson, G. Divergent Effects of Acute and Prolonged Interleukin 33 Exposure on Mast Cell IgE-Mediated Functions. Front. Immunol. 2019, 10, 1361. [Google Scholar] [CrossRef]
- Irani, A.M.; Schwartz, L.B. Human mast cell heterogeneity. Allergy Proc. 1994, 15, 303–308. [Google Scholar] [CrossRef]
- Moon, T.C.; St Laurent, C.D.; Morris, K.E.; Marcet, C.; Yoshimura, T.; Sekar, Y.; Befus, A.D. Advances in mast cell biology: New understanding of heterogeneity and function. Mucosal Immunol. 2010, 3, 111–128. [Google Scholar] [CrossRef]
- Wang, Z.; Guhl, S.; Franke, K.; Artuc, M.; Zuberbier, T.; Babina, M. IL-33 and MRGPRX2-Triggered Activation of Human Skin Mast Cells-Elimination of Receptor Expression on Chronic Exposure, but Reinforced Degranulation on Acute Priming. Cells 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Wang, Z.; Franke, K.; Guhl, S.; Artuc, M.; Zuberbier, T. Yin-yang of IL-33 in human skin mast cells: Reduced degranulation, but augmented histamine synthesis through p38 activation. J. Investig. Dermatol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Wang, Z.; Artuc, M.; Guhl, S.; Zuberbier, T. MRGPRX2 is negatively targeted by SCF and IL-4 to diminish pseudo-allergic stimulation of skin mast cells in culture. Exp. Dermatol. 2018, 27, 1298–1303. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Guhl, S.; Stärke, A.; Kirchhof, L.; Zuberbier, T.; Henz, B.M. Comparative cytokine profile of human skin mast cells from two compartments—Strong resemblance with monocytes at baseline but induction of IL-5 by IL-4 priming. J. Leukoc. Biol. 2004, 75, 244–252. [Google Scholar] [CrossRef] [PubMed]
- Babina, M.; Guhl, S.; Artuc, M.; Zuberbier, T. Allergic FcεRI- and pseudo-allergic MRGPRX2-triggered mast cell activation routes are independent and inversely regulated by SCF. Allergy 2018, 73, 256–260. [Google Scholar] [CrossRef]
- Hazzan, T.; Guhl, S.; Artuc, M.; Franke, K.; Worm, M.; Zuberbier, T.; Babina, M. An efficient method for gene knock-down by RNA interference in human skin mast cells. Exp. Dermatol. 2017, 26, 1136–1139. [Google Scholar] [CrossRef] [PubMed]
- Guhl, S.; Babina, M.; Neou, A.; Zuberbier, T.; Artuc, M. Mast cell lines HMC-1 and LAD2 in comparison with mature human skin mast cells—Drastically reduced levels of tryptase and chymase in mast cell lines. Exp. Dermatol. 2010, 19, 845–847. [Google Scholar] [CrossRef] [PubMed]
- Guhl, S.; Artuc, M.; Neou, A.; Babina, M.; Zuberbier, T. Long-term cultured human skin mast cells are suitable for pharmacological studies of anti-allergic drugs due to high responsiveness to FcεRI cross-linking. Biosci. Biotechnol. Biochem. 2011, 75, 382–384. [Google Scholar] [CrossRef] [PubMed]
- Guhl, S.; Hartmann, K.; Tapkenhinrichs, S.; Smorodchenko, A.; Grützkau, A.; Henz, B.M.; Zuberbier, T. Ultraviolet irradiation induces apoptosis in human immature, but not in skin mast cells. J. Investig. Dermatol. 2003, 121, 837–844. [Google Scholar] [CrossRef]
- Kim, J.; Guhl, S.; Babina, M.; Zuberbier, T.; Artuc, M. Integration of the human dermal mast cell into the organotypic co-culture skin model. Methods Mol. Biol. 2014, 1192, 69–85. [Google Scholar] [CrossRef]
- Kumari, V.; Babina, M.; Hazzan, T.; Worm, M. Thymic stromal lymphopoietin induction by skin irritation is independent of tumour necrosis factor-α, but supported by interleukin-1. Br. J. Dermatol. 2015, 172, 951–960. [Google Scholar] [CrossRef]
- Babina, M.; Guhl, S.; Artuc, M.; Trivedi, N.N.; Zuberbier, T. Phenotypic variability in human skin mast cells. Exp. Dermatol. 2016, 25, 434–439. [Google Scholar] [CrossRef]
- Butterfield, J.H.; Weiler, D.; Dewald, G.; Gleich, G.J. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leuk. Res. 1988, 12, 345–355. [Google Scholar] [CrossRef]
- Motakis, E.; Guhl, S.; Ishizu, Y.; Itoh, M.; Kawaji, H.; de Hoon, M.; Lassmann, T.; Carninci, P.; Hayashizaki, Y.; Zuberbier, T.; et al. Redefinition of the human mast cell transcriptome by deep-CAGE sequencing. Blood 2014, 123, e58–e67. [Google Scholar] [CrossRef]
- Galluzzi, L.; Bravo-San Pedro, J.M.; Vitale, I.; Aaronson, S.A.; Abrams, J.M.; Adam, D.; Alnemri, E.S.; Altucci, L.; Andrews, D.; Annicchiarico-Petruzzelli, M.; et al. Essential versus accessory aspects of cell death: Recommendations of the NCCD 2015. Cell Death Differ. 2015, 22, 58–73. [Google Scholar] [CrossRef]
- Collins, J.A.; Schandi, C.A.; Young, K.K.; Vesely, J.; Willingham, M.C. Major DNA fragmentation is a late event in apoptosis. J. Histochem. Cytochem. 1997, 45, 923–934. [Google Scholar] [CrossRef]
- Chaix, A.; Lopez, S.; Voisset, E.; Gros, L.; Dubreuil, P.; De Sepulveda, P. Mechanisms of STAT protein activation by oncogenic KIT mutants in neoplastic mast cells. J. Biol. Chem. 2011, 286, 5956–5966. [Google Scholar] [CrossRef]
- Guhl, S.; Neou, A.; Artuc, M.; Zuberbier, T.; Babina, M. Skin mast cells develop non-synchronized changes in typical lineage characteristics upon culture. Exp. Dermatol. 2014, 23, 933–935. [Google Scholar] [CrossRef]
- Forrest, A.R.; Kawaji, H.; Rehli, M.; Baillie, J.K.; de Hoon, M.J.; Haberle, V.; Lassmann, T.; Kulakovskiy, I.V.; Lizio, M.; Itoh, M.; et al. A promoter-level mammalian expression atlas. Nature 2014, 507, 462–470. [Google Scholar] [CrossRef]
- Metcalfe, D.D. Mast cells and mastocytosis. Blood 2008, 112, 946–956. [Google Scholar] [CrossRef]
- Fang, Y.; Larsson, L.; Bruhns, P.; Xiang, Z. Apoptosis of mouse mast cells is reciprocally regulated by the IgG receptors FcγRIIB and FcγRIIIA. Allergy 2012, 67, 1233–1240. [Google Scholar] [CrossRef]
- Takai, T. TSLP expression: Cellular sources, triggers, and regulatory mechanisms. Allergol. Int. 2012, 61, 3–17. [Google Scholar] [CrossRef]
- Leyva-Castillo, J.M.; Hener, P.; Jiang, H.; Li, M. TSLP produced by keratinocytes promotes allergen sensitization through skin and thereby triggers atopic march in mice. J. Investig. Dermatol. 2013, 133, 154–163. [Google Scholar] [CrossRef]
- Varricchi, G.; Pecoraro, A.; Marone, G.; Criscuolo, G.; Spadaro, G.; Genovese, A. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front. Immunol. 2018, 9, 1595. [Google Scholar] [CrossRef]
- Lai, Y.; Altemeier, W.A.; Vandree, J.; Piliponsky, A.M.; Johnson, B.; Appel, C.L.; Frevert, C.W.; Hyde, D.M.; Ziegler, S.F.; Smith, D.E.; et al. Increased density of intraepithelial mast cells in patients with exercise-induced bronchoconstriction regulated through epithelially derived thymic stromal lymphopoietin and IL-33. J. Allergy Clin. Immunol. 2014, 133, 1448–1455. [Google Scholar] [CrossRef]
- Wong, C.K.; Hu, S.; Cheung, P.F.; Lam, C.W. Thymic stromal lymphopoietin induces chemotactic and prosurvival effects in eosinophils: Implications in allergic inflammation. Am. J. Respir. Cell Mol. Biol. 2010, 43, 305–315. [Google Scholar] [CrossRef]
- Scheeren, F.A.; van Lent, A.U.; Nagasawa, M.; Weijer, K.; Spits, H.; Legrand, N.; Blom, B. Thymic stromal lymphopoietin induces early human B-cell proliferation and differentiation. Eur. J. Immunol. 2010, 40, 955–965. [Google Scholar] [CrossRef]
- Tormo, A.J.; Gauchat, J.F. A novel role for STAT5 in DC: Controlling the Th2-response. JAK-STAT 2013, 2, e25352. [Google Scholar] [CrossRef]
- Kabata, H.; Moro, K.; Fukunaga, K.; Suzuki, Y.; Miyata, J.; Masaki, K.; Betsuyaku, T.; Koyasu, S.; Asano, K. Thymic stromal lymphopoietin induces corticosteroid resistance in natural helper cells during airway inflammation. Nat. Commun. 2013, 4, 2675. [Google Scholar] [CrossRef]
- Bell, B.D.; Kitajima, M.; Larson, R.P.; Stoklasek, T.A.; Dang, K.; Sakamoto, K.; Wagner, K.U.; Kaplan, D.H.; Reizis, B.; Hennighausen, L.; et al. The transcription factor STAT5 is critical in dendritic cells for the development of TH2 but not TH1 responses. Nat. Immunol. 2013, 14, 364–371. [Google Scholar] [CrossRef]
- Yao, W.; Zhang, Y.; Jabeen, R.; Nguyen, E.T.; Wilkes, D.S.; Tepper, R.S.; Kaplan, M.H.; Zhou, B. Interleukin-9 is required for allergic airway inflammation mediated by the cytokine TSLP. Immunity 2013, 38, 360–372. [Google Scholar] [CrossRef]
- Takahashi, N.; Sugaya, M.; Suga, H.; Oka, T.; Kawaguchi, M.; Miyagaki, T.; Fujita, H.; Sato, S. Thymic Stromal Chemokine TSLP Acts through Th2 Cytokine Production to Induce Cutaneous T-cell Lymphoma. Cancer Res. 2016, 76, 6241–6252. [Google Scholar] [CrossRef]
- Liu, S.; Verma, M.; Michalec, L.; Liu, W.; Sripada, A.; Rollins, D.; Good, J.; Ito, Y.; Chu, H.; Gorska, M.M.; et al. Steroid resistance of airway type 2 innate lymphoid cells from patients with severe asthma: The role of thymic stromal lymphopoietin. J. Allergy Clin. Immunol. 2018, 141, 257–268. [Google Scholar] [CrossRef]
- Shan, L.; Redhu, N.S.; Saleh, A.; Halayko, A.J.; Chakir, J.; Gounni, A.S. Thymic stromal lymphopoietin receptor-mediated IL-6 and CC/CXC chemokines expression in human airway smooth muscle cells: Role of MAPKs (ERK1/2, p38, and JNK) and STAT3 pathways. J. Immunol. 2010, 184, 7134–7143. [Google Scholar] [CrossRef]
- Shelburne, C.P.; McCoy, M.E.; Piekorz, R.; Sexl, V.; Roh, K.H.; Jacobs-Helber, S.M.; Gillespie, S.R.; Bailey, D.P.; Mirmonsef, P.; Mann, M.N.; et al. Stat5 expression is critical for mast cell development and survival. Blood 2003, 102, 1290–1297. [Google Scholar] [CrossRef]
- Barnstein, B.O.; Li, G.; Wang, Z.; Kennedy, S.; Chalfant, C.; Nakajima, H.; Bunting, K.D.; Ryan, J.J. Stat5 expression is required for IgE-mediated mast cell function. J. Immunol. 2006, 177, 3421–3426. [Google Scholar] [CrossRef]
- Li, G.; Miskimen, K.L.; Wang, Z.; Xie, X.Y.; Brenzovich, J.; Ryan, J.J.; Tse, W.; Moriggl, R.; Bunting, K.D. STAT5 requires the N-domain for suppression of miR15/16, induction of bcl-2, and survival signaling in myeloproliferative disease. Blood 2010, 115, 1416–1424. [Google Scholar] [CrossRef]
- Pullen, N.A.; Barnstein, B.O.; Falanga, Y.T.; Wang, Z.; Suzuki, R.; Tamang, T.D.; Khurana, M.C.; Harry, E.A.; Draber, P.; Bunting, K.D.; et al. Novel mechanism for Fc{epsilon}RI-mediated signal transducer and activator of transcription 5 (STAT5) tyrosine phosphorylation and the selective influence of STAT5B over mast cell cytokine production. J. Biol. Chem. 2012, 287, 2045–2054. [Google Scholar] [CrossRef]
- Li, Y.; Qi, X.; Liu, B.; Huang, H. The STAT5-GATA2 pathway is critical in basophil and mast cell differentiation and maintenance. J. Immunol. 2015, 194, 4328–4338. [Google Scholar] [CrossRef]
- Tobío, A.; Bandara, G.; Morris, D.A.; Kim, D.K.; O’Connell, M.P.; Komarow, H.D.; Carter, M.C.; Smrz, D.; Metcalfe, D.D.; Olivera, A. Oncogenic D816V-KIT signaling in mast cells causes persistent IL-6 production. Haematologica 2019. [Google Scholar] [CrossRef]
- Liu, J.; Lin, A. Role of JNK activation in apoptosis: A double-edged sword. Cell Res. 2005, 15, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Dhanasekaran, D.N.; Reddy, E.P. JNK signaling in apoptosis. Oncogene 2008, 27, 6245–6251. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Tsukada, J.; Higashi, T.; Mizobe, T.; Matsuura, A.; Mouri, F.; Sawamukai, N.; Ra, C.; Tanaka, Y. Growth suppression of human mast cells expressing constitutively active c-kit receptors by JNK inhibitor SP600125. Genes Cells 2006, 11, 983–992. [Google Scholar] [CrossRef] [PubMed]
- Mekori, Y.A.; Gilfillan, A.M.; Akin, C.; Hartmann, K.; Metcalfe, D.D. Human mast cell apoptosis is regulated through Bcl-2 and Bcl-XL. J. Clin. Immunol. 2001, 21, 171–174. [Google Scholar] [CrossRef] [PubMed]
- Alfredsson, J.; Puthalakath, H.; Martin, H.; Strasser, A.; Nilsson, G. Proapoptotic Bcl-2 family member Bim is involved in the control of mast cell survival and is induced together with Bcl-XL upon IgE-receptor activation. Cell Death Differ. 2005, 12, 136–144. [Google Scholar] [CrossRef] [PubMed]
- Rådinger, M.; Smrž, D.; Metcalfe, D.D.; Gilfillan, A.M. Glycogen synthase kinase-3β is a prosurvival signal for the maintenance of human mast cell homeostasis. J. Immunol. 2011, 187, 5587–5595. [Google Scholar] [CrossRef] [PubMed]
- Lilla, J.N.; Chen, C.C.; Mukai, K.; BenBarak, M.J.; Franco, C.B.; Kalesnikoff, J.; Yu, M.; Tsai, M.; Piliponsky, A.M.; Galli, S.J. Reduced mast cell and basophil numbers and function in Cpa3-Cre; Mcl-1fl/fl mice. Blood 2011, 118, 6930–6938. [Google Scholar] [CrossRef]
- Reinhart, R.; Rohner, L.; Wicki, S.; Fux, M.; Kaufmann, T. BH3 mimetics efficiently induce apoptosis in mouse basophils and mast cells. Cell Death Differ. 2018, 25, 204–216. [Google Scholar] [CrossRef]
- Aichberger, K.J.; Mayerhofer, M.; Gleixner, K.V.; Krauth, M.T.; Gruze, A.; Pickl, W.F.; Wacheck, V.; Selzer, E.; Müllauer, L.; Agis, H.; et al. Identification of MCL1 as a novel target in neoplastic mast cells in systemic mastocytosis: Inhibition of mast cell survival by MCL1 antisense oligonucleotides and synergism with PKC412. Blood 2007, 109, 3031–3041. [Google Scholar] [CrossRef]
- Yoshimoto, G.; Miyamoto, T.; Jabbarzadeh-Tabrizi, S.; Iino, T.; Rocnik, J.L.; Kikushige, Y.; Mori, Y.; Shima, T.; Iwasaki, H.; Takenaka, K.; et al. FLT3-ITD up-regulates MCL-1 to promote survival of stem cells in acute myeloid leukemia via FLT3-ITD-specific STAT5 activation. Blood 2009, 114, 5034–5043. [Google Scholar] [CrossRef]
- Malin, S.; McManus, S.; Cobaleda, C.; Novatchkova, M.; Delogu, A.; Bouillet, P.; Strasser, A.; Busslinger, M. Role of STAT5 in controlling cell survival and immunoglobulin gene recombination during pro-B cell development. Nat. Immunol. 2010, 11, 171–179. [Google Scholar] [CrossRef]
- Sathe, P.; Delconte, R.B.; Souza-Fonseca-Guimaraes, F.; Seillet, C.; Chopin, M.; Vandenberg, C.J.; Rankin, L.C.; Mielke, L.A.; Vikstrom, I.; Kolesnik, T.B.; et al. Innate immunodeficiency following genetic ablation of Mcl1 in natural killer cells. Nat. Commun. 2014, 5, 4539. [Google Scholar] [CrossRef]
- Dumon, S.; Santos, S.C.; Debierre-Grockiego, F.; Gouilleux-Gruart, V.; Cocault, L.; Boucheron, C.; Mollat, P.; Gisselbrecht, S.; Gouilleux, F. IL-3 dependent regulation of Bcl-xL gene expression by STAT5 in a bone marrow derived cell line. Oncogene 1999, 18, 4191–4199. [Google Scholar] [CrossRef]
- de Groot, R.P.; Raaijmakers, J.A.; Lammers, J.W.; Koenderman, L. STAT5-Dependent CyclinD1 and Bcl-xL expression in Bcr-Abl-transformed cells. Mol. Cell Biol. Res. Commun. 2000, 3, 299–305. [Google Scholar] [CrossRef]
- Kim, S.M.; Park, H.S.; Jun, D.Y.; Woo, H.J.; Woo, M.H.; Yang, C.H.; Kim, Y.H. Mollugin induces apoptosis in human Jurkat T cells through endoplasmic reticulum stress-mediated activation of JNK and caspase-12 and subsequent activation of mitochondria-dependent caspase cascade regulated by Bcl-xL. Toxicol. Appl. Pharmacol. 2009, 241, 210–220. [Google Scholar] [CrossRef]
- Shajahan, A.N.; Dobbin, Z.C.; Hickman, F.E.; Dakshanamurthy, S.; Clarke, R. Tyrosine-phosphorylated caveolin-1 (Tyr-14) increases sensitivity to paclitaxel by inhibiting BCL2 and BCLxL proteins via c-Jun N-terminal kinase (JNK). J. Biol. Chem. 2012, 287, 17682–17692. [Google Scholar] [CrossRef]
- Tait, S.W.; Green, D.R. Mitochondrial regulation of cell death. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hazzan, T.; Eberle, J.; Worm, M.; Babina, M. Thymic Stromal Lymphopoietin Interferes with the Apoptosis of Human Skin Mast Cells by a Dual Strategy Involving STAT5/Mcl-1 and JNK/Bcl-xL. Cells 2019, 8, 829. https://doi.org/10.3390/cells8080829
Hazzan T, Eberle J, Worm M, Babina M. Thymic Stromal Lymphopoietin Interferes with the Apoptosis of Human Skin Mast Cells by a Dual Strategy Involving STAT5/Mcl-1 and JNK/Bcl-xL. Cells. 2019; 8(8):829. https://doi.org/10.3390/cells8080829
Chicago/Turabian StyleHazzan, Tarek, Jürgen Eberle, Margitta Worm, and Magda Babina. 2019. "Thymic Stromal Lymphopoietin Interferes with the Apoptosis of Human Skin Mast Cells by a Dual Strategy Involving STAT5/Mcl-1 and JNK/Bcl-xL" Cells 8, no. 8: 829. https://doi.org/10.3390/cells8080829
APA StyleHazzan, T., Eberle, J., Worm, M., & Babina, M. (2019). Thymic Stromal Lymphopoietin Interferes with the Apoptosis of Human Skin Mast Cells by a Dual Strategy Involving STAT5/Mcl-1 and JNK/Bcl-xL. Cells, 8(8), 829. https://doi.org/10.3390/cells8080829