NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction


Abstract
1. Introduction
2. OA Pathogenesis
3. General Function and Regulation of NF-κB
4. Significance of NF-κB in OA Pathogenesis
5. Chondrocyte Catabolism Regulated by NF-κB
5.1. The Regulation of Matrix-Degrading Enzymes by NF-κB
5.2. Factors That Regulate NF-κB Activity via Direct Interaction
5.3. Factors That Activate NF-κB under OA Conditions
5.4. Factors That Inhibit NF-κB in OA Conditions
6. Epigenetics Associated with NF-κB in OA
6.1. Histone Deacetylases (HDACs)
6.2. MicroRNAs
7. Chondrocyte Apoptosis Regulated by NF-κB
8. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Hunter, D.J.; Schofield, D.; Callander, E. The individual and socioeconomic impact of osteoarthritis. Nat. Rev. Rheumatol. 2014, 10, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Bliddal, H.; Leeds, A.R.; Christensen, R. Osteoarthritis, obesity and weight loss: Evidence, hypotheses and horizons—A scoping review. Obes. Rev. 2014, 15, 578–586. [Google Scholar] [CrossRef] [PubMed]
- Shane Anderson, A.; Loeser, R.F. Why is osteoarthritis an age-related disease? Best Pract. Res. Clin. Rheumatol. 2010, 24, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wei, X.; Zhou, J.; Wei, L. The age-related changes in cartilage and osteoarthritis. Biomed. Res. Int. 2013, 2013, 916530. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ouyang, H.; Dass, C.R.; Xu, J. Current research on pharmacologic and regenerative therapies for osteoarthritis. Bone Res. 2016, 4, 15040. [Google Scholar] [CrossRef] [PubMed]
- Le Graverand-Gastineau, M.P. Disease modifying osteoarthritis drugs: Facing development challenges and choosing molecular targets. Curr. Drug Targets 2010, 11, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, J.; Fitzpatrick, J.; Perera, N.K.P.; Orchard, J. Osteoarthritis—A systematic review of long-term safety implications for osteoarthritis of the knee. BMC Musculoskelet Disord. 2019, 20, 151. [Google Scholar] [CrossRef] [PubMed]
- Jones, I.A.; Togashi, R.; Wilson, M.L.; Heckmann, N.; Vangsness, C.T., Jr. Intra-articular treatment options for knee osteoarthritis. Nat. Rev. Rheumatol. 2019, 15, 77–90. [Google Scholar] [CrossRef]
- Hashimoto, M.; Nakasa, T.; Hikata, T.; Asahara, H. Molecular network of cartilage homeostasis and osteoarthritis. Med. Res. Rev. 2008, 28, 464–481. [Google Scholar] [CrossRef]
- Goldring, M.B.; Marcu, K.B. Cartilage homeostasis in health and rheumatic diseases. Arthritis Res. Ther. 2009, 11, 224. [Google Scholar] [CrossRef]
- Chen, L.X.; Lin, L.; Wang, H.J.; Wei, X.L.; Fu, X.; Zhang, J.Y.; Yu, C.L. Suppression of early experimental osteoarthritis by in vivo delivery of the adenoviral vector-mediated NF-kappaBp65-specific siRNA. Osteoarthritis Cartilage 2008, 16, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Marcu, K.B.; Otero, M.; Olivotto, E.; Borzi, R.M.; Goldring, M.B. NF-kappaB signaling: Multiple angles to target OA. Curr. Drug Targets 2010, 11, 599–613. [Google Scholar] [CrossRef] [PubMed]
- Nam, J.; Aguda, B.D.; Rath, B.; Agarwal, S. Biomechanical thresholds regulate inflammation through the NF-kappaB pathway: Experiments and modeling. PLoS ONE 2009, 4, e5262. [Google Scholar] [CrossRef] [PubMed]
- Gu, R.; Liu, N.; Luo, S.; Huang, W.; Zha, Z.; Yang, J. MicroRNA-9 regulates the development of knee osteoarthritis through the NF-kappaB1 pathway in chondrocytes. Medicine 2016, 95, e4315. [Google Scholar] [CrossRef] [PubMed]
- Roman-Blas, J.A.; Jimenez, S.A. NF-kappaB as a potential therapeutic target in osteoarthritis and rheumatoid arthritis. Osteoarthritis Cartilage 2006, 14, 839–848. [Google Scholar] [CrossRef]
- Choi, M.C.; MaruYama, T.; Chun, C.H.; Park, Y. Alleviation of Murine Osteoarthritis by Cartilage-Specific Deletion of IkappaBzeta. Arthritis Rheumatol. 2018, 70, 1440–1449. [Google Scholar] [CrossRef] [PubMed]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell Physiol. 2007, 213, 626–634. [Google Scholar] [CrossRef]
- Bian, Q.; Wang, Y.J.; Liu, S.F.; Li, Y.P. Osteoarthritis: Genetic factors, animal models, mechanisms, and therapies. Front. Biosci. 2012, 4, 74–100. [Google Scholar] [CrossRef]
- Burr, D.B.; Gallant, M.A. Bone remodelling in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 665–673. [Google Scholar] [CrossRef]
- Poole, A.R.; Kobayashi, M.; Yasuda, T.; Laverty, S.; Mwale, F.; Kojima, T.; Sakai, T.; Wahl, C.; El-Maadawy, S.; Webb, G.; et al. Type II collagen degradation and its regulation in articular cartilage in osteoarthritis. Ann. Rheum. Dis. 2002, 61 (Suppl. 2), 78–81. [Google Scholar] [CrossRef]
- Huang, K.; Wu, L.D. Aggrecanase and aggrecan degradation in osteoarthritis: A review. J. Int. Med. Res. 2008, 36, 1149–1160. [Google Scholar] [CrossRef] [PubMed]
- Little, C.B.; Barai, A.; Burkhardt, D.; Smith, S.M.; Fosang, A.J.; Werb, Z.; Shah, M.; Thompson, E.W. Matrix metalloproteinase 13-deficient mice are resistant to osteoarthritic cartilage erosion but not chondrocyte hypertrophy or osteophyte development. Arthritis Rheum. 2009, 60, 3723–3733. [Google Scholar] [CrossRef] [PubMed]
- Glasson, S.S.; Askew, R.; Sheppard, B.; Carito, B.; Blanchet, T.; Ma, H.L.; Flannery, C.R.; Peluso, D.; Kanki, K.; Yang, Z.; et al. Deletion of active ADAMTS5 prevents cartilage degradation in a murine model of osteoarthritis. Nature 2005, 434, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Stanton, H.; Rogerson, F.M.; East, C.J.; Golub, S.B.; Lawlor, K.E.; Meeker, C.T.; Little, C.B.; Last, K.; Farmer, P.J.; Campbell, I.K.; et al. ADAMTS5 is the major aggrecanase in mouse cartilage in vivo and in vitro. Nature 2005, 434, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Neuhold, L.A.; Killar, L.; Zhao, W.; Sung, M.L.; Warner, L.; Kulik, J.; Turner, J.; Wu, W.; Billinghurst, C.; Meijers, T.; et al. Postnatal expression in hyaline cartilage of constitutively active human collagenase-3 (MMP-13) induces osteoarthritis in mice. J. Clin. Investig. 2001, 107, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Mueller, M.B.; Tuan, R.S. Anabolic/Catabolic balance in pathogenesis of osteoarthritis: Identifying molecular targets. PM R 2011, 3, S3–S11. [Google Scholar] [CrossRef]
- Mathiessen, A.; Conaghan, P.G. Synovitis in osteoarthritis: Current understanding with therapeutic implications. Arthritis Res. Ther. 2017, 19, 18. [Google Scholar] [CrossRef] [PubMed]
- De Lange-Brokaar, B.J.; Ioan-Facsinay, A.; van Osch, G.J.; Zuurmond, A.M.; Schoones, J.; Toes, R.E.; Huizinga, T.W.; Kloppenburg, M. Synovial inflammation, immune cells and their cytokines in osteoarthritis: A review. Osteoarthritis Cartilage 2012, 20, 1484–1499. [Google Scholar] [CrossRef]
- Kim, H.A.; Cho, M.L.; Choi, H.Y.; Yoon, C.S.; Jhun, J.Y.; Oh, H.J.; Kim, H.Y. The catabolic pathway mediated by Toll-like receptors in human osteoarthritic chondrocytes. Arthritis Rheum. 2006, 54, 2152–2163. [Google Scholar] [CrossRef]
- Rigoglou, S.; Papavassiliou, A.G. The NF-kappaB signalling pathway in osteoarthritis. Int. J. Biochem. Cell Biol. 2013, 45, 2580–2584. [Google Scholar] [CrossRef]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Pulai, J.I.; Chen, H.; Im, H.J.; Kumar, S.; Hanning, C.; Hegde, P.S.; Loeser, R.F. NF-kappa B mediates the stimulation of cytokine and chemokine expression by human articular chondrocytes in response to fibronectin fragments. J. Immunol. 2005, 174, 5781–5788. [Google Scholar] [CrossRef] [PubMed]
- Berenbaum, F. Osteoarthritis as an inflammatory disease (osteoarthritis is not osteoarthrosis!). Osteoarthritis Cartilage 2013, 21, 16–21. [Google Scholar] [CrossRef] [PubMed]
- Krenn, V.; Morawietz, L.; Burmester, G.R.; Kinne, R.W.; Mueller-Ladner, U.; Muller, B.; Haupl, T. Synovitis score: Discrimination between chronic low-grade and high-grade synovitis. Histopathology 2006, 49, 358–364. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.L.; Brandt, K.D.; Ehlich, J.W.; Braunstein, E.M.; Shelbourne, K.D.; Heck, D.A.; Kalasinski, L.A. Synovial inflammation in patients with early osteoarthritis of the knee. J. Rheumatol. 1990, 17, 1662–1669. [Google Scholar] [PubMed]
- Loeuille, D.; Chary-Valckenaere, I.; Champigneulle, J.; Rat, A.C.; Toussaint, F.; Pinzano-Watrin, A.; Goebel, J.C.; Mainard, D.; Blum, A.; Pourel, J.; et al. Macroscopic and microscopic features of synovial membrane inflammation in the osteoarthritic knee: Correlating magnetic resonance imaging findings with disease severity. Arthritis Rheum. 2005, 52, 3492–3501. [Google Scholar] [CrossRef] [PubMed]
- Benito, M.J.; Veale, D.J.; FitzGerald, O.; van den Berg, W.B.; Bresnihan, B. Synovial tissue inflammation in early and late osteoarthritis. Ann. Rheum. Dis. 2005, 64, 1263–1267. [Google Scholar] [CrossRef]
- Tak, P.P.; Firestein, G.S. NF-kappaB: A key role in inflammatory diseases. J. Clin. Investig. 2001, 107, 7–11. [Google Scholar] [CrossRef]
- Niederberger, E.; Geisslinger, G. The IKK-NF-kappaB pathway: A source for novel molecular drug targets in pain therapy? FASEB J. 2008, 22, 3432–3442. [Google Scholar] [CrossRef]
- Goldring, M.B.; Otero, M. Inflammation in osteoarthritis. Curr. Opin. Rheumatol. 2011, 23, 471–478. [Google Scholar] [CrossRef]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Miagkov, A.V.; Kovalenko, D.V.; Brown, C.E.; Didsbury, J.R.; Cogswell, J.P.; Stimpson, S.A.; Baldwin, A.S.; Makarov, S.S. NF-kappaB activation provides the potential link between inflammation and hyperplasia in the arthritic joint. Proc. Natl. Acad. Sci. USA 1998, 95, 13859–13864. [Google Scholar] [CrossRef] [PubMed]
- Olivotto, E.; Otero, M.; Marcu, K.B.; Goldring, M.B. Pathophysiology of osteoarthritis: Canonical NF-kappaB/IKKbeta-dependent and kinase-independent effects of IKKalpha in cartilage degradation and chondrocyte differentiation. RMD Open 2015, 1, e000061. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-kappaB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef] [PubMed]
- Courtois, G.; Gilmore, T.D. Mutations in the NF-kappaB signaling pathway: Implications for human disease. Oncogene 2006, 25, 6831–6843. [Google Scholar] [CrossRef]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-kappaB: Its role in health and disease. J. Mol. Med. 2004, 82, 434–448. [Google Scholar] [CrossRef]
- Herrington, F.D.; Carmody, R.J.; Goodyear, C.S. Modulation of NF-kappaB Signaling as a Therapeutic Target in Autoimmunity. J. Biomol. Screen. 2016, 21, 223–242. [Google Scholar] [CrossRef]
- Oeckinghaus, A.; Ghosh, S. The NF-kappaB family of transcription factors and its regulation. Cold Spring Harb Perspect. Biol. 2009, 1, a000034. [Google Scholar] [CrossRef]
- Hoffmann, A.; Baltimore, D. Circuitry of nuclear factor kappaB signaling. Immunol. Rev. 2006, 210, 171–186. [Google Scholar] [CrossRef]
- Karin, M.; Ben-Neriah, Y. Phosphorylation meets ubiquitination: The control of NF-[kappa]B activity. Annu. Rev. Immunol. 2000, 18, 621–663. [Google Scholar] [CrossRef]
- Hayden, M.S.; Ghosh, S. Shared principles in NF-kappaB signaling. Cell 2008, 132, 344–362. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Yang, X.D.; Lamb, A.; Chen, L.F. Posttranslational modifications of NF-kappaB: Another layer of regulation for NF-kappaB signaling pathway. Cell. Signal. 2010, 22, 1282–1290. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.F.; Williams, S.A.; Mu, Y.; Nakano, H.; Duerr, J.M.; Buckbinder, L.; Greene, W.C. NF-kappaB RelA phosphorylation regulates RelA acetylation. Mol. Cell. Biol. 2005, 25, 7966–7975. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef]
- Yamamoto, M.; Yamazaki, S.; Uematsu, S.; Sato, S.; Hemmi, H.; Hoshino, K.; Kaisho, T.; Kuwata, H.; Takeuchi, O.; Takeshige, K.; et al. Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IkappaBzeta. Nature 2004, 430, 218–222. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, S.; Yamazaki, S.; Takeshige, K.; Muta, T. Crucial roles of binding sites for NF-kappaB and C/EBPs in IkappaB-zeta-mediated transcriptional activation. Biochem. J. 2007, 405, 605–615. [Google Scholar] [CrossRef] [PubMed]
- Nogai, H.; Wenzel, S.S.; Hailfinger, S.; Grau, M.; Kaergel, E.; Seitz, V.; Wollert-Wulf, B.; Pfeifer, M.; Wolf, A.; Frick, M.; et al. IkappaB-zeta controls the constitutive NF-kappaB target gene network and survival of ABC DLBCL. Blood 2013, 122, 2242–2250. [Google Scholar] [CrossRef] [PubMed]
- Nolan, G.P.; Fujita, T.; Bhatia, K.; Huppi, C.; Liou, H.C.; Scott, M.L.; Baltimore, D. The bcl-3 proto-oncogene encodes a nuclear I kappa B-like molecule that preferentially interacts with NF-kappa B p50 and p52 in a phosphorylation-dependent manner. Mol. Cell. Biol. 1993, 13, 3557–3566. [Google Scholar] [CrossRef]
- Sun, S.C. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat. Rev. Immunol. 2017, 17, 545–558. [Google Scholar] [CrossRef]
- Shih, V.F.; Tsui, R.; Caldwell, A.; Hoffmann, A. A single NFkappaB system for both canonical and non-canonical signaling. Cell Res. 2011, 21, 86–102. [Google Scholar] [CrossRef]
- Espin-Palazon, R.; Traver, D. The NF-kappaB family: Key players during embryonic development and HSC emergence. Exp. Hematol. 2016, 44, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Sha, W.C.; Bronson, R.T.; Ghosh, S.; Baltimore, D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-kappa B. Nature 1995, 376, 167–170. [Google Scholar] [CrossRef] [PubMed]
- Sha, W.C.; Liou, H.C.; Tuomanen, E.I.; Baltimore, D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 1995, 80, 321–330. [Google Scholar] [CrossRef]
- Hwang, H.S.; Kim, H.A. Chondrocyte Apoptosis in the Pathogenesis of Osteoarthritis. Int. J. Mol. Sci. 2015, 16, 26035–26054. [Google Scholar] [CrossRef] [PubMed]
- Heinegard, D.; Saxne, T. The role of the cartilage matrix in osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Fang, H.; Beier, F. Mouse models of osteoarthritis: Modelling risk factors and assessing outcomes. Nat. Rev. Rheumatol. 2014, 10, 413–421. [Google Scholar] [CrossRef]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Zafarullah, M. Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1) and nuclear factor kappa B (NF-kappa B) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol. 2002, 21, 251–262. [Google Scholar] [CrossRef]
- Mengshol, J.A.; Vincenti, M.P.; Coon, C.I.; Barchowsky, A.; Brinckerhoff, C.E. Interleukin-1 induction of collagenase 3 (matrix metalloproteinase 13) gene expression in chondrocytes requires p38, c-Jun N-terminal kinase, and nuclear factor kappaB: Differential regulation of collagenase 1 and collagenase 3. Arthritis Rheum. 2000, 43, 801–811. [Google Scholar] [CrossRef]
- Yan, H.; Duan, X.; Pan, H.; Holguin, N.; Rai, M.F.; Akk, A.; Springer, L.E.; Wickline, S.A.; Sandell, L.J.; Pham, C.T. Suppression of NF-kappaB activity via nanoparticle-based siRNA delivery alters early cartilage responses to injury. Proc. Natl. Acad. Sci. USA 2016, 113, E6199–E6208. [Google Scholar] [CrossRef]
- Murahashi, Y.; Yano, F.; Kobayashi, H.; Makii, Y.; Iba, K.; Yamashita, T.; Tanaka, S.; Saito, T. Intra-articular administration of IkappaBalpha kinase inhibitor suppresses mouse knee osteoarthritis via downregulation of the NF-kappaB/HIF-2alpha axis. Sci. Rep. 2018, 8, 16475. [Google Scholar] [CrossRef] [PubMed]
- Bond, M.; Fabunmi, R.P.; Baker, A.H.; Newby, A.C. Synergistic upregulation of metalloproteinase-9 by growth factors and inflammatory cytokines: An absolute requirement for transcription factor NF-kappa B. FEBS Lett. 1998, 435, 29–34. [Google Scholar] [CrossRef]
- Yan, C.; Wang, H.; Aggarwal, B.; Boyd, D.D. A novel homologous recombination system to study 92 kDa type IV collagenase transcription demonstrates that the NF-kappaB motif drives the transition from a repressed to an activated state of gene expression. FASEB J. 2004, 18, 540–541. [Google Scholar] [CrossRef]
- Farina, A.R.; Tacconelli, A.; Vacca, A.; Maroder, M.; Gulino, A.; Mackay, A.R. Transcriptional up-regulation of matrix metalloproteinase-9 expression during spontaneous epithelial to neuroblast phenotype conversion by SK-N-SH neuroblastoma cells, involved in enhanced invasivity, depends upon GT-box and nuclear factor kappaB elements. Cell Growth Differ. 1999, 10, 353–367. [Google Scholar] [PubMed]
- Kobayashi, H.; Hirata, M.; Saito, T.; Itoh, S.; Chung, U.I.; Kawaguchi, H. Transcriptional induction of ADAMTS5 protein by nuclear factor-kappaB (NF-kappaB) family member RelA/p65 in chondrocytes during osteoarthritis development. J. Biol. Chem. 2013, 288, 28620–28629. [Google Scholar] [CrossRef] [PubMed]
- Vincenti, M.P.; Coon, C.I.; Brinckerhoff, C.E. Nuclear factor kappaB/p50 activates an element in the distal matrix metalloproteinase 1 promoter in interleukin-1beta-stimulated synovial fibroblasts. Arthritis Rheum. 1998, 41, 1987–1994. [Google Scholar] [CrossRef]
- O’Kane, C.M.; Elkington, P.T.; Jones, M.D.; Caviedes, L.; Tovar, M.; Gilman, R.H.; Stamp, G.; Friedland, J.S. STAT3, p38 MAPK, and NF-kappaB drive unopposed monocyte-dependent fibroblast MMP-1 secretion in tuberculosis. Am. J. Respir. Cell Mol. Biol. 2010, 43, 465–474. [Google Scholar] [CrossRef]
- Vuolteenaho, K.; Moilanen, T.; Knowles, R.G.; Moilanen, E. The role of nitric oxide in osteoarthritis. Scand. J. Rheumatol. 2007, 36, 247–258. [Google Scholar] [CrossRef]
- Ulivi, V.; Giannoni, P.; Gentili, C.; Cancedda, R.; Descalzi, F. p38/NF-kB-dependent expression of COX-2 during differentiation and inflammatory response of chondrocytes. J. Cell. Biochem. 2008, 104, 1393–1406. [Google Scholar] [CrossRef]
- Allport, V.C.; Slater, D.M.; Newton, R.; Bennett, P.R. NF-kappaB and AP-1 are required for cyclo-oxygenase 2 gene expression in amnion epithelial cell line (WISH). Mol. Hum. Reprod. 2000, 6, 561–565. [Google Scholar] [CrossRef]
- Lianxu, C.; Hongti, J.; Changlong, Y. NF-kappaBp65-specific siRNA inhibits expression of genes of COX-2, NOS-2 and MMP-9 in rat IL-1beta-induced and TNF-alpha-induced chondrocytes. Osteoarthritis Cartilage 2006, 14, 367–376. [Google Scholar] [CrossRef]
- Latourte, A.; Cherifi, C.; Maillet, J.; Ea, H.K.; Bouaziz, W.; Funck-Brentano, T.; Cohen-Solal, M.; Hay, E.; Richette, P. Systemic inhibition of IL-6/Stat3 signalling protects against experimental osteoarthritis. Ann. Rheum. Dis. 2016, 76, 748–755. [Google Scholar] [CrossRef] [PubMed]
- de Andres, M.C.; Imagawa, K.; Hashimoto, K.; Gonzalez, A.; Roach, H.I.; Goldring, M.B.; Oreffo, R.O. Loss of methylation in CpG sites in the NF-kappaB enhancer elements of inducible nitric oxide synthase is responsible for gene induction in human articular chondrocytes. Arthritis Rheum. 2013, 65, 732–742. [Google Scholar] [CrossRef] [PubMed]
- Abramson, S.B. Osteoarthritis and nitric oxide. Osteoarthritis Cartilage 2008, 16 (Suppl. 2), S15–S20. [Google Scholar] [CrossRef]
- Van de Loo, F.A.; Arntz, O.J.; van Enckevort, F.H.; van Lent, P.L.; van den Berg, W.B. Reduced cartilage proteoglycan loss during zymosan-induced gonarthritis in NOS2-deficient mice and in anti-interleukin-1-treated wild-type mice with unabated joint inflammation. Arthritis Rheum. 1998, 41, 634–646. [Google Scholar] [CrossRef]
- Pelletier, J.P.; Jovanovic, D.V.; Lascau-Coman, V.; Fernandes, J.C.; Manning, P.T.; Connor, J.R.; Currie, M.G.; Martel-Pelletier, J. Selective inhibition of inducible nitric oxide synthase reduces progression of experimental osteoarthritis in vivo: Possible link with the reduction in chondrocyte apoptosis and caspase 3 level. Arthritis Rheum. 2000, 43, 1290–1299. [Google Scholar] [CrossRef]
- Saito, T.; Fukai, A.; Mabuchi, A.; Ikeda, T.; Yano, F.; Ohba, S.; Nishida, N.; Akune, T.; Yoshimura, N.; Nakagawa, T.; et al. Transcriptional regulation of endochondral ossification by HIF-2alpha during skeletal growth and osteoarthritis development. Nat. Med. 2010, 16, 678–686. [Google Scholar] [CrossRef]
- Yang, S.; Kim, J.; Ryu, J.H.; Oh, H.; Chun, C.H.; Kim, B.J.; Min, B.H.; Chun, J.S. Hypoxia-inducible factor-2alpha is a catabolic regulator of osteoarthritic cartilage destruction. Nat. Med. 2010, 16, 687–693. [Google Scholar] [CrossRef]
- Fujioka, S.; Niu, J.; Schmidt, C.; Sclabas, G.M.; Peng, B.; Uwagawa, T.; Li, Z.; Evans, D.B.; Abbruzzese, J.L.; Chiao, P.J. NF-kappaB and AP-1 connection: Mechanism of NF-kappaB-dependent regulation of AP-1 activity. Mol. Cell. Biol. 2004, 24, 7806–7819. [Google Scholar] [CrossRef]
- Grall, F.; Gu, X.; Tan, L.; Cho, J.Y.; Inan, M.S.; Pettit, A.R.; Thamrongsak, U.; Choy, B.K.; Manning, C.; Akbarali, Y.; et al. Responses to the proinflammatory cytokines interleukin-1 and tumor necrosis factor alpha in cells derived from rheumatoid synovium and other joint tissues involve nuclear factor kappaB-mediated induction of the Ets transcription factor ESE-1. Arthritis Rheum. 2003, 48, 1249–1260. [Google Scholar] [CrossRef]
- Wu, J.; Duan, R.; Cao, H.; Field, D.; Newnham, C.M.; Koehler, D.R.; Zamel, N.; Pritchard, M.A.; Hertzog, P.; Post, M.; et al. Regulation of epithelium-specific Ets-like factors ESE-1 and ESE-3 in airway epithelial cells: Potential roles in airway inflammation. Cell Res. 2008, 18, 649–663. [Google Scholar] [CrossRef] [PubMed]
- Pi, Y.; Zhang, X.; Shao, Z.; Zhao, F.; Hu, X.; Ao, Y. Intra-articular delivery of anti-Hif-2alpha siRNA by chondrocyte-homing nanoparticles to prevent cartilage degeneration in arthritic mice. Gene Ther. 2015, 22, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Hirata, M.; Kugimiya, F.; Fukai, A.; Saito, T.; Yano, F.; Ikeda, T.; Mabuchi, A.; Sapkota, B.R.; Akune, T.; Nishida, N.; et al. C/EBPbeta and RUNX2 cooperate to degrade cartilage with MMP-13 as the target and HIF-2alpha as the inducer in chondrocytes. Hum. Mol. Genet. 2012, 21, 1111–1123. [Google Scholar] [CrossRef] [PubMed]
- Muddasani, P.; Norman, J.C.; Ellman, M.; van Wijnen, A.J.; Im, H.J. Basic fibroblast growth factor activates the MAPK and NFkappaB pathways that converge on Elk-1 to control production of matrix metalloproteinase-13 by human adult articular chondrocytes. J. Biol. Chem. 2007, 282, 31409–31421. [Google Scholar] [CrossRef]
- Rudders, S.; Gaspar, J.; Madore, R.; Voland, C.; Grall, F.; Patel, A.; Pellacani, A.; Perrella, M.A.; Libermann, T.A.; Oettgen, P. ESE-1 is a novel transcriptional mediator of inflammation that interacts with NF-kappa B to regulate the inducible nitric-oxide synthase gene. J. Biol. Chem. 2001, 276, 3302–3309. [Google Scholar] [CrossRef] [PubMed]
- Grall, F.T.; Prall, W.C.; Wei, W.; Gu, X.; Cho, J.Y.; Choy, B.K.; Zerbini, L.F.; Inan, M.S.; Goldring, S.R.; Gravallese, E.M.; et al. The Ets transcription factor ESE-1 mediates induction of the COX-2 gene by LPS in monocytes. FEBS J. 2005, 272, 1676–1687. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Plumb, D.A.; Tsuchimochi, K.; Dragomir, C.L.; Hashimoto, K.; Peng, H.; Olivotto, E.; Bevilacqua, M.; Tan, L.; Yang, Z.; et al. E74-like factor 3 (ELF3) impacts on matrix metalloproteinase 13 (MMP13) transcriptional control in articular chondrocytes under proinflammatory stress. J. Biol. Chem. 2012, 287, 3559–3572. [Google Scholar] [CrossRef]
- Wondimu, E.B.; Culley, K.L.; Quinn, J.; Chang, J.; Dragomir, C.L.; Plumb, D.A.; Goldring, M.B.; Otero, M. Elf3 Contributes to Cartilage Degradation in vivo in a Surgical Model of Post-Traumatic Osteoarthritis. Sci. Rep. 2018, 8, 6438. [Google Scholar] [CrossRef]
- Van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthritis Cartilage 2012, 20, 223–232. [Google Scholar] [CrossRef]
- Dreier, R. Hypertrophic differentiation of chondrocytes in osteoarthritis: The developmental aspect of degenerative joint disorders. Arthritis Res. Ther. 2010, 12, 216. [Google Scholar] [CrossRef]
- Sun, M.M.; Beier, F. Chondrocyte hypertrophy in skeletal development, growth, and disease. Birth Defects Res. Part C Embryo Today Rev. 2014, 102, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.; Balbin, M.; Santos, F.; Fernandez, M.; Ferrando, S.; Lopez, J.M. Different bone growth rates are associated with changes in the expression pattern of types II and X collagens and collagenase 3 in proximal growth plates of the rat tibia. J. Bone Miner. Res. 2000, 15, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Shlopov, B.V.; Lie, W.R.; Mainardi, C.L.; Cole, A.A.; Chubinskaya, S.; Hasty, K.A. Osteoarthritic lesions: Involvement of three different collagenases. Arthritis Rheum. 1997, 40, 2065–2074. [Google Scholar] [CrossRef] [PubMed]
- Tchetina, E.V.; Squires, G.; Poole, A.R. Increased type II collagen degradation and very early focal cartilage degeneration is associated with upregulation of chondrocyte differentiation related genes in early human articular cartilage lesions. J. Rheumatol. 2005, 32, 876–886. [Google Scholar] [PubMed]
- Pfander, D.; Kortje, D.; Zimmermann, R.; Weseloh, G.; Kirsch, T.; Gesslein, M.; Cramer, T.; Swoboda, B. Vascular endothelial growth factor in articular cartilage of healthy and osteoarthritic human knee joints. Ann. Rheum. Dis. 2001, 60, 1070–1073. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Marcu, K.B.; Goldring, M.B.; Otero, M. Phenotypic instability of chondrocytes in osteoarthritis: On a path to hypertrophy. Ann. N. Y. Acad. Sci. 2019, 1442, 17–34. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Zhong, L.; van Blitterswijk, C.A.; Post, J.N.; Karperien, M. T cell factor 4 is a pro-catabolic and apoptotic factor in human articular chondrocytes by potentiating nuclear factor kappaB signaling. J. Biol. Chem. 2013, 288, 17552–17558. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Sun, C.; Zhang, S.; Xu, X.; Zhai, L.; Wang, Y.; Wang, S.; Liu, Z.; Cheng, H.; Xiao, M.; et al. Sam68 Promotes NF-kappaB Activation and Apoptosis Signaling in Articular Chondrocytes during Osteoarthritis. Inflamm. Res. 2015, 64, 895–902. [Google Scholar] [CrossRef]
- Tao, R.; Xu, X.; Sun, C.; Wang, Y.; Wang, S.; Liu, Z.; Zhai, L.; Cheng, H.; Xiao, M.; Zhang, D. KPNA2 interacts with P65 to modulate catabolic events in osteoarthritis. Exp. Mol. Pathol. 2015, 99, 245–252. [Google Scholar] [CrossRef]
- Okamoto, K.; Iwai, Y.; Oh-Hora, M.; Yamamoto, M.; Morio, T.; Aoki, K.; Ohya, K.; Jetten, A.M.; Akira, S.; Muta, T.; et al. IkappaBzeta regulates T(H)17 development by cooperating with ROR nuclear receptors. Nature 2010, 464, 1381–1385. [Google Scholar] [CrossRef]
- Ma, B.; Hottiger, M.O. Crosstalk between Wnt/beta-Catenin and NF-kappaB Signaling Pathway during Inflammation. Front. Immunol. 2016, 7, 378. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, P.; Baltimore, D. Sam68 is required for both NF-kappaB activation and apoptosis signaling by the TNF receptor. Mol. Cell. 2011, 43, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Sun, X.; Zheng, W.; Wier, E.M.; Hodgson, A.; Tran, D.Q.; Richard, S.; Wan, F. Sam68 modulates the promoter specificity of NF-kappaB and mediates expression of CD25 in activated T cells. Nat. Commun. 2013, 4, 1909. [Google Scholar] [CrossRef] [PubMed]
- Liang, P.; Zhang, H.; Wang, G.; Li, S.; Cong, S.; Luo, Y.; Zhang, B. KPNB1, XPO7 and IPO8 mediate the translocation ofNF-kappaB/p65 into the nucleus. Traffic 2013, 14, 1132–1143. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.; Hu, X.; Dai, L.; Zhang, X.; Ren, B.; Shi, W.; Liu, Z.; Duan, X.; Zhang, J.; Fu, X.; et al. Inhibition of transforming growth factor beta-activated kinase 1 prevents inflammation-related cartilage degradation in osteoarthritis. Sci. Rep. 2016, 6, 34497. [Google Scholar] [CrossRef]
- Klatt, A.R.; Klinger, G.; Neumuller, O.; Eidenmuller, B.; Wagner, I.; Achenbach, T.; Aigner, T.; Bartnik, E. TAK1 downregulation reduces IL-1beta induced expression of MMP13, MMP1 and TNF-alpha. Biomed. Pharmacother. 2006, 60, 55–61. [Google Scholar] [CrossRef]
- Chang, S.H.; Mori, D.; Kobayashi, H.; Mori, Y.; Nakamoto, H.; Okada, K.; Taniguchi, Y.; Sugita, S.; Yano, F.; Chung, U.I.; et al. Excessive mechanical loading promotes osteoarthritis through the gremlin-1-NF-kappaB pathway. Nat. Commun. 2019, 10, 1442. [Google Scholar] [CrossRef]
- Dossumbekova, A.; Anghelina, M.; Madhavan, S.; He, L.; Quan, N.; Knobloch, T.; Agarwal, S. Biomechanical signals inhibit IKK activity to attenuate NF-kappaB transcription activity in inflamed chondrocytes. Arthritis Rheum. 2007, 56, 3284–3296. [Google Scholar] [CrossRef]
- Madhavan, S.; Anghelina, M.; Sjostrom, D.; Dossumbekova, A.; Guttridge, D.C.; Agarwal, S. Biomechanical signals suppress TAK1 activation to inhibit NF-kappaB transcriptional activation in fibrochondrocytes. J. Immunol. 2007, 179, 6246–6254. [Google Scholar] [CrossRef]
- Knobloch, T.J.; Madhavan, S.; Nam, J.; Agarwal, S., Jr.; Agarwal, S. Regulation of chondrocytic gene expression by biomechanical signals. Crit. Rev. Eukaryot. Gene Expr. 2008, 18, 139–150. [Google Scholar] [CrossRef]
- Azamar-Llamas, D.; Hernandez-Molina, G.; Ramos-Avalos, B.; Furuzawa-Carballeda, J. Adipokine Contribution to the Pathogenesis of Osteoarthritis. Mediators Inflamm. 2017, 2017, 5468023. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, A.; Vuolteenaho, K.; Nieminen, R.; Moilanen, T.; Moilanen, E. Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from OA patients. Clin. Exp. Rheumatol. 2011, 29, 57–64. [Google Scholar] [PubMed]
- Yaykasli, K.O.; Hatipoglu, O.F.; Yaykasli, E.; Yildirim, K.; Kaya, E.; Ozsahin, M.; Uslu, M.; Gunduz, E. Leptin induces ADAMTS-4, ADAMTS-5, and ADAMTS-9 genes expression by mitogen-activated protein kinases and NF-kB signaling pathways in human chondrocytes. Cell Biol. Int. 2015, 39, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Tong, K.M.; Chen, C.P.; Huang, K.C.; Shieh, D.C.; Cheng, H.C.; Tzeng, C.Y.; Chen, K.H.; Chiu, Y.C.; Tang, C.H. Adiponectin increases MMP-3 expression in human chondrocytes through AdipoR1 signaling pathway. J. Cell. Biochem. 2011, 112, 1431–1440. [Google Scholar] [CrossRef] [PubMed]
- Vuolteenaho, K.; Koskinen, A.; Kukkonen, M.; Nieminen, R.; Paivarinta, U.; Moilanen, T.; Moilanen, E. Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage--mediator role of NO in leptin-induced PGE2, IL-6, and IL-8 production. Mediators Inflamm. 2009, 2009, 345838. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Wang, X.; Pan, H.; Yang, H.; Li, X.; Zhang, K.; Wang, H.; Zheng, Z.; Liu, H.; Wang, J. Resistin promotes CCL4 expression through toll-like receptor-4 and activation of the p38-MAPK and NF-kappaB signaling pathways: Implications for intervertebral disc degeneration. Osteoarthritis Cartilage 2017, 25, 341–350. [Google Scholar] [CrossRef]
- Laiguillon, M.C.; Houard, X.; Bougault, C.; Gosset, M.; Nourissat, G.; Sautet, A.; Jacques, C.; Berenbaum, F.; Sellam, J. Expression and function of visfatin (Nampt), an adipokine-enzyme involved in inflammatory pathways of osteoarthritis. Arthritis Res. Ther. 2014, 16, R38. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Ryu, J.H.; Oh, H.; Jeon, J.; Kwak, J.S.; Kim, J.H.; Kim, H.A.; Chun, C.H.; Chun, J.S. NAMPT (visfatin), a direct target of hypoxia-inducible factor-2alpha, is an essential catabolic regulator of osteoarthritis. Ann. Rheum. Dis. 2015, 74, 595–602. [Google Scholar] [CrossRef]
- Budak, E.; Fernandez Sanchez, M.; Bellver, J.; Cervero, A.; Simon, C.; Pellicer, A. Interactions of the hormones leptin, ghrelin, adiponectin, resistin, and PYY3-36 with the reproductive system. Fertil. Steril. 2006, 85, 1563–1581. [Google Scholar] [CrossRef]
- Qu, R.; Chen, X.; Wang, W.; Qiu, C.; Ban, M.; Guo, L.; Vasilev, K.; Chen, J.; Li, W.; Zhao, Y. Ghrelin protects against osteoarthritis through interplay with Akt and NF-kappaB signaling pathways. FASEB J. 2018, 32, 1044–1058. [Google Scholar] [CrossRef]
- Kahles, F.; Findeisen, H.M.; Bruemmer, D. Osteopontin: A novel regulator at the cross roads of inflammation, obesity and diabetes. Mol. Metab. 2014, 3, 384–393. [Google Scholar] [CrossRef]
- De Fusco, C.; Messina, A.; Monda, V.; Viggiano, E.; Moscatelli, F.; Valenzano, A.; Esposito, T.; Sergio, C.; Cibelli, G.; Monda, M.; et al. Osteopontin: Relation between Adipose Tissue and Bone Homeostasis. Stem. Cells Int. 2017, 2017, 4045238. [Google Scholar] [CrossRef]
- Nomiyama, T.; Perez-Tilve, D.; Ogawa, D.; Gizard, F.; Zhao, Y.; Heywood, E.B.; Jones, K.L.; Kawamori, R.; Cassis, L.A.; Tschop, M.H.; et al. Osteopontin mediates obesity-induced adipose tissue macrophage infiltration and insulin resistance in mice. J. Clin. Investig. 2007, 117, 2877–2888. [Google Scholar] [CrossRef]
- Nakazeki, F.; Nishiga, M.; Horie, T.; Nishi, H.; Nakashima, Y.; Baba, O.; Kuwabara, Y.; Nishino, T.; Nakao, T.; Ide, Y.; et al. Loss of periostin ameliorates adipose tissue inflammation and fibrosis in vivo. Sci. Rep. 2018, 8, 8553. [Google Scholar] [CrossRef]
- Bonnet, N.; Garnero, P.; Ferrari, S. Periostin action in bone. Mol. Cell. Endocrinol. 2016, 432, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.G.; Li, K.H.; Zeng, K.B.; Tu, M.; Xu, M.; Lei, G.H. Elevated osteopontin level of synovial fluid and articular cartilage is associated with disease severity in knee osteoarthritis patients. Osteoarthritis Cartilage 2010, 18, 82–87. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Gao, S.; Lei, G. Association of osteopontin with osteoarthritis. Rheumatol. Int. 2014, 34, 1627–1631. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Jiang, W.; Wang, H.; Deng, Z.; Zeng, C.; Tu, M.; Li, L.; Xiao, W.; Gao, S.; Luo, W.; et al. Osteopontin Promotes Expression of Matrix Metalloproteinase 13 through NF-kappaB Signaling in Osteoarthritis. Biomed. Res. Int. 2016, 2016, 6345656. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.; Zhang, F.J.; Tian, J.; Tu, M.; Xiong, Y.L.; Luo, W.; Li, Y.S.; Song, B.B.; Gao, S.G.; Lei, G.H. Osteopontin inhibits HIF-2alpha mRNA expression in osteoarthritic chondrocytes. Exp. Ther. Med. 2015, 9, 2415–2419. [Google Scholar] [CrossRef] [PubMed]
- Matsui, Y.; Iwasaki, N.; Kon, S.; Takahashi, D.; Morimoto, J.; Matsui, Y.; Denhardt, D.T.; Rittling, S.; Minami, A.; Uede, T. Accelerated development of aging-associated and instability-induced osteoarthritis in osteopontin-deficient mice. Arthritis Rheum. 2009, 60, 2362–2371. [Google Scholar] [CrossRef] [PubMed]
- Chijimatsu, R.; Kunugiza, Y.; Taniyama, Y.; Nakamura, N.; Tomita, T.; Yoshikawa, H. Expression and pathological effects of periostin in human osteoarthritis cartilage. BMC Musculoskelet Disord. 2015, 16, 215. [Google Scholar] [CrossRef] [PubMed]
- Wojdasiewicz, P.; Poniatowski, L.A.; Szukiewicz, D. The role of inflammatory and anti-inflammatory cytokines in the pathogenesis of osteoarthritis. Mediators Inflamm. 2014, 2014, 561459. [Google Scholar] [CrossRef] [PubMed]
- Mabey, T.; Honsawek, S. Cytokines as biochemical markers for knee osteoarthritis. World J. Orthop. 2015, 6, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Frey, S.; Derer, A.; Messbacher, M.E.; Baeten, D.L.; Bugatti, S.; Montecucco, C.; Schett, G.; Hueber, A.J. The novel cytokine interleukin-36alpha is expressed in psoriatic and rheumatoid arthritis synovium. Ann. Rheum. Dis. 2013, 72, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Conde, J.; Scotece, M.; Abella, V.; Lois, A.; Lopez, V.; Garcia-Caballero, T.; Pino, J.; Gomez-Reino, J.J.; Gomez, R.; Lago, F.; et al. IL-36alpha: A novel cytokine involved in the catabolic and inflammatory response in chondrocytes. Sci. Rep. 2015, 5, 16674. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chubinskaya, S.; Esposito, A.; Jin, X.; Tagliafierro, L.; Loeser, R.; Hakimiyan, A.A.; Longobardi, L.; Ozkan, H.; Spagnoli, A. TGF-beta type 2 receptor-mediated modulation of the IL-36 family can be therapeutically targeted in osteoarthritis. Sci. Transl. Med. 2019, 11, eaan2585. [Google Scholar] [CrossRef] [PubMed]
- Van der Kraan, P.M. Differential Role of Transforming Growth Factor-beta in an Osteoarthritic or a Healthy Joint. J. Bone Metab. 2018, 25, 65–72. [Google Scholar] [CrossRef] [PubMed]
- Shen, J.; Li, S.; Chen, D. TGF-beta signaling and the development of osteoarthritis. Bone Res. 2014, 2. [Google Scholar] [CrossRef]
- Shen, J.; Li, J.; Wang, B.; Jin, H.; Wang, M.; Zhang, Y.; Yang, Y.; Im, H.J.; O’Keefe, R.; Chen, D. Deletion of the transforming growth factor beta receptor type II gene in articular chondrocytes leads to a progressive osteoarthritis-like phenotype in mice. Arthritis Rheum. 2013, 65, 3107–3119. [Google Scholar] [CrossRef]
- Wang, Q.; Tan, Q.Y.; Xu, W.; Qi, H.B.; Chen, D.; Zhou, S.; Ni, Z.H.; Kuang, L.; Guo, J.Y.; Huang, J.L.; et al. Cartilage-specific deletion of Alk5 gene results in a progressive osteoarthritis-like phenotype in mice. Osteoarthritis Cartilage 2017, 25, 1868–1879. [Google Scholar] [CrossRef]
- Garcia-Arnandis, I.; Guillen, M.I.; Gomar, F.; Pelletier, J.P.; Martel-Pelletier, J.; Alcaraz, M.J. High mobility group box 1 potentiates the pro-inflammatory effects of interleukin-1beta in osteoarthritic synoviocytes. Arthritis Res. Ther. 2010, 12, R165. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Sun, W.; Gao, R.; Su, Y.; Umehara, H.; Dong, L.; Gong, F. The role of high mobility group box chromosomal protein 1 in rheumatoid arthritis. Rheumatology 2013, 52, 1739–1747. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Taniguchi, N.; Kawahara, K.; Yone, K.; Hashiguchi, T.; Yamakuchi, M.; Goto, M.; Inoue, K.; Yamada, S.; Ijiri, K.; Matsunaga, S.; et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003, 48, 971–981. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Lei, J.; Zhuang, Y.; Zhang, K.; Lu, D. Overexpression of HMGB1 A-box reduced IL-1beta-induced MMP expression and the production of inflammatory mediators in human chondrocytes. Exp. Cell Res. 2016, 349, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Chockalingam, P.S.; Varadarajan, U.; Sheldon, R.; Fortier, E.; LaVallie, E.R.; Morris, E.A.; Yaworsky, P.J.; Majumdar, M.K. Involvement of protein kinase Czeta in interleukin-1beta induction of ADAMTS-4 and type 2 nitric oxide synthase via NF-kappaB signaling in primary human osteoarthritic chondrocytes. Arthritis Rheum. 2007, 56, 4074–4083. [Google Scholar] [CrossRef] [PubMed]
- LaVallie, E.R.; Chockalingam, P.S.; Collins-Racie, L.A.; Freeman, B.A.; Keohan, C.C.; Leitges, M.; Dorner, A.J.; Morris, E.A.; Majumdar, M.K.; Arai, M. Protein kinase Czeta is up-regulated in osteoarthritic cartilage and is required for activation of NF-kappaB by tumor necrosis factor and interleukin-1 in articular chondrocytes. J. Biol. Chem. 2006, 281, 24124–24137. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Yang, B.; Li, Y.; Zhang, S.; Li, Z. Blocking of the P2X7 receptor inhibits the activation of the MMP-13 and NF-kappaB pathways in the cartilage tissue of rats with osteoarthritis. Int. J. Mol. Med. 2016, 38, 1922–1932. [Google Scholar] [CrossRef]
- Choi, M.C.; Choi, W.H. Mithramycin A Alleviates Osteoarthritic Cartilage Destruction by Inhibiting HIF-2alpha Expression. Int. J. Mol. Sci. 2018, 19, 1411. [Google Scholar] [CrossRef]
- Liang, S.; Lv, Z.T.; Zhang, J.M.; Wang, Y.T.; Dong, Y.H.; Wang, Z.G.; Chen, K.; Cheng, P.; Yang, Q.; Guo, F.J.; et al. Necrostatin-1 Attenuates Trauma-Induced Mouse Osteoarthritis and IL-1beta Induced Apoptosis via HMGB1/TLR4/SDF-1 in Primary Mouse Chondrocytes. Front. Pharmacol. 2018, 9, 1378. [Google Scholar] [CrossRef]
- Deng, Y.; Lu, J.; Li, W.; Wu, A.; Zhang, X.; Tong, W.; Ho, K.K.; Qin, L.; Song, H.; Mak, K.K. Reciprocal inhibition of YAP/TAZ and NF-kappaB regulates osteoarthritic cartilage degradation. Nat. Commun. 2018, 9, 4564. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, Y.; Qu, R.; Chen, X.; Wang, W.; Qiu, C.; Liu, B.; Pan, X.; Liu, L.; Vasilev, K.; et al. Cortistatin binds to TNF-alpha receptors and protects against osteoarthritis. EBioMedicine 2019, 41, 556–570. [Google Scholar] [CrossRef] [PubMed]
- Uchimura, T.; Foote, A.T.; Smith, E.L.; Matzkin, E.G.; Zeng, L. Insulin-Like Growth Factor II (IGF-II) Inhibits IL-1beta-Induced Cartilage Matrix Loss and Promotes Cartilage Integrity in Experimental Osteoarthritis. J. Cell. Biochem. 2015, 116, 2858–2869. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Conde, J.; Scotece, M.; Abella, V.; Lois, A.; Lopez, V.; Pino, J.; Gomez, R.; Gomez-Reino, J.J.; Gualillo, O. SERPINE2 Inhibits IL-1alpha-Induced MMP-13 Expression in Human Chondrocytes: Involvement of ERK/NF-kappaB/AP-1 Pathways. PLoS ONE 2015, 10, e0135979. [Google Scholar] [CrossRef] [PubMed]
- Yang, E.; Zheng, H.; Peng, H.; Ding, Y. Lentivirus-induced knockdown of LRP1 induces osteoarthritic-like effects and increases susceptibility to apoptosis in chondrocytes via the nuclear factor-kappaB pathway. Exp. Ther. Med. 2015, 10, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.S.; Park, J.K.; Kang, E.H.; Lee, Y.K.; Kim, T.K.; Chung, J.H.; Zimmerer, J.M.; Carson, W.E.; Song, Y.W.; Lee, Y.J. Cytokine signaling-1 suppressor is inducible by IL-1beta and inhibits the catabolic effects of IL-1beta in chondrocytes: Its implication in the paradoxical joint-protective role of IL-1beta. Arthritis Res. Ther. 2013, 15, R191. [Google Scholar] [CrossRef]
- Khan, N.M.; Haqqi, T.M. Epigenetics in osteoarthritis: Potential of HDAC inhibitors as therapeutics. Pharmacol. Res. 2018, 128, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Peffers, M.J.; Balaskas, P.; Smagul, A. Osteoarthritis year in review 2017: Genetics and epigenetics. Osteoarthritis Cartilage 2018, 26, 304–311. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS ONE 2012, 7, e46364. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 2004, 23, 2369–2380. [Google Scholar] [CrossRef]
- Kawahara, T.L.; Michishita, E.; Adler, A.S.; Damian, M.; Berber, E.; Lin, M.; McCord, R.A.; Ongaigui, K.C.; Boxer, L.D.; Chang, H.Y.; et al. SIRT6 links histone H3 lysine 9 deacetylation to NF-kappaB-dependent gene expression and organismal life span. Cell 2009, 136, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, T.; Sasaki, H.; Takayama, K.; Ishida, K.; Matsumoto, T.; Kubo, S.; Matsuzaki, T.; Nishida, K.; Kurosaka, M.; Kuroda, R. The overexpression of SIRT1 inhibited osteoarthritic gene expression changes induced by interleukin-1beta in human chondrocytes. J. Orthop. Res. 2013, 31, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Nishida, K.; Matsushita, T.; Takayama, K.; Tanaka, T.; Miyaji, N.; Ibaraki, K.; Araki, D.; Kanzaki, N.; Matsumoto, T.; Kuroda, R. Intraperitoneal injection of the SIRT1 activator SRT1720 attenuates the progression of experimental osteoarthritis in mice. Bone Joint Res. 2018, 7, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Moon, M.H.; Jeong, J.K.; Lee, Y.J.; Seol, J.W.; Jackson, C.J.; Park, S.Y. SIRT1, a class III histone deacetylase, regulates TNF-alpha-induced inflammation in human chondrocytes. Osteoarthritis Cartilage 2013, 21, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Matsuzaki, T.; Matsushita, T.; Takayama, K.; Matsumoto, T.; Nishida, K.; Kuroda, R.; Kurosaka, M. Disruption of Sirt1 in chondrocytes causes accelerated progression of osteoarthritis under mechanical stress and during ageing in mice. Ann. Rheum. Dis. 2014, 73, 1397–1404. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Chen, L.; Wang, Y.; Li, W.; Lin, Y.; Yu, D.; Zhang, L.; Li, F.; Pan, Z. Overexpression of Sirtuin 6 suppresses cellular senescence and NF-kappaB mediated inflammatory responses in osteoarthritis development. Sci. Rep. 2015, 5, 17602. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Y.; Lotz, M.; Terkeltaub, R.; Liu-Bryan, R. Modulation of matrix metabolism by ATP-citrate lyase in articular chondrocytes. J. Biol. Chem. 2018, 293, 12259–12270. [Google Scholar] [CrossRef] [PubMed]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Luscher, B.; Hottiger, M.O. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar] [CrossRef]
- Zhong, H.M.; Ding, Q.H.; Chen, W.P.; Luo, R.B. Vorinostat, a HDAC inhibitor, showed anti-osteoarthritic activities through inhibition of iNOS and MMP expression, p38 and ERK phosphorylation and blocking NF-kappaB nuclear translocation. Int. Immunopharmacol. 2013, 17, 329–335. [Google Scholar] [CrossRef]
- Cheng, C.; Shan, W.; Huang, W.; Ding, Z.; Cui, G.; Liu, F.; Lu, W.; Xu, J.; He, W.; Yin, Z. ACY-1215 exhibits anti-inflammatory and chondroprotective effects in human osteoarthritis chondrocytes via inhibition of STAT3 and NF-kappaB signaling pathways. Biomed. Pharmacother. 2019, 109, 2464–2471. [Google Scholar] [CrossRef]
- Cai, D.; Yin, S.; Yang, J.; Jiang, Q.; Cao, W. Histone deacetylase inhibition activates Nrf2 and protects against osteoarthritis. Arthritis Res. Ther. 2015, 17, 269. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.P.; Bao, J.P.; Hu, P.F.; Feng, J.; Wu, L.D. Alleviation of osteoarthritis by Trichostatin A, a histone deacetylase inhibitor, in experimental osteoarthritis. Mol. Biol. Rep. 2010, 37, 3967–3972. [Google Scholar] [CrossRef] [PubMed]
- Miyaki, S.; Asahara, H. Macro view of microRNA function in osteoarthritis. Nat. Rev. Rheumatol. 2012, 8, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Endisha, H.; Rockel, J.; Jurisica, I.; Kapoor, M. The complex landscape of microRNAs in articular cartilage: Biology, pathology, and therapeutic targets. JCI Insight 2018, 3. [Google Scholar] [CrossRef]
- Xu, B.; Li, Y.Y.; Ma, J.; Pei, F.X. Roles of microRNA and signaling pathway in osteoarthritis pathogenesis. J. Zhejiang Univ. Sci. B 2016, 17, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, N.; Rasheed, Z.; Ramamurthy, S.; Anbazhagan, A.N.; Voss, F.R.; Haqqi, T.M. MicroRNA-27b regulates the expression of matrix metalloproteinase 13 in human osteoarthritis chondrocytes. Arthritis Rheum. 2010, 62, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Liang, Z.J.; Zhuang, H.; Wang, G.X.; Li, Z.; Zhang, H.T.; Yu, T.Q.; Zhang, B.D. MiRNA-140 is a negative feedback regulator of MMP-13 in IL-1beta-stimulated human articular chondrocyte C28/I2 cells. Inflamm. Res. 2012, 61, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Taganov, K.D.; Boldin, M.P.; Chang, K.J.; Baltimore, D. NF-kappaB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. USA 2006, 103, 12481–12486. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, K.; Nakasa, T.; Miyaki, S.; Ishikawa, M.; Deie, M.; Adachi, N.; Yasunaga, Y.; Asahara, H.; Ochi, M. Expression of MicroRNA-146a in osteoarthritis cartilage. Arthritis Rheum. 2009, 60, 1035–1041. [Google Scholar] [CrossRef]
- Kang, D.; Shin, J.; Cho, Y.; Kim, H.S.; Gu, Y.R.; Kim, H.; You, K.T.; Chang, M.J.; Chang, C.B.; Kang, S.B.; et al. Stress-activated miR-204 governs senescent phenotypes of chondrocytes to promote osteoarthritis development. Sci. Transl. Med. 2019, 11, eaar6659. [Google Scholar] [CrossRef]
- Yang, X.; Guan, Y.; Tian, S.; Wang, Y.; Sun, K.; Chen, Q. Mechanical and IL-1beta Responsive miR-365 Contributes to Osteoarthritis Development by Targeting Histone Deacetylase 4. Int. J. Mol. Sci. 2016, 17, 436. [Google Scholar] [CrossRef] [PubMed]
- Rasheed, Z.; Al-Shobaili, H.A.; Rasheed, N.; Mahmood, A.; Khan, M.I. MicroRNA-26a-5p regulates the expression of inducible nitric oxide synthase via activation of NF-kappaB pathway in human osteoarthritis chondrocytes. Arch. Biochem. Biophys. 2016, 594, 61–67. [Google Scholar] [CrossRef] [PubMed]
- Mao, G.; Wu, P.; Zhang, Z.; Zhang, Z.; Liao, W.; Li, Y.; Kang, Y. MicroRNA-92a-3p Regulates Aggrecanase-1 and Aggrecanase-2 Expression in Chondrogenesis and IL-1beta-Induced Catabolism in Human Articular Chondrocytes. Cell. Physiol. Biochem. 2017, 44, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zhang, Z.; Chen, W.; Huang, G.; He, A.; Hou, C.; Long, Y.; Yang, Z.; Zhang, Z.; Liao, W. MicroRNA-320 regulates matrix metalloproteinase-13 expression in chondrogenesis and interleukin-1beta-induced chondrocyte responses. Osteoarthritis Cartilage 2016, 24, 932–941. [Google Scholar] [CrossRef] [PubMed]
- Park, S.J.; Cheon, E.J.; Kim, H.A. MicroRNA-558 regulates the expression of cyclooxygenase-2 and IL-1beta-induced catabolic effects in human articular chondrocytes. Osteoarthritis Cartilage 2013, 21, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Wei, Z.J.; Liu, J.; Qin, J. miR-138 suppressed the progression of osteoarthritis mainly through targeting p65. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 2177–2184. [Google Scholar] [PubMed]
- Ding, Y.; Wang, L.; Zhao, Q.; Wu, Z.; Kong, L. MicroRNA93 inhibits chondrocyte apoptosis and inflammation in osteoarthritis by targeting the TLR4/NFkappaB signaling pathway. Int. J. Mol. Med. 2019, 43, 779–790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Cao, X.; Li, J.; Zhao, G. MiR-210 inhibits NF-kappaB signaling pathway by targeting DR6 in osteoarthritis. Sci. Rep. 2015, 5, 12775. [Google Scholar] [CrossRef]
- Yin, X.; Wang, J.Q.; Yan, S.Y. Reduced miR26a and miR26b expression contributes to the pathogenesis of osteoarthritis via the promotion of p65 translocation. Mol. Med. Rep. 2017, 15, 551–558. [Google Scholar] [CrossRef]
- Chen, Q.; Wu, S.; Wu, Y.; Chen, L.; Pang, Q. MiR-149 suppresses the inflammatory response of chondrocytes in osteoarthritis by down-regulating the activation of TAK1/NF-kappaB. Biomed. Pharmacother. 2018, 101, 763–768. [Google Scholar] [CrossRef]
- Zhong, J.H.; Li, J.; Liu, C.F.; Liu, N.; Bian, R.X.; Zhao, S.M.; Yan, S.Y.; Zhang, Y.B. Effects of microRNA-146a on the proliferation and apoptosis of human osteoarthritis chondrocytes by targeting TRAF6 through the NF-kappaB signalling pathway. Biosci. Rep. 2017, 37. [Google Scholar] [CrossRef] [PubMed]
- Gu, S.X.; Li, X.; Hamilton, J.L.; Chee, A.; Kc, R.; Chen, D.; An, H.S.; Kim, J.S.; Oh, C.D.; Ma, Y.Z.; et al. MicroRNA-146a reduces IL-1 dependent inflammatory responses in the intervertebral disc. Gene 2015, 555, 80–87. [Google Scholar] [CrossRef]
- Li, X.; Gibson, G.; Kim, J.S.; Kroin, J.; Xu, S.; van Wijnen, A.J.; Im, H.J. MicroRNA-146a is linked to pain-related pathophysiology of osteoarthritis. Gene 2011, 480, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Zhao, J.; Jing, W.; Yan, S.; Wang, X.; Xiao, C.; Ma, B. Role of miR-146a in human chondrocyte apoptosis in response to mechanical pressure injury in vitro. Int. J. Mol. Med. 2014, 34, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Huang, J.; Dai, L.; Yu, D.; Chen, Q.; Zhang, X.; Dai, K. miR-146a, an IL-1beta responsive miRNA, induces vascular endothelial growth factor and chondrocyte apoptosis by targeting Smad4. Arthritis Res. Ther. 2012, 14, R75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wang, C.; Zhao, J.; Xu, J.; Geng, Y.; Dai, L.; Huang, Y.; Fu, S.C.; Dai, K.; Zhang, X. miR-146a facilitates osteoarthritis by regulating cartilage homeostasis via targeting Camk2d and Ppp3r2. Cell Death Dis. 2017, 8, e2734. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.M.; Fuller, C.J.; Whittles, C.E.; Sharif, M. Chondrocyte death by apoptosis is associated with cartilage matrix degradation. Osteoarthritis Cartilage 2007, 15, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Blanco, F.J.; Guitian, R.; Vazquez-Martul, E.; de Toro, F.J.; Galdo, F. Osteoarthritis chondrocytes die by apoptosis. A possible pathway for osteoarthritis pathology. Arthritis Rheum. 1998, 41, 284–289. [Google Scholar] [CrossRef]
- Hashimoto, S.; Ochs, R.L.; Komiya, S.; Lotz, M. Linkage of chondrocyte apoptosis and cartilage degradation in human osteoarthritis. Arthritis Rheum. 1998, 41, 1632–1638. [Google Scholar] [CrossRef]
- Mistry, D.; Oue, Y.; Chambers, M.G.; Kayser, M.V.; Mason, R.M. Chondrocyte death during murine osteoarthritis. Osteoarthritis Cartilage 2004, 12, 131–141. [Google Scholar] [CrossRef]
- D’Lima, D.; Hermida, J.; Hashimoto, S.; Colwell, C.; Lotz, M. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006, 54, 1814–1821. [Google Scholar] [CrossRef] [PubMed]
- Zamli, Z.; Sharif, M. Chondrocyte apoptosis: A cause or consequence of osteoarthritis? Int. J. Rheum. Dis. 2011, 14, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Beg, A.A.; Baltimore, D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science 1996, 274, 782–784. [Google Scholar] [CrossRef] [PubMed]
- Van Antwerp, D.J.; Martin, S.J.; Kafri, T.; Green, D.R.; Verma, I.M. Suppression of TNF-alpha-induced apoptosis by NF-kappaB. Science 1996, 274, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.G.; Hsu, H.; Goeddel, D.V.; Karin, M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell 1996, 87, 565–576. [Google Scholar] [CrossRef]
- Wang, Y.; Li de, L.; Zhang, X.B.; Duan, Y.H.; Wu, Z.H.; Hao, D.S.; Chen, B.S.; Qiu, G.X. Increase of TNFalpha-stimulated osteoarthritic chondrocytes apoptosis and decrease of matrix metalloproteinases 9 by NF-kappaB inhibition. Biomed. Environ. Sci. 2013, 26, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Ijiri, K.; Zerbini, L.F.; Peng, H.; Otu, H.H.; Tsuchimochi, K.; Otero, M.; Dragomir, C.; Walsh, N.; Bierbaum, B.E.; Mattingly, D.; et al. Differential expression of GADD45beta in normal and osteoarthritic cartilage: Potential role in homeostasis of articular chondrocytes. Arthritis Rheum. 2008, 58, 2075–2087. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Yong, Y.; Choi, S.W.; Kim, J.H.; Lee, J.E.; Kim, D.W. Constitutive RelA activation mediated by Nkx3.2 controls chondrocyte viability. Nat. Cell Biol. 2007, 9, 287–298. [Google Scholar] [CrossRef]
- Ryu, J.H.; Shin, Y.; Huh, Y.H.; Yang, S.; Chun, C.H.; Chun, J.S. Hypoxia-inducible factor-2alpha regulates Fas-mediated chondrocyte apoptosis during osteoarthritic cartilage destruction. Cell Death Differ. 2012, 19, 440–450. [Google Scholar] [CrossRef]
- Kobayashi, H.; Chang, S.H.; Mori, D.; Itoh, S.; Hirata, M.; Hosaka, Y.; Taniguchi, Y.; Okada, K.; Mori, Y.; Yano, F.; et al. Biphasic regulation of chondrocytes by Rela through induction of anti-apoptotic and catabolic target genes. Nat. Commun. 2016, 7, 13336. [Google Scholar] [CrossRef]
- Kim, J.R.; Yoo, J.J.; Kim, H.A. Therapeutics in Osteoarthritis Based on an Understanding of Its Molecular Pathogenesis. Int. J. Mol. Sci. 2018, 19, 674. [Google Scholar] [CrossRef] [PubMed]



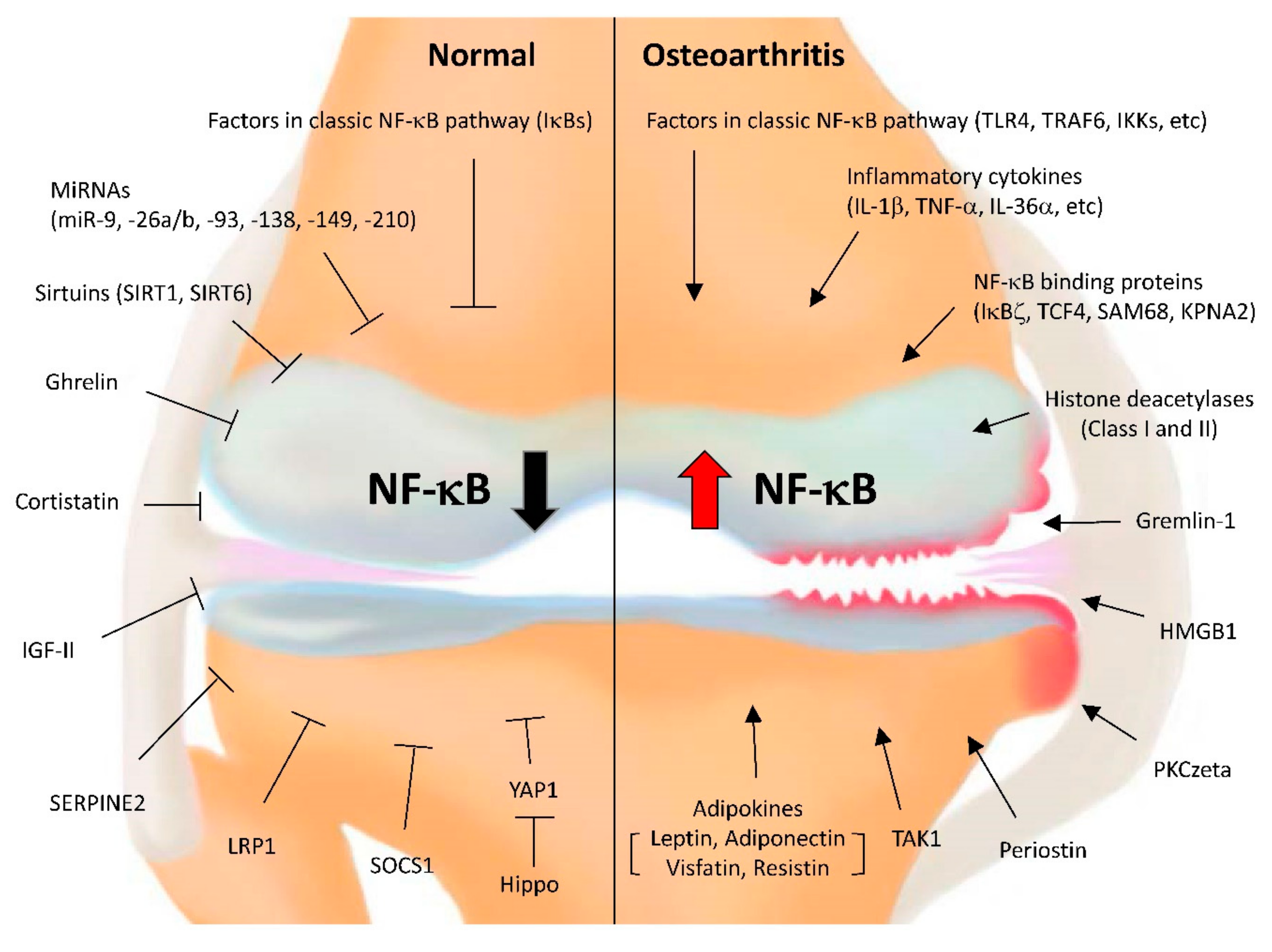{kind=link}
{kind=link}
| miRNA(s) | Regulation by NF-κB | Target Gene(s) | Function(s) in Chondrocytes | Reference |
|---|---|---|---|---|
| miR-365 | Increased | HDAC4 | Promotes catabolism | [191] |
| miR-204 | Increased | Multiple genes in PG biosynthesis pathway | Promotes OA development | [190] |
| miR-27b,-140 | Increased | MMP13 | Inhibits catabolism | [186,187] |
| miR-320 | Decreased | MMP13 | Inhibits catabolism | [194] |
| miR-92a-3p | Decreased | ADAMTS4/5 | Inhibits catabolism | [193] |
| miR-9 | ND | NF-κB p105/50 | Directly inhibits NF-κB | [14] |
| miR-138 | ND | NF-κB p65 | Directly inhibits NF-κB | [196] |
| miR-93 | ND | TLR4 | Inhibits NF-κB upstream | [197] |
| miR-210 | ND | DR6 | Inhibits NF-κB upstream | [198] |
| miR-26a/b | ND | KPNA3 | Inhibits NF-κB upstream | [199] |
| miR-149 | ND | TAK1 | Inhibits NF-κB upstream | [200] |
| miR-146a | Increased | TRAF6/IRAK1 | Inhibits NF-κB upstream | [189,201] |
| Smad4 | Promotes OA development | [204,205,206] | ||
| miR-26a-5p | Decreased | iNOS | Inhibits NF-κB downstream | [192] |
| miR-558 | Decreased | COX2 | Inhibits NF-κB downstream | [195] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, M.-C.; Jo, J.; Park, J.; Kang, H.K.; Park, Y. NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells 2019, 8, 734. https://doi.org/10.3390/cells8070734
Choi M-C, Jo J, Park J, Kang HK, Park Y. NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells. 2019; 8(7):734. https://doi.org/10.3390/cells8070734
Chicago/Turabian StyleChoi, Moon-Chang, Jiwon Jo, Jonggwan Park, Hee Kyoung Kang, and Yoonkyung Park. 2019. "NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction" Cells 8, no. 7: 734. https://doi.org/10.3390/cells8070734
APA StyleChoi, M.-C., Jo, J., Park, J., Kang, H. K., & Park, Y. (2019). NF-κB Signaling Pathways in Osteoarthritic Cartilage Destruction. Cells, 8(7), 734. https://doi.org/10.3390/cells8070734