Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts

 ,
, 
 and
and 
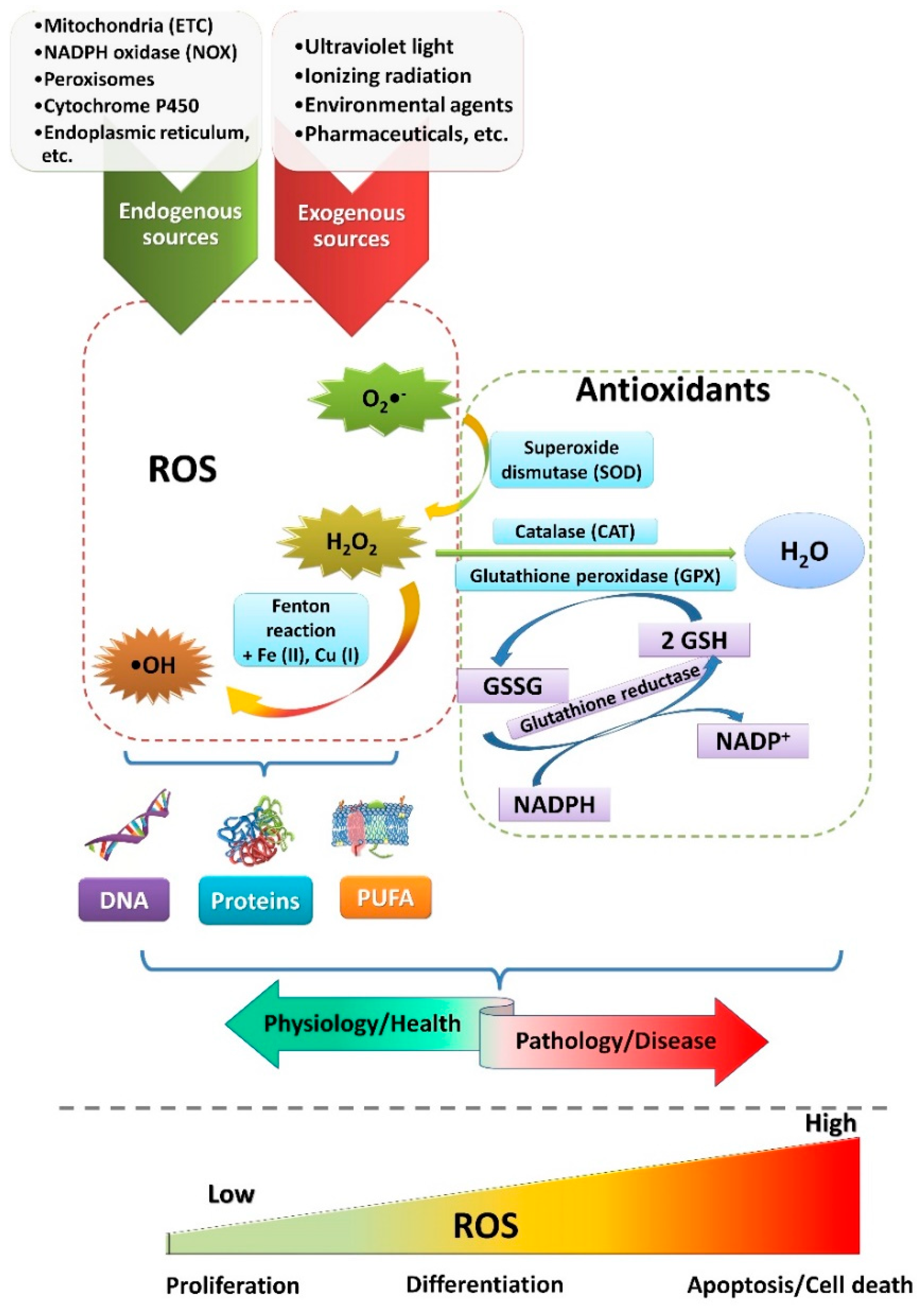{kind=link}
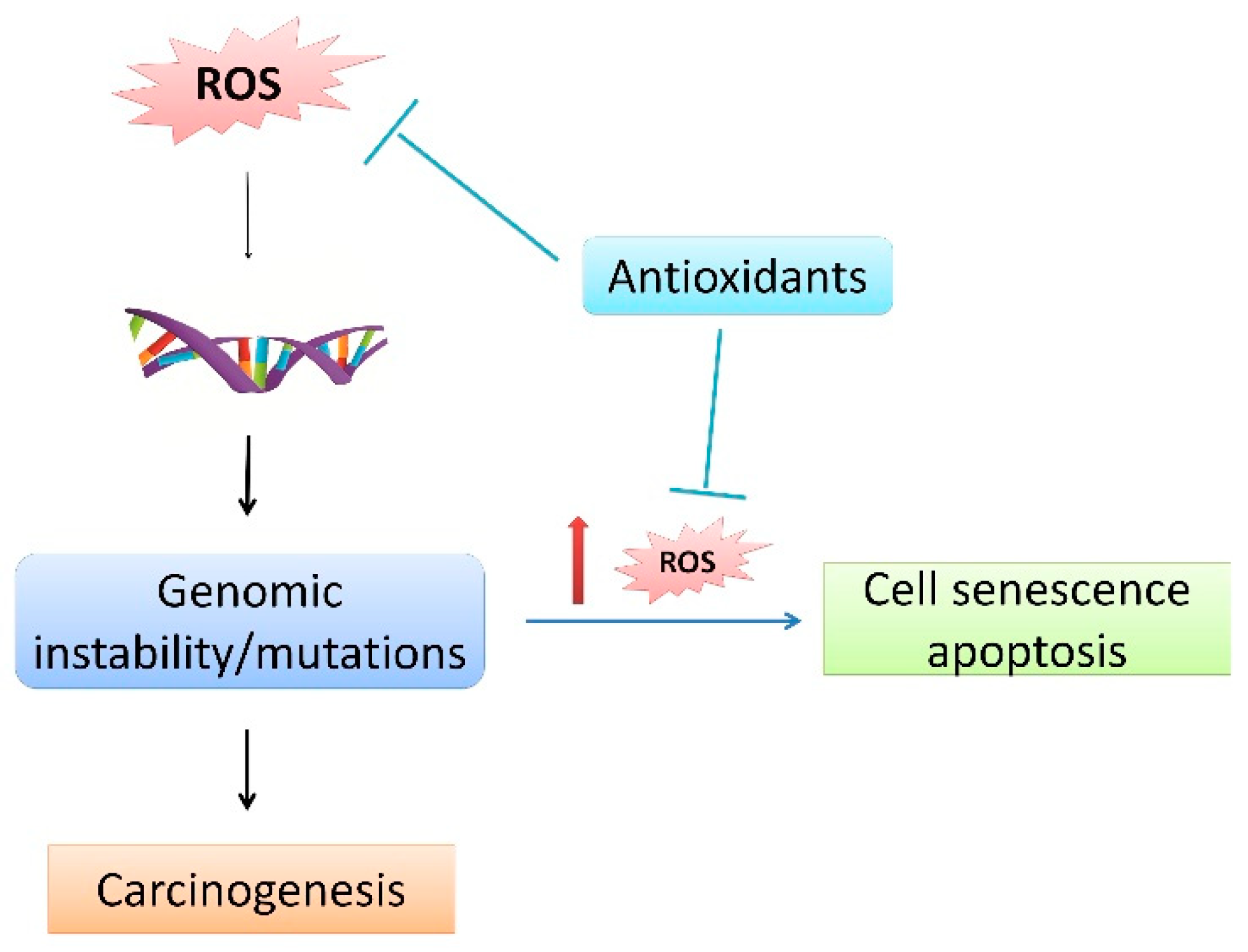{kind=link}
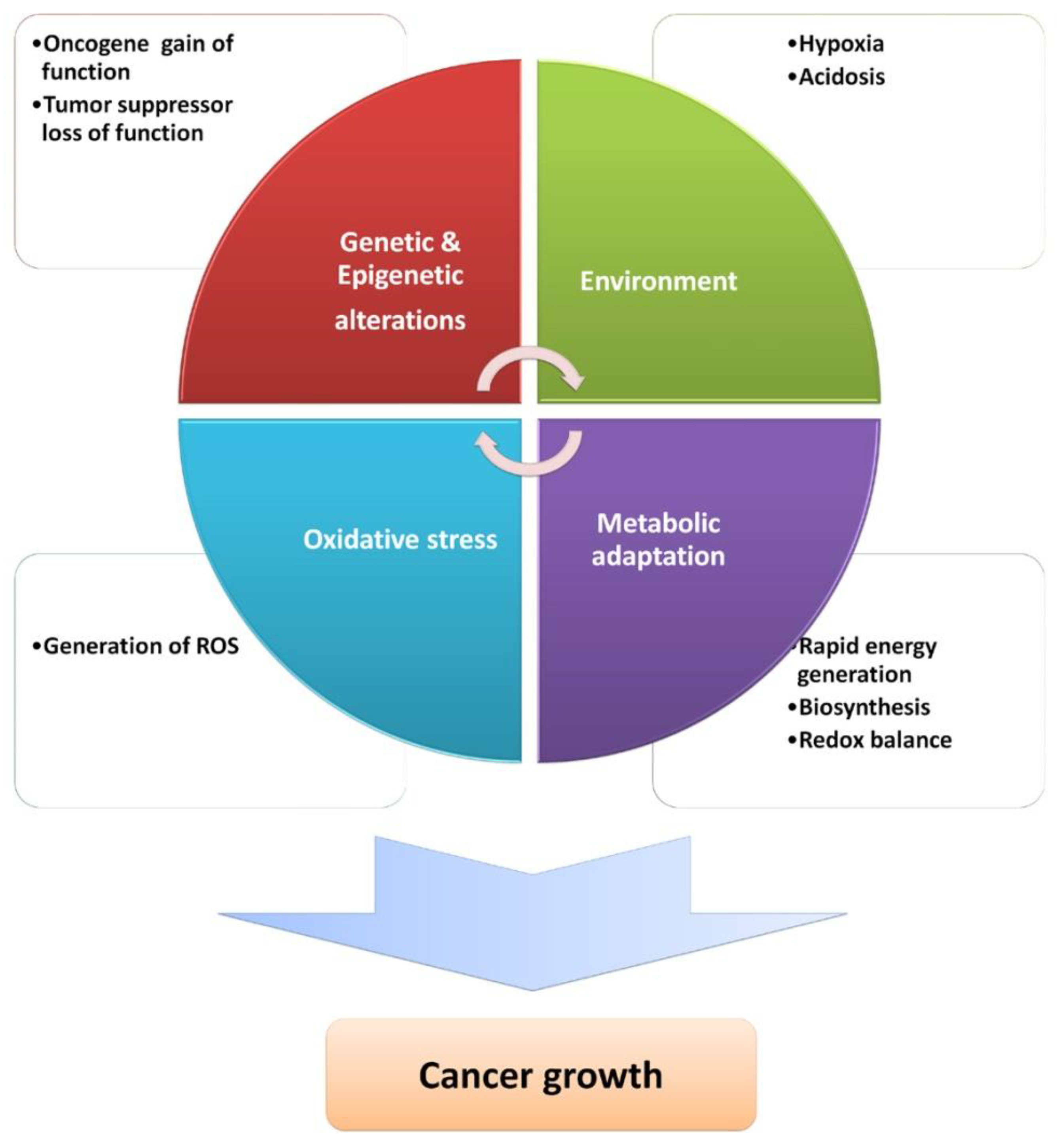{kind=link}
Abstract
1. Introduction
2. ROS Sources and Functions
3. Cell/Tissue-Specific Roles of ROS in (Patho)physiology
3.1. Stem Cells
3.2. Immune Cells
3.3. Central Nervous System (CNS)
3.4. Other Examples of Cell/Tissue-Specific Roles of ROS
4. ROS in Carcinogenesis
5. ROS Implications in Therapy
5.1. ROS and Anticancer Therapy
5.2. ROS and Regenerative Medicine
6. Conclusions and Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sies, H. Oxidative stress: Introductory remarks. In Oxidative Stress; Sies, H., Ed.; Academic Press: London, UK, 1985; pp. 1–7. [Google Scholar]
- Sies, H. On the history of oxidative stress: Concept and some aspects of current development. Curr. Opin. Toxicol. 2018, 7, 122–126. [Google Scholar] [CrossRef]
- Niki, E. Oxidative stress and antioxidants: Distress or eustress? Free Radic. Biol. Med. 2018, 124, 564. [Google Scholar] [CrossRef]
- Egea, J.; Fabregat, I.; Frapart, Y.M.; Ghezzi, P.; Görlach, A.; Kietzmann, T.; Kubaichuk, K.; Knaus, U.G.; Lopez, M.G.; Olaso-Gonzalez, G.; et al. European contribution to the study of ROS: A summary of the findings and prospects for the future from the COST action BM1203 (EU-ROS). Redox Biol. 2017, 13, 94–162. [Google Scholar] [CrossRef] [PubMed]
- Frijhoff, J.; Winyard, P.G.; Zarkovic, N.; Davies, S.S.; Stocker, R.; Cheng, D.; Knight, A.R.; Taylor, E.L.; Oettrich, J.; Ruskovska, T.; et al. Clinical Relevance of Biomarkers of Oxidative Stress. Antioxid. Redox Signal. 2015, 23, 1144–1170. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Shao, L.; Spitz, D.R. Reactive oxygen species in normal and tumor stem cells. Adv. Cancer Res. 2014, 122, 1–67. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Siems, W.; Siems, R.; Zarkovic, N. Oxidative stress and antioxidants in carcinogenesis and integrative therapy of cancer. Curr. Pharm. Des. 2014, 20, 6529–6542. [Google Scholar] [CrossRef] [PubMed]
- Mirończuk-Chodakowska, I.; Witkowska, A.M.; Zujko, M.E. Endogenous non-enzymatic antioxidants in the human body. Adv. Med. Sci. 2018, 63, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Schieber, M.; Chandel, N.S. ROS function in redox signaling and oxidative stress. Curr. Biol. 2014, 24, R453–R462. [Google Scholar] [CrossRef] [PubMed]
- Lubos, E.; Loscalzo, J.; Handy, D.E. Glutathione Peroxidase-1 in Health and Disease: From Molecular Mechanisms to Therapeutic Opportunities. Antioxid. Redox Signal. 2011, 15, 1957–1997. [Google Scholar] [CrossRef]
- Liu, Z.; Zhou, T.; Ziegler, A.C.; Dimitrion, P.; Zuo, L. Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications. Oxid. Med. Cell. Longev. 2017, 2017, 2525967. [Google Scholar] [CrossRef]
- Kattoor, A.J.; Pothineni, N.V.K.; Palagiri, D.; Mehta, J.L. Oxidative Stress in Atherosclerosis. Curr. Atheroscler. Rep. 2017, 19, 42. [Google Scholar] [CrossRef]
- Lennicke, C.; Rahn, J.; Lichtenfels, R.; Wessjohann, L.A.; Seliger, B. Hydrogen peroxide—Production, fate and role in redox signaling of tumor cells. Cell Commun. Signal. 2015, 13, 1–19. [Google Scholar] [CrossRef]
- Forman, H.J.; Ursini, F.; Maiorino, M. An overview of mechanisms of redox signaling. J. Mol. Cell. Cardiol. 2014, 73, 2–9. [Google Scholar] [CrossRef]
- Di Marzo, N.; Chisci, E.; Giovannoni, R. The Role of Hydrogen Peroxide in Redox-Dependent Signaling: Homeostatic and Pathological Responses in Mammalian Cells. Cells 2018, 7, 156. [Google Scholar] [CrossRef]
- Veal, E.A.; Day, A.M.; Morgan, B.A. Hydrogen Peroxide Sensing and Signaling. Mol. Cell 2007, 26, 1–14. [Google Scholar] [CrossRef]
- Bienert, G.P.; Chaumont, F. Aquaporin-facilitated transmembrane diffusion of hydrogen peroxide. Biochim. Biophys. Acta 2014, 1840, 1596–1604. [Google Scholar] [CrossRef]
- Rodrigues, C.; Mósca, A.F.; Martins, A.P.; Nobre, T.; Prista, C.; Antunes, F.; Cipak Gasparovic, A.; Soveral, G. Rat Aquaporin-5 Is pH-Gated Induced by Phosphorylation and Is Implicated in Oxidative Stress. Int. J. Mol. Sci. 2016, 17, 2090. [Google Scholar] [CrossRef]
- Prata, C.; Hrelia, S.; Fiorentini, D. Peroxiporins in cancer. Int. J. Mol. Sci. 2019, 20, 1371. [Google Scholar] [CrossRef]
- Milkovic, L.; Cipak Gasparovic, A.; Zarkovic, N. Overview on major lipid peroxidation bioactive factor 4-hydroxynonenal as pluripotent growth-regulating factor. Free Radic. Res. 2015, 49, 850–860. [Google Scholar] [CrossRef]
- Hanschmann, E.-M.; Godoy, J.R.; Berndt, C.; Hudemann, C.; Lillig, C.H. Thioredoxins, Glutaredoxins, and Peroxiredoxins—Molecular Mechanisms and Health Significance: From Cofactors to Antioxidants to Redox Signaling. Antioxid. Redox Signal. 2013, 19, 1539–1605. [Google Scholar] [CrossRef]
- Laurindo, F.R.M. Chapter 10-Redox Cellular Signaling Pathways in Endothelial Dysfunction and Vascular Disease. In Endothelium and Cardiovascular Diseases; Da Luz, P.L., Libby, P., Chagas, A.C.P., Laurindo, F.R.M., Eds.; Academic Press (An imprint of Elsevier): London, UK, 2018; pp. 127–145. [Google Scholar]
- Zhang, J.; Wang, X.; Vikash, V.; Ye, Q.; Wu, D.; Liu, Y.; Dong, W. ROS and ROS-Mediated Cellular Signaling. Oxid. Med. Cell. Longev. 2016, 2016, 4350965. [Google Scholar] [CrossRef]
- Milkovic, L.; Zarkovic, N.; Saso, L. Controversy about pharmacological modulation of Nrf2 for cancer therapy. Redox Biol. 2017, 12, 727–732. [Google Scholar] [CrossRef]
- Lingappan, K. NF-κB in Oxidative Stress. Curr. Opin. Toxicol. 2018, 7, 81–86. [Google Scholar] [CrossRef]
- Morgan, M.J.; Liu, Z. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef]
- Kitagishi, Y.; Matsuda, S. Redox regulation of tumor suppressor PTEN in cancer and aging (Review). Int. J. Mol. Med. 2013, 31, 511–515. [Google Scholar] [CrossRef]
- Filomeni, G.; De Zio, D.; Cecconi, F. Oxidative stress and autophagy: The clash between damage and metabolic needs. Cell Death Differ. 2015, 22, 377–388. [Google Scholar] [CrossRef]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Liang, D. A Salutary Role of Reactive Oxygen Species in Intercellular Tunnel-Mediated Communication. Front. Cell Dev. Biol. 2018, 6. [Google Scholar] [CrossRef]
- Covarrubias, L.; Hernández-García, D.; Schnabel, D.; Salas-Vidal, E.; Castro-Obregón, S. Function of reactive oxygen species during animal development: Passive or active? Dev. Biol. 2008, 320, 1–11. [Google Scholar] [CrossRef]
- Lopes, A.S.; Lane, M.; Thompson, J.G. Oxygen consumption and ROS production are increased at the time of fertilization and cell cleavage in bovine zygotes. Hum. Reprod. 2010, 25, 2762–2773. [Google Scholar] [CrossRef]
- Wong, J.L.; Créton, R.; Wessel, G.M. The oxidative burst at fertilization is dependent upon activation of the dual oxidase Udx1. Dev. Cell 2004, 7, 801–814. [Google Scholar] [CrossRef]
- Han, Y.; Ishibashi, S.; Iglesias-Gonzalez, J.; Chen, Y.; Love, N.R.; Amaya, E. Ca2+-Induced Mitochondrial ROS Regulate the Early Embryonic Cell Cycle. Cell Rep. 2018, 22, 218–231. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem cells and the impact of ROS signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef]
- Liang, R.; Ghaffari, S. Stem cells, redox signaling, and stem cell aging. Antioxid. Redox Signal. 2014, 20, 1902–1916. [Google Scholar] [CrossRef]
- Ryu, J.M.; Lee, H.J.; Jung, Y.H.; Lee, K.H.; Kim, D.I.; Kim, J.Y.; Ko, S.H.; Choi, G.E.; Chai, I.I.; Song, E.J.; et al. Regulation of Stem Cell Fate by ROS-mediated Alteration of Metabolism. Int. J. Stem Cells 2015, 8, 24–35. [Google Scholar] [CrossRef]
- Jang, Y.-Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef]
- Le Belle, J.E.; Orozco, N.M.; Paucar, A.A.; Saxe, J.P.; Mottahedeh, J.; Pyle, A.D.; Wu, H.; Kornblum, H.I. Proliferative neural stem cells have high endogenous ROS levels that regulate self-renewal and neurogenesis in a PI3K/Akt-dependant manner. Cell Stem Cell 2011, 8, 59–71. [Google Scholar] [CrossRef]
- Hu, C.; Zhao, L.; Peng, C.; Li, L. Regulation of the mitochondrial reactive oxygen species: Strategies to control mesenchymal stem cell fates ex vivo and in vivo. J. Cell. Mol. Med. 2018, 22, 5196–5207. [Google Scholar] [CrossRef]
- Tatapudy, S.; Aloisio, F.; Barber, D.; Nystul, T. Cell fate decisions: Emerging roles for metabolic signals and cell morphology. EMBO Rep. 2017, 18, 2105–2118. [Google Scholar] [CrossRef]
- Roos, D. Chronic granulomatous disease. Br. Med. Bull. 2016, 118, 50–63. [Google Scholar] [CrossRef]
- Nathan, C.; Cunningham-Bussel, A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat. Rev. Immunol. 2013, 13, 349–361. [Google Scholar] [CrossRef]
- Roy, J.; Galano, J.M.; Durand, T.; Le Guennec, J.Y.; Lee, J.C.Y. Physiological role of reactive oxygen species as promoters of natural defenses. FASEB J. 2017, 31, 3729–3745. [Google Scholar] [CrossRef]
- Cobley, J.N.; Fiorello, M.L.; Bailey, D.M. 13 reasons why the brain is susceptible to oxidative stress. Redox Biol. 2018, 15, 490–503. [Google Scholar] [CrossRef]
- Salim, S. Oxidative Stress and the Central Nervous System. J. Pharmacol. Exp. Ther. 2016, 360, 201–205. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. A Cellular Perspective on Brain Energy Metabolism and Functional Imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain energy metabolism: Focus on Astrocyte-neuron metabolic cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef]
- Bélanger, M.; Magistretti, P.J. The role of astroglia in neuroprotection. Dialogues Clin. Neurosci. 2009, 11, 281–295. [Google Scholar]
- Fernandez-Fernandez, S.; Almeida, A.; Bolaños, J.P. Antioxidant and bioenergetic coupling between neurons and astrocytes. Biochem. J. 2012, 443, 3–11. [Google Scholar] [CrossRef]
- Wilson, C.; González-Billault, C. Regulation of cytoskeletal dynamics by redox signaling and oxidative stress: Implications for neuronal development and trafficking. Front. Cell. Neurosci. 2015, 9, 381. [Google Scholar] [CrossRef]
- Oswald, M.C.W.; Garnham, N.; Sweeney, S.T.; Landgraf, M. Regulation of neuronal development and function by ROS. FEBS Lett. 2018, 592, 679–691. [Google Scholar] [CrossRef]
- Dickinson, B.C.; Peltier, J.; Stone, D.; Schaffer, D.V.; Chang, C.J. Nox2 redox signaling maintains essential cell populations in the brain. Nat. Chem. Biol. 2011, 7, 106–112. [Google Scholar] [CrossRef]
- Lopez-Fabuel, I.; Le Douce, J.; Logan, A.; James, A.M.; Bonvento, G.; Murphy, M.P.; Almeida, A.; Bolaños, J.P. Complex I assembly into supercomplexes determines differential mitochondrial ROS production in neurons and astrocytes. Proc. Natl. Acad. Sci. USA 2016, 113, 13063–13068. [Google Scholar] [CrossRef]
- Vicente-Gutierrez, C.; Bonora, N.; Bobo-Jimenez, V.; Jimenez-Blasco, D.; Lopez-Fabuel, I.; Fernandez, E.; Josephine, C.; Bonvento, G.; Enriquez, J.A.; Almeida, A.; et al. Astrocytic mitochondrial ROS modulate brain metabolism and mouse behaviour. Nat. Metab. 2019, 1, 201–211. [Google Scholar] [CrossRef]
- Ishimoto, Y.; Tanaka, T.; Yoshida, Y.; Inagi, R. Physiological and pathophysiological role of reactive oxygen species and reactive nitrogen species in the kidney. Clin. Exp. Pharmacol. Physiol. 2018, 45, 1097–1105. [Google Scholar] [CrossRef]
- Muñoz, M.; Martínez, M.P.; López-Oliva, M.E.; Rodríguez, C.; Corbacho, C.; Carballido, J.; García-Sacristán, A.; Hernández, M.; Rivera, L.; Sáenz-Medina, J.; et al. Hydrogen peroxide derived from NADPH oxidase 4- and 2 contributes to the endothelium-dependent vasodilatation of intrarenal arteries. Redox Biol. 2018, 19, 92–104. [Google Scholar] [CrossRef]
- Schürmann, C.; Rezende, F.; Kruse, C.; Yasar, Y.; Löwe, O.; Fork, C.; van de Sluis, B.; Bremer, R.; Weissmann, N.; Shah, A.M.; et al. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur. Heart J. 2015, 36, 3447–3456. [Google Scholar] [CrossRef]
- Chen, S.; Su, Y.; Wang, J. ROS-mediated platelet generation: A microenvironment-dependent manner for megakaryocyte proliferation, differentiation, and maturation. Cell Death Dis. 2013, 4, e722. [Google Scholar] [CrossRef]
- Motohashi, H.; Kimura, M.; Fujita, R.; Inoue, A.; Pan, X.; Takayama, M.; Katsuoka, F.; Aburatani, H.; Bresnick, E.H.; Yamamoto, M. NF-E2 domination over Nrf2 promotes ROS accumulation and megakaryocytic maturation. Blood 2010, 115, 677–686. [Google Scholar] [CrossRef]
- Canli, Ö.; Nicolas, A.M.; Gupta, J.; Finkelmeier, F.; Goncharova, O.; Pesic, M.; Neumann, T.; Horst, D.; Löwer, M.; Sahin, U.; et al. Myeloid Cell-Derived Reactive Oxygen Species Induce Epithelial Mutagenesis. Cancer Cell 2017, 32, 869–883.e5. [Google Scholar] [CrossRef]
- Holmström, K.M.; Finkel, T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat. Rev. Mol. Cell Biol. 2014, 15, 411–421. [Google Scholar] [CrossRef]
- Harris, I.S.; Treloar, A.E.; Inoue, S.; Sasaki, M.; Gorrini, C.; Lee, K.C.; Yung, K.Y.; Brenner, D.; Knobbe-Thomsen, C.B.; Cox, M.A.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Sotgia, F.; Lisanti, M.P. Metastasis and Oxidative Stress: Are Antioxidants a Metabolic Driver of Progression? Cell Metab. 2015, 22, 956–958. [Google Scholar] [CrossRef]
- Fiaschi, T.; Chiarugi, P. Oxidative stress, tumor microenvironment, and metabolic reprogramming: A diabolic liaison. Int. J. Cell Biol. 2012, 2012, 762825. [Google Scholar] [CrossRef]
- Gào, X.; Schöttker, B. Reduction–oxidation pathways involved in cancer development: A systematic review of literature reviews. Oncotarget 2017, 8, 51888–51906. [Google Scholar] [CrossRef]
- Liou, G.-Y.; Storz, P. Reactive oxygen species in cancer. Free Radic. Res. 2010, 44, 479–496. [Google Scholar] [CrossRef]
- Warburg, O. The Metabolism of Carcinoma Cells. J. Cancer Res. 1925, 9, 148–163. [Google Scholar] [CrossRef]
- Phan, L.M.; Yeung, S.-C.J.; Lee, M.-H. Cancer metabolic reprogramming: Importance, main features, and potentials for precise targeted anti-cancer therapies. Cancer Biol. Med. 2014, 11, 1–19. [Google Scholar] [CrossRef]
- Zheng, J. Energy metabolism of cancer: Glycolysis versus oxidative phosphorylation (Review). Oncol. Lett. 2012, 4, 1151–1157. [Google Scholar] [CrossRef]
- Gatenby, R.A.; Gillies, R.J. Why do cancers have high aerobic glycolysis? Nat. Rev. Cancer 2004, 4, 891–899. [Google Scholar] [CrossRef]
- Jose, C.; Bellance, N.; Rossignol, R. Choosing between glycolysis and oxidative phosphorylation: A tumor’s dilemma? Biochim. Biophys. Acta 2011, 1807, 552–561. [Google Scholar] [CrossRef]
- Yu, L.; Lu, M.; Jia, D.; Ma, J.; Ben-Jacob, E.; Levine, H.; Kaipparettu, B.A.; Onuchic, J.N. Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res. 2017, 77, 1564–1574. [Google Scholar] [CrossRef]
- Semenza, G.L. Targeting HIF-1 for cancer therapy. Nat. Rev. Cancer 2003, 3, 721–732. [Google Scholar] [CrossRef]
- Galanis, A.; Pappa, A.; Giannakakis, A.; Lanitis, E.; Dangaj, D.; Sandaltzopoulos, R. Reactive oxygen species and HIF-1 signalling in cancer. Cancer Lett. 2008, 266, 12–20. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, X.; Liu, Y.; Dong, S.; Wen, Z.; He, W.; Zhang, S.; Huang, Q.; Shi, M. ROS signaling under metabolic stress: Cross-talk between AMPK and AKT pathway. Mol. Cancer 2017, 16, 79. [Google Scholar] [CrossRef]
- Jeon, S.-M. Regulation and function of AMPK in physiology and diseases. Exp. Mol. Med. 2016, 48, e245. [Google Scholar] [CrossRef]
- Sundararaman, A.; Amirtham, U.; Rangarajan, A. Calcium-Oxidant Signaling Network Regulates AMP-activated Protein Kinase (AMPK) Activation upon Matrix Deprivation. J. Biol. Chem. 2016, 291, 14410–14429. [Google Scholar] [CrossRef]
- Zadra, G.; Batista, J.L.; Loda, M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 2015, 13, 1059–1072. [Google Scholar] [CrossRef]
- Faubert, B.; Vincent, E.E.; Poffenberger, M.C.; Jones, R.G. The AMP-activated protein kinase (AMPK) and cancer: Many faces of a metabolic regulator. Cancer Lett. 2015, 356, 165–170. [Google Scholar] [CrossRef]
- Panieri, E.; Santoro, M.M. ROS homeostasis and metabolism: A dangerous liason in cancer cells. Cell Death Dis. 2016, 7, e2253. [Google Scholar] [CrossRef]
- Ghanbari Movahed, Z.; Rastegari-Pouyani, M.; Mohammadi, M.H.; Mansouri, K. Cancer cells change their glucose metabolism to overcome increased ROS: One step from cancer cell to cancer stem cell? Biomed. Pharmacother. 2019, 112, 108690. [Google Scholar] [CrossRef]
- Kerins, M.J.; Ooi, A. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci. Rep. 2018, 8, 12846. [Google Scholar] [CrossRef]
- Ramirez, L.Y.; Huestis, S.E.; Yap, T.Y.; Zyzanski, S.; Drotar, D.; Kodish, E. Potential chemotherapy side effects: What do oncologists tell parents? Pediatr. Blood Cancer 2009, 52, 497–502. [Google Scholar] [CrossRef]
- Brenneisen, P.; Reichert, A.S. Nanotherapy and Reactive Oxygen Species (ROS) in Cancer: A Novel Perspective. Antioxidants 2018, 7, 31. [Google Scholar] [CrossRef]
- Liu, X.; Zhao, P.; Wang, X.; Wang, L.; Zhu, Y.; Gao, W. Triptolide Induces Glioma Cell Autophagy and Apoptosis via Upregulating the ROS/JNK and Downregulating the Akt/mTOR Signaling Pathways. Front. Oncol. 2019, 9, 387. [Google Scholar] [CrossRef]
- Yang, D.; Zhang, H.; Wu, J.; Ma, R.; Li, Z.; Wang, K.; Yang, F. The role of chamaejasmine in cellular apoptosis and autophagy in MG-63 cells. Biosci. Rep. 2019, 39, BSR20181707. [Google Scholar] [CrossRef]
- Kim, S.-H.; Kim, K.-Y.; Park, S.-G.; Yu, S.-N.; Kim, Y.-W.; Nam, H.-W.; An, H.-H.; Kim, Y.-W.; Ahn, S.-C. Mitochondrial ROS activates ERK/autophagy pathway as a protected mechanism against deoxypodophyllotoxin-induced apoptosis. Oncotarget 2017, 8, 111581–111596. [Google Scholar] [CrossRef]
- Jeong, A.; Kim, J.-H.; Lee, H.-J.; Kim, S.-H. Reactive oxygen species dependent phosphorylation of the liver kinase B1/AMP activated protein kinase/acetyl-CoA carboxylase signaling is critically involved in apoptotic effect of lambertianic acid in hepatocellular carcinoma cells. Oncotarget 2017, 8, 70116–70129. [Google Scholar] [CrossRef]
- Wang, J.; Luo, B.; Li, X.; Lu, W.; Yang, J.; Hu, Y.; Huang, P.; Wen, S. Inhibition of cancer growth in vitro and in vivo by a novel ROS-modulating agent with ability to eliminate stem-like cancer cells. Cell Death Dis. 2017, 8, e2887. [Google Scholar] [CrossRef]
- Lee, J.; Cho, Y.S.; Jung, H.; Choi, I. Pharmacological Regulation of Oxidative Stress in Stem Cells. Oxid. Med. Cell. Longev. 2018, 2018, 4081890. [Google Scholar] [CrossRef]
- Mouthuy, P.-A.; Snelling, S.J.B.; Dakin, S.G.; Milković, L.; Gašparović, A.Č.; Carr, A.J.; Žarković, N. Biocompatibility of implantable materials: An oxidative stress viewpoint. Biomaterials 2016, 109, 55–68. [Google Scholar] [CrossRef]
- Dziubla, T.; Butterfield, D.A. (Eds.) Oxidative Stress and Biomaterials, 1st ed.; Academic Press (An imprint of Elsevier): London, UK, 2016. [Google Scholar]
- Mrakovcic, L.; Wildburger, R.; Jaganjac, M.; Cindric, M.; Cipak, A.; Borovic-Sunjic, S.; Waeg, G.; Milankovic, A.M.; Zarkovic, N. Lipid peroxidation product 4-hydroxynonenal as factor of oxidative homeostasis supporting bone regeneration with bioactive glasses. Acta Biochim. Pol. 2010, 57, 173–178. [Google Scholar] [CrossRef] [PubMed]
- Milkovic, L.; Hoppe, A.; Detsch, R.; Boccaccini, A.R.; Zarkovic, N. Effects of Cu-doped 45S5 bioactive glass on the lipid peroxidation-associated growth of human osteoblast-like cells in vitro. J. Biomed. Mater. Res. A 2013. [Google Scholar] [CrossRef]
- Ambrożewicz, E.; Muszyńska, M.; Tokajuk, G.; Grynkiewicz, G.; Žarković, N.; Skrzydlewska, E. Beneficial Effects of Vitamins K and D3 on Redox Balance of Human Osteoblasts Cultured with Hydroxyapatite-Based Biomaterials. Cells 2019, 8, 325. [Google Scholar] [CrossRef] [PubMed]
- Sena, L.A.; Chandel, N.S. Physiological roles of mitochondrial reactive oxygen species. Mol. Cell 2012, 48, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Hourihan, J.M.; Moronetti Mazzeo, L.E.; Fernández-Cárdenas, L.P.; Blackwell, T.K. Cysteine Sulfenylation Directs IRE-1 to Activate the SKN-1/Nrf2 Antioxidant Response. Mol. Cell 2016, 63, 553–566. [Google Scholar] [CrossRef] [PubMed]
- Bauer, G.; Zarkovic, N. Revealing mechanisms of selective, concentration-dependent potentials of 4-hydroxy-2-nonenal to induce apoptosis in cancer cells through inactivation of membrane-associated catalase. Free Radic. Biol. Med. 2015, 81, 128–144. [Google Scholar] [CrossRef] [PubMed]



© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milkovic, L.; Cipak Gasparovic, A.; Cindric, M.; Mouthuy, P.-A.; Zarkovic, N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells 2019, 8, 793. https://doi.org/10.3390/cells8080793
Milkovic L, Cipak Gasparovic A, Cindric M, Mouthuy P-A, Zarkovic N. Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells. 2019; 8(8):793. https://doi.org/10.3390/cells8080793
Chicago/Turabian StyleMilkovic, Lidija, Ana Cipak Gasparovic, Marina Cindric, Pierre-Alexis Mouthuy, and Neven Zarkovic. 2019. "Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts" Cells 8, no. 8: 793. https://doi.org/10.3390/cells8080793
APA StyleMilkovic, L., Cipak Gasparovic, A., Cindric, M., Mouthuy, P.-A., & Zarkovic, N. (2019). Short Overview of ROS as Cell Function Regulators and Their Implications in Therapy Concepts. Cells, 8(8), 793. https://doi.org/10.3390/cells8080793