Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the α7 Nicotinic Receptor and Promotes Neurotoxicity


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Preparation of Aβ Peptides
Synthetic Peptides: Aβ42
2.2. Mouse Neuroblastoma Cell Culture and Transient Transfection
2.3. Radioligand Assay
2.4. Competition Radioligand Assay
2.5. Human Neuroblastoma SH-SY5Y Culture and Differentiation
2.6. Calcium Imaging
2.7. Electrophysiology
2.8. Modelling of Aβ:α7nAChR and Iso-Aβ:α7nAChR Interaction
2.9. Neurotoxicity Measurements
2.10. Statistical Methods Used for Data Analysis
3. Results
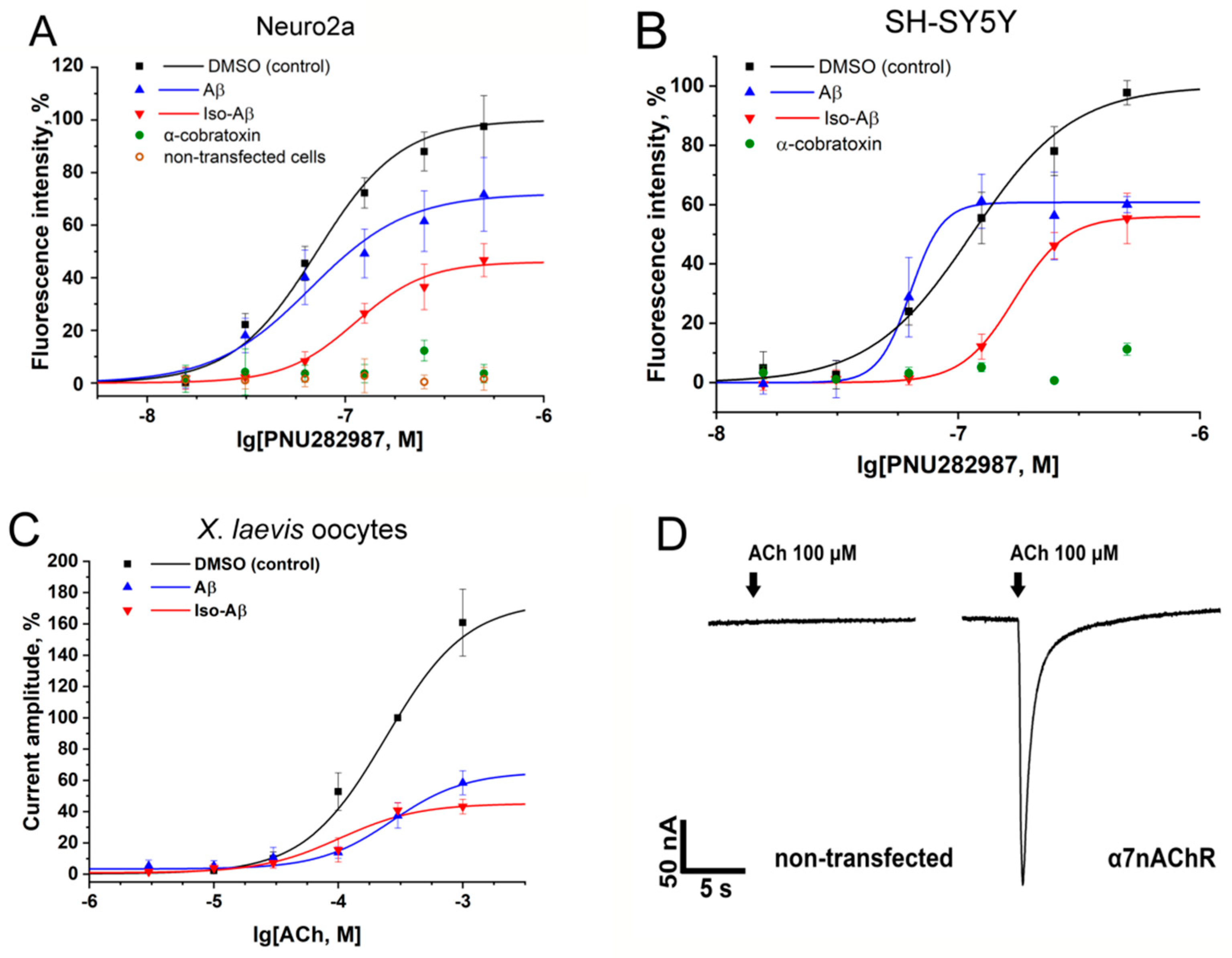
3.1. Effects of Aβ42 and Iso-Aβ42 on Functional Activity of α7nAChR in N2a and SH-SY5Y Cells and in Xenopus Laevis Oocytes
3.2. Aβ42 and Iso-Aβ42 Do not Compete with α-Bungarotoxin for Binding to the α7nAChR or to Acetylcholine-Binding Proteins (AChBPs)
3.3. The Aβ42-Induced Reduction of α7nAChR Representation is Neutralized by the Isomerization of Asp7
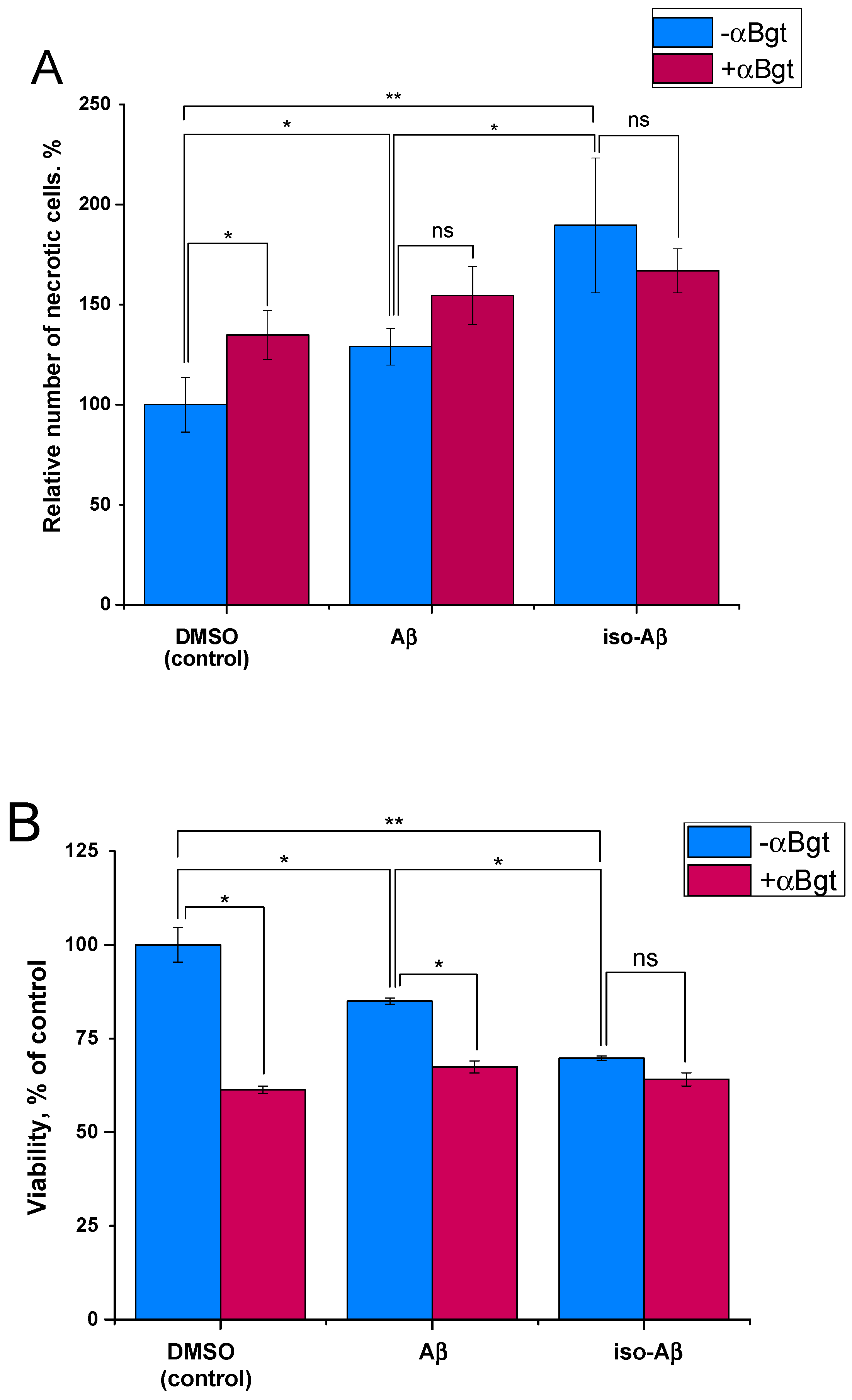
3.4. Comparison of the Toxicity of Aβ42and Iso-Aβ42 toward SH-SY5Y Cells
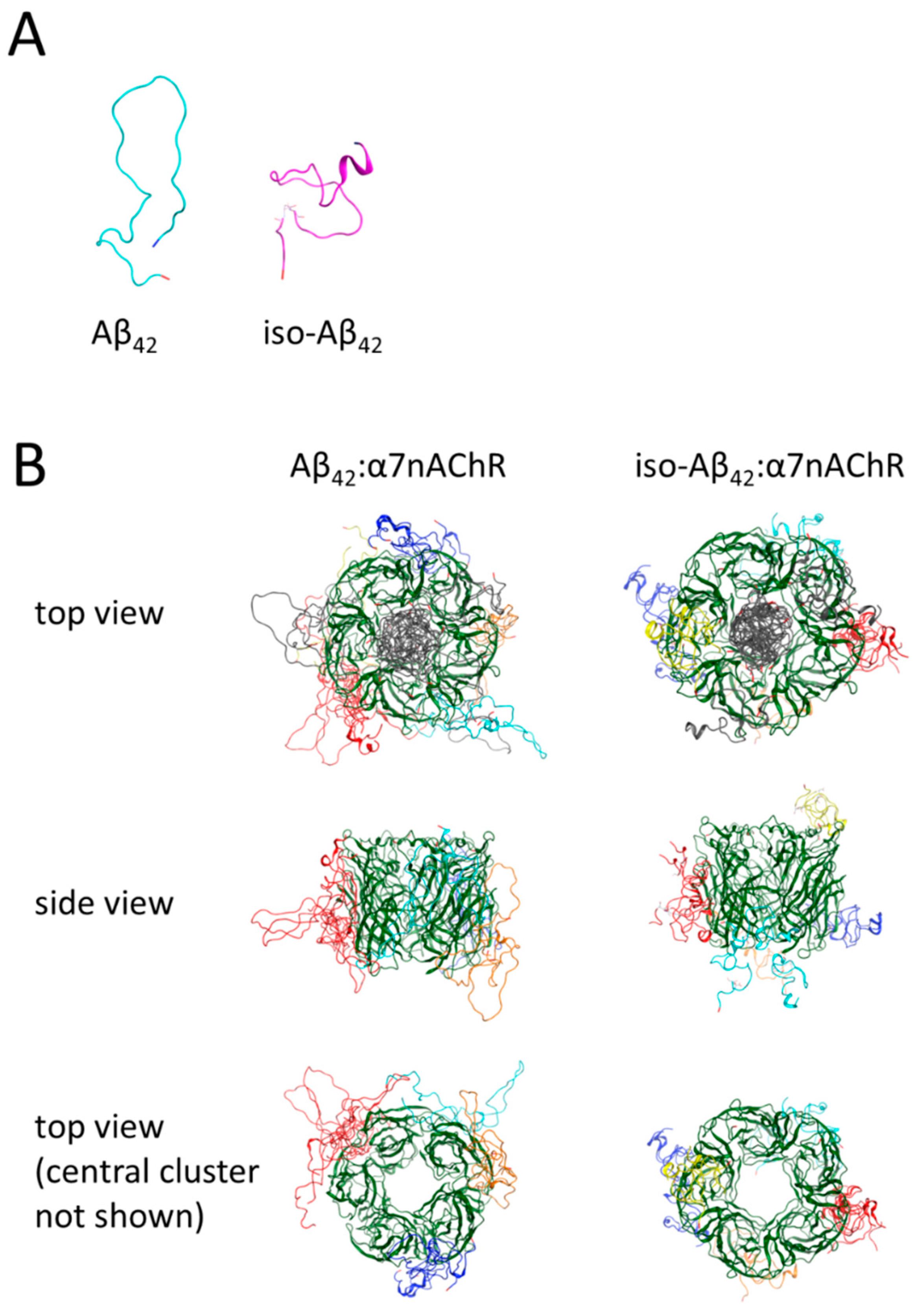
3.5. Molecular Modeling of the Binding Sites for Iso-Aβ42 and Aβ42 in the α7nAChR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Whitehouse, P.J.; Martino, A.M.; Marcus, K.A.; Zweig, R.M.; Singer, H.S.; Price, D.L.; Kellar, K.J. Reductions in Acetylcholine and Nicotine Binding in Several Degenerative Diseases. Arch. Neurol. 1988, 45, 722–724. [Google Scholar] [CrossRef] [PubMed]
- Herholz, K. Acetylcholine esterase activity in mild cognitive impairment and Alzheimer’s disease. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Appel, S.H. A unifying hypothesis for the cause of amyotrophic lateral sclerosis, parkinsonism, and alzheimer disease. Ann. Neurol. 1981, 10, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64 Suppl 9, 7–10. [Google Scholar]
- Stahl, S.M. The New Cholinesterase Inhibitors for Alzheimer’s Disease, Part 2: Illustrating Their Mechanisms of Action: (Brainstorms). J. Clin. Psychiatry 2000, 61, 813–814. [Google Scholar] [CrossRef] [PubMed]
- Albuquerque, E.X.; Pereira, E.F.R.; Alkondon, M.; Rogers, S.W. Mammalian Nicotinic Acetylcholine Receptors: From Structure to Function. Physiol. Rev. 2009, 89, 73–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotti, C.; Clementi, F. Neuronal nicotinic receptors: From structure to pathology. Prog. Neurobiol. 2004, 74, 363–396. [Google Scholar] [CrossRef] [PubMed]
- Gotti, C.; Fornasari, D.; Clementi, F. Human neuronal nicotinic receptors. Prog. Neurobiol. 1997, 53, 199–237. [Google Scholar] [CrossRef]
- Hogg, R.C.; Raggenbass, M.; Bertrand, D. Nicotinic acetylcholine receptors: From structure to brain function. In Reviews of Physiology, Biochemistry and Pharmacology; Springer: Berlin/Heidelberg, Germany, 2003; Volume 147, pp. 1–46. [Google Scholar]
- Gotti, C.; Zoli, M.; Clementi, F. Brain nicotinic acetylcholine receptors: Native subtypes and their relevance. Trends Pharmacol. Sci. 2006, 27, 482–491. [Google Scholar] [CrossRef]
- Oz, M.; Petroianu, G.; Lorke, D.E. α7-Nicotinic Acetylcholine Receptors: New Therapeutic Avenues in Alzheimer’s Disease. In Nicotinic Acetylcholine Receptor Technologies; Li, M.D., Ed.; Neuromethods; Springer: New York, NY, USA, 2016; pp. 149–169. ISBN 978-1-4939-3768-4. [Google Scholar]
- Nagele, R.G.; D’Andrea, M.R.; Anderson, W.J.; Wang, H.-Y. Intracellular accumulation of β-amyloid1–42 in neurons is facilitated by the α7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002, 110, 199–211. [Google Scholar] [CrossRef]
- Wang, H.Y.; Lee, D.H.; Davis, C.B.; Shank, R.P. Amyloid peptide Abeta(1-42) binds selectively and with picomolar affinity to alpha7 nicotinic acetylcholine receptors. J. Neurochem. 2000, 75, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Bencherif, M.; Lippiello, P.M. Alpha7 neuronal nicotinic receptors: The missing link to understanding Alzheimer’s etiopathology? Med. Hypotheses 2010, 74, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Prusiner, S.B. A Unifying Role for Prions in Neurodegenerative Diseases. Science 2012, 336, 1511–1513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jucker, M.; Walker, L.C. Self-propagation of pathogenic protein aggregates in neurodegenerative diseases. Nature 2013, 501, 45. [Google Scholar] [CrossRef] [PubMed]
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784. [Google Scholar] [CrossRef] [PubMed]
- Barykin, E.P.; Mitkevich, V.A.; Kozin, S.A.; Makarov, A.A. Amyloid β Modification: A Key to the Sporadic Alzheimer’s Disease? Front. Genet. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Watanabe, A.; Ogawara, M.; Mori, H.; Shirasawa, T. Isoaspartate formation and neurodegeneration in Alzheimer’s disease. Arch Biochem Biophys 2000, 381, 225–234. [Google Scholar] [CrossRef]
- Kozin, S.A.; Mitkevich, V.A.; Makarov, A.A. Amyloid-β containing isoaspartate 7 as potential biomarker and drug target in Alzheimer’s disease. Mendeleev Commun. 2016, 26, 269–275. [Google Scholar] [CrossRef]
- Kozin, S.A.; Barykin, E.P.; Mitkevich, V.A.; Makarov, A.A. Anti-amyloid Therapy of Alzheimer’s Disease: Current State and Prospects. Biochem. Mosc. 2018, 83, 1057–1067. [Google Scholar] [CrossRef]
- Roher, A.E.; Lowenson, J.D.; Clarke, S.; Wolkow, C.; Wang, R.; Cotter, R.J.; Reardon, I.M.; Zürcher-Neely, H.A.; Heinrikson, R.L.; Ball, M.J. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J. Biol. Chem. 1993, 268, 3072–3083. [Google Scholar]
- Moro, M.L.; Phillips, A.S.; Gaimster, K.; Paul, C.; Mudher, A.; Nicoll, J.A.R.; Boche, D. Pyroglutamate and Isoaspartate modified Amyloid-Beta in ageing and Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6. [Google Scholar] [CrossRef] [PubMed]
- Kozin, S.A.; Cheglakov, I.B.; Ovsepyan, A.A.; Telegin, G.B.; Tsvetkov, P.O.; Lisitsa, A.V.; Makarov, A.A. Peripherally Applied Synthetic Peptide isoAsp7-Aβ(1-42) Triggers Cerebral β-Amyloidosis. Neurotox. Res. 2013, 24, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Barykin, E.P.; Petrushanko, I.Y.; Kozin, S.A.; Telegin, G.B.; Chernov, A.S.; Lopina, O.D.; Radko, S.P.; Mitkevich, V.A.; Makarov, A.A. Phosphorylation of the Amyloid-Beta Peptide Inhibits Zinc-Dependent Aggregation, Prevents Na,K-ATPase Inhibition, and Reduces Cerebral Plaque Deposition. Front. Mol. Neurosci. 2018, 11. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.; Matta, J.A.; Lord, B.; Harrington, A.W.; Sutton, S.W.; Davini, W.B.; Bredt, D.S. Brain α7 Nicotinic Acetylcholine Receptor Assembly Requires NACHO. Neuron 2016, 89, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Tsetlin, V.I.; Hucho, F. Snake and snail toxins acting on nicotinic acetylcholine receptors: Fundamental aspects and medical applications. FEBS Lett. 2004, 557, 9–13. [Google Scholar] [CrossRef]
- Encinas, M.; Iglesias, M.; Liu, Y.; Wang, H.; Muhaisen, A.; Ceña, V.; Gallego, C.; Comella, J.X. Sequential Treatment of SH-SY5Y Cells with Retinoic Acid and Brain-Derived Neurotrophic Factor Gives Rise to Fully Differentiated, Neurotrophic Factor-Dependent, Human Neuron-Like Cells. J. Neurochem. 2000, 75, 991–1003. [Google Scholar] [CrossRef] [PubMed]
- Shelukhina, I.; Zhmak, M.; Lobanov, A.; Ivanov, I.; Garifulina, A.; Kravchenko, I.; Rasskazova, E.; Salmova, M.; Tukhovskaya, E.; Rykov, V.; et al. Azemiopsin, a Selective Peptide Antagonist of Muscle Nicotinic Acetylcholine Receptor: Preclinical Evaluation as a Local Muscle Relaxant. Toxins 2018, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Shelukhina, I.; Spirova, E.; Kudryavtsev, D.; Ojomoko, L.; Werner, M.; Methfessel, C.; Hollmann, M.; Tsetlin, V. Calcium imaging with genetically encoded sensor Case12: Facile analysis of α7/α9 nAChR mutants. PLoS ONE 2017, 12, e0181936. [Google Scholar] [CrossRef] [PubMed]
- Sitzia, F.; Brown, J.T.; Randall, A.; Dunlop, J. Voltage- and Temperature-Dependent Allosteric Modulation of α7 Nicotinic Receptors by PNU120596. Front. Pharmacol. 2011, 2. [Google Scholar] [CrossRef] [PubMed]
- Hajós, M.; Hurst, R.S.; Hoffmann, W.E.; Krause, M.; Wall, T.M.; Higdon, N.R.; Groppi, V.E. The Selective α7 Nicotinic Acetylcholine Receptor Agonist PNU-282987 [N-[(3R)-1-Azabicyclo[2.2.2]oct-3-yl]-4-chlorobenzamide Hydrochloride] Enhances GABAergic Synaptic Activity in Brain Slices and Restores Auditory Gating Deficits in Anesthetized Rats. J. Pharmacol. Exp. Ther. 2005, 312, 1213–1222. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, A.A.; Anashkina, A.A.; Makarov, A.A. Left-handed polyproline-II helix revisited: Proteins causing proteopathies. J. Biomol. Struct. Dyn. 2017, 35, 2701–2713. [Google Scholar] [CrossRef] [PubMed]
- Jayaram, B.; Bhushan, K.; Shenoy, S.R.; Narang, P.; Bose, S.; Agrawal, P.; Sahu, D.; Pandey, V. Bhageerath: An energy based web enabled computer software suite for limiting the search space of tertiary structures of small globular proteins. Nucleic Acids Res. 2006, 34, 6195–6204. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anashkina, A.A.; Kravatsky, Y.; Kuznetsov, E.; Makarov, A.A.; Adzhubei, A.A. Meta-server for automatic analysis, scoring and ranking of docking models. Bioinformatics 2018, 34, 297–299. [Google Scholar] [CrossRef] [PubMed]
- Guan, Z.-Z.; Yu, W.-F.; Shan, K.-R.; Nordman, T.; Olsson, J.; Nordberg, A. Loss of nicotinic receptors induced by beta-amyloid peptides in PC12 cells: Possible mechanism involving lipid peroxidation. J. Neurosci. Res. 2003, 71, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Inestrosa, N.C.; Godoy, J.A.; Vargas, J.Y.; Arrazola, M.S.; Rios, J.A.; Carvajal, F.J.; Serrano, F.G.; Farias, G.G. Nicotine Prevents Synaptic Impairment Induced by Amyloid-β Oligomers Through α7-Nicotinic Acetylcholine Receptor Activation. NeuroMolecular Med. 2013, 15, 549–569. [Google Scholar] [CrossRef] [PubMed]
- Bitan, G. Structural Study of Metastable Amyloidogenic Protein Oligomers by Photo-Induced Cross-Linking of Unmodified Proteins. In Methods in Enzymology; Amyloid, Prions, and Other Protein Aggregates, Part C; Academic Press: Cambridge, MA, USA, 2006; Volume 413, pp. 217–236. [Google Scholar]
- Corradi, J.; Bouzat, C. Understanding the Bases of Function and Modulation of α7 Nicotinic Receptors: Implications for Drug Discovery. Mol. Pharmacol. 2016, 90, 288–299. [Google Scholar] [CrossRef]
- Spurny, R.; Debaveye, S.; Farinha, A.; Veys, K.; Vos, A.M.; Gossas, T.; Atack, J.; Bertrand, S.; Bertrand, D.; Danielson, U.H.; et al. Molecular blueprint of allosteric binding sites in a homologue of the agonist-binding domain of the α7 nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. 2015, 112, E2543–E2552. [Google Scholar] [CrossRef]
- Gay, E.A.; Giniatullin, R.; Skorinkin, A.; Yakel, J.L. Aromatic residues at position 55 of rat α7 nicotinic acetylcholine receptors are critical for maintaining rapid desensitization. J. Physiol. 2008, 586, 1105–1115. [Google Scholar] [CrossRef]
- Magdesian, M.H.; Nery, A.A.; Martins, A.H.B.; Juliano, M.A.; Juliano, L.; Ulrich, H.; Ferreira, S.T. Peptide blockers of the inhibition of neuronal nicotinic acetylcholine receptors by amyloid beta. J. Biol. Chem. 2005, 280, 31085–31090. [Google Scholar] [CrossRef]
- Birks, J.S. Cholinesterase inhibitors for Alzheimer’s disease. Cochrane Database Syst. Rev. 2006. [Google Scholar] [CrossRef]
- Nordberg, A. Nicotinic receptor abnormalities of Alzheimer’s disease: Therapeutic implications. Biol. Psychiatry 2001, 49, 200–210. [Google Scholar] [CrossRef]
- Liu, Q.; Kawai, H.; Berg, D.K. beta -Amyloid peptide blocks the response of alpha 7-containing nicotinic receptors on hippocampal neurons. Proc. Natl. Acad. Sci. USA 2001, 98, 4734–4739. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Petrushanko, I.Y.; Yegorov, Y.E.; Simonenko, O.V.; Vishnyakova, K.S.; Kulikova, A.A.; Tsvetkov, P.O.; Makarov, A.A.; Kozin, S.A. Isomerization of Asp7 leads to increased toxic effect of amyloid-beta42 on human neuronal cells. Cell Death Dis 2013, 4, e939. [Google Scholar] [CrossRef] [PubMed]
- Lyukmanova, E.N.; Shulepko, M.A.; Kudryavtsev, D.; Bychkov, M.L.; Kulbatskii, D.S.; Kasheverov, I.E.; Astapova, M.V.; Feofanov, A.V.; Thomsen, M.S.; Mikkelsen, J.D.; et al. Human Secreted Ly-6/uPAR Related Protein-1 (SLURP-1) Is a Selective Allosteric Antagonist of α7 Nicotinic Acetylcholine Receptor. PLoS ONE 2016, 11, e0149733. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Katz, M.; Gerzanich, V.; Anand, R.; Lindstrom, J. Human alpha 7 acetylcholine receptor: cloning of the alpha 7 subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional alpha 7 homomers expressed in Xenopus oocytes. Mol. Pharmacol. 1994, 45, 546–554. [Google Scholar] [PubMed]
- Zheng, X.; Xie, Z.; Zhu, Z.; Liu, Z.; Wang, Y.; Wei, L.; Yang, H.; Yang, H.; Liu, Y.; Bi, J. Methyllycaconitine Alleviates Amyloid-β Peptides-Induced Cytotoxicity in SH-SY5Y Cells. PLoS ONE 2014, 9, e111536. [Google Scholar] [CrossRef] [PubMed]
- Spirova, E.N.; Ivanov, I.A.; Kasheverov, I.E.; Kudryavtsev, D.S.; Shelukhina, I.V.; Garifulina, A.I.; Son, L.V.; Lummis, S.C.R.; Malca-Garcia, G.R.; Bussmann, R.W.; et al. Curare alkaloids from Matis Dart Poison: Comparison with d-tubocurarine in interactions with nicotinic, 5-HT3 serotonin and GABAA receptors. PLoS ONE 2019, 14, e0210182. [Google Scholar] [CrossRef]
- Manetti, D.; Garifulina, A.; Bartolucci, G.; Bazzicalupi, C.; Bellucci, C.; Chiaramonte, N.; Dei, S.; Di Cesare Mannelli, L.; Ghelardini, C.; Gratteri, P.; et al. New Rigid Nicotine Analogues, Carrying a Norbornane Moiety, Are Potent Agonists of α7 and α3* Nicotinic Receptors. J. Med. Chem. 2019, 62, 1887–1901. [Google Scholar] [CrossRef]
- Kryukova, E.V.; Ivanov, I.A.; Lebedev, D.S.; Spirova, E.N.; Egorova, N.S.; Zouridakis, M.; Kasheverov, I.E.; Tzartos, S.J.; Tsetlin, V.I. Orthosteric and/or Allosteric Binding of α-Conotoxins to Nicotinic Acetylcholine Receptors and Their Models. Mar. Drugs 2018, 16. [Google Scholar] [CrossRef]
- Durek, T.; Shelukhina, I.V.; Tae, H.-S.; Thongyoo, P.; Spirova, E.N.; Kudryavtsev, D.S.; Kasheverov, I.E.; Faure, G.; Corringer, P.-J.; Craik, D.J.; et al. Interaction of Synthetic Human SLURP-1 with the Nicotinic Acetylcholine Receptors. Sci. Rep. 2017, 7, 16606. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.K.; Wang, J.; Papke, R.L. Investigation of the molecular mechanism of the α7 nicotinic acetylcholine receptor positive allosteric modulator PNU-120596 provides evidence for two distinct desensitized states. Mol. Pharmacol. 2011, 80, 1013–1032. [Google Scholar] [CrossRef] [PubMed]
- Hurst, R.S.; Hajós, M.; Raggenbass, M.; Wall, T.M.; Higdon, N.R.; Lawson, J.A.; Rutherford-Root, K.L.; Berkenpas, M.B.; Hoffmann, W.E.; Piotrowski, D.W.; et al. A novel positive allosteric modulator of the alpha7 neuronal nicotinic acetylcholine receptor: In vitro and in vivo characterization. J. Neurosci. Off. J. Soc. Neurosci. 2005, 25, 4396–4405. [Google Scholar] [CrossRef] [PubMed]
- Grønlien, J.H.; Håkerud, M.; Ween, H.; Thorin-Hagene, K.; Briggs, C.A.; Gopalakrishnan, M.; Malysz, J. Distinct profiles of alpha7 nAChR positive allosteric modulation revealed by structurally diverse chemotypes. Mol. Pharmacol. 2007, 72, 715–724. [Google Scholar] [CrossRef] [PubMed]
- Ospina, J.A.; Broide, R.S.; Acevedo, D.; Robertson, R.T.; Leslie, F.M. Calcium Regulation of Agonist Binding to α7-Type Nicotinic Acetylcholine Receptors in Adult and Fetal Rat Hippocampus. J. Neurochem. 1998, 70, 1061–1068. [Google Scholar] [CrossRef] [PubMed]
- Khiroug, L.; Giniatullin, R.; Klein, R.C.; Fayuk, D.; Yakel, J.L. Functional Mapping and Ca2+ Regulation of Nicotinic Acetylcholine Receptor Channels in Rat Hippocampal CA1 Neurons. J. Neurosci. 2003, 23, 9024–9031. [Google Scholar] [CrossRef] [PubMed]
- Liu, Q.-S.; Berg, D.K. Extracellular Calcium Regulates Responses of Both α3- and α7-Containing Nicotinic Receptors on Chick Ciliary Ganglion Neurons. J. Neurophysiol. 1999, 82, 1124–1132. [Google Scholar] [CrossRef] [PubMed]
- Maatuk, N.; Samson, A.O. Modeling the binding mechanism of Alzheimer’s Aβ1–42 to nicotinic acetylcholine receptors based on similarity with snake α-neurotoxins. NeuroToxicology 2013, 34, 236–242. [Google Scholar] [CrossRef]
- Lyukmanova, E.N.; Shenkarev, Z.O.; Shulepko, M.A.; Mineev, K.S.; D’Hoedt, D.; Kasheverov, I.E.; Filkin, S.Y.; Krivolapova, A.P.; Janickova, H.; Dolezal, V.; et al. NMR Structure and Action on Nicotinic Acetylcholine Receptors of Water-soluble Domain of Human LYNX1. J. Biol. Chem. 2011, 286, 10618–10627. [Google Scholar] [CrossRef] [Green Version]
- D’Andrea, M.R.; Nagele, R.G. Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer’s disease pyramidal neurons. Curr. Pharm. Des. 2006, 12, 677–684. [Google Scholar] [CrossRef]
- Nichols, W.A.; Henderson, B.J.; Yu, C.; Parker, R.L.; Richards, C.I.; Lester, H.A.; Miwa, J.M. Lynx1 Shifts α4β2 Nicotinic Receptor Subunit Stoichiometry by Affecting Assembly in the Endoplasmic Reticulum. J. Biol. Chem. 2014, 289, 31423–31432. [Google Scholar] [CrossRef] [PubMed]
- George, A.A.; Bloy, A.; Miwa, J.M.; Lindstrom, J.M.; Lukas, R.J.; Whiteaker, P. Isoform-specific mechanisms of α3β4*-nicotinic acetylcholine receptor modulation by the prototoxin lynx1. FASEB J. 2017, 31, 1398–1420. [Google Scholar] [CrossRef] [PubMed]
- Xue, M.; Zhu, L.; Zhang, J.; Qiu, J.; Du, G.; Qiao, Z.; Jin, G.; Gao, F.; Zhang, Q. Low Dose Nicotine Attenuates Aβ Neurotoxicity through Activation Early Growth Response Gene 1 Pathway. PLoS ONE 2015, 10, e0120267. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Bakshi, K.; Shen, C.; Frankfurt, M.; Trocme-Thibierge, C.; Morain, P. S 24795 limits beta-amyloid-alpha7 nicotinic receptor interaction and reduces Alzheimer’s disease-like pathologies. Biol. Psychiatry 2010, 67, 522–530. [Google Scholar] [CrossRef] [PubMed]
- Messi, M.L.; Renganathan, M.; Grigorenko, E.; Delbono, O. Activation of α 7 nicotinic acetylcholine receptor promotes survival of spinal cord motoneurons. FEBS Lett. 1997, 411, 32–38. [Google Scholar] [CrossRef]
- Dajas-Bailador, F.A.; Lima, P.A.; Wonnacott, S. The α7 nicotinic acetylcholine receptor subtype mediates nicotine protection against NMDA excitotoxicity in primary hippocampal cultures through a Ca2+ dependent mechanism. Neuropharmacology 2000, 39, 2799–2807. [Google Scholar] [CrossRef]
- Ren, K.; Puig, V.; Papke, R.L.; Itoh, Y.; Hughes, J.A.; Meyer, E.M. Multiple calcium channels and kinases mediate α7 nicotinic receptor neuroprotection in PC12 cells. J. Neurochem. 2005, 94, 926–933. [Google Scholar] [CrossRef]
- Barykin, E.P.; Petrushanko, I.Y.; Burnysheva, K.M.; Makarov, A.A.; Mitkevich, V.A. Isomerization of Asp7 increases the toxic effects of amyloid beta and its phosphorylated form in SH-SY5Y neuroblastoma cells. Mol. Biol. 2016, 50, 863–869. [Google Scholar]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Calcium signals induced by amyloid β peptide and their consequences in neurons and astrocytes in culture. Biochim. Biophys. Acta 2004, 1742, 81–87. [Google Scholar] [CrossRef]
- Abramov, A.Y.; Canevari, L.; Duchen, M.R. Changes in Intracellular Calcium and Glutathione in Astrocytes as the Primary Mechanism of Amyloid Neurotoxicity. J. Neurosci. 2003, 23, 5088–5095. [Google Scholar] [CrossRef] [Green Version]
- Demuro, A.; Parker, I.; Stutzmann, G.E. Calcium Signaling and Amyloid Toxicity in Alzheimer Disease. J. Biol. Chem. 2010, 285, 12463–12468. [Google Scholar] [CrossRef] [PubMed] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cell Line | Treatment | EC50 Value, nM |
|---|---|---|
| N2a | Control | 71.9 ± 8.4 |
| Aβ42 | 66.8 ± 8.6 | |
| iso-Aβ42 | 112.6 ± 4.3 | |
| SH-SY5Y | Control | 114 ± 7.0 |
| Aβ42 | 63.4 ± 2.3 | |
| iso-Aβ42 | 171 ± 3.3 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barykin, E.P.; Garifulina, A.I.; Kruykova, E.V.; Spirova, E.N.; Anashkina, A.A.; Adzhubei, A.A.; Shelukhina, I.V.; Kasheverov, I.E.; Mitkevich, V.A.; Kozin, S.A.; et al. Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the α7 Nicotinic Receptor and Promotes Neurotoxicity. Cells 2019, 8, 771. https://doi.org/10.3390/cells8080771
Barykin EP, Garifulina AI, Kruykova EV, Spirova EN, Anashkina AA, Adzhubei AA, Shelukhina IV, Kasheverov IE, Mitkevich VA, Kozin SA, et al. Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the α7 Nicotinic Receptor and Promotes Neurotoxicity. Cells. 2019; 8(8):771. https://doi.org/10.3390/cells8080771
Chicago/Turabian StyleBarykin, Evgeny P., Alexandra I. Garifulina, Elena V. Kruykova, Ekaterina N. Spirova, Anastasia A. Anashkina, Alexei A. Adzhubei, Irina V. Shelukhina, Igor E. Kasheverov, Vladimir A. Mitkevich, Sergey A. Kozin, and et al. 2019. "Isomerization of Asp7 in Beta-Amyloid Enhances Inhibition of the α7 Nicotinic Receptor and Promotes Neurotoxicity" Cells 8, no. 8: 771. https://doi.org/10.3390/cells8080771