HIPK2 Phosphorylates the Microtubule-Severing Enzyme Spastin at S268 for Abscission

 , and
, and 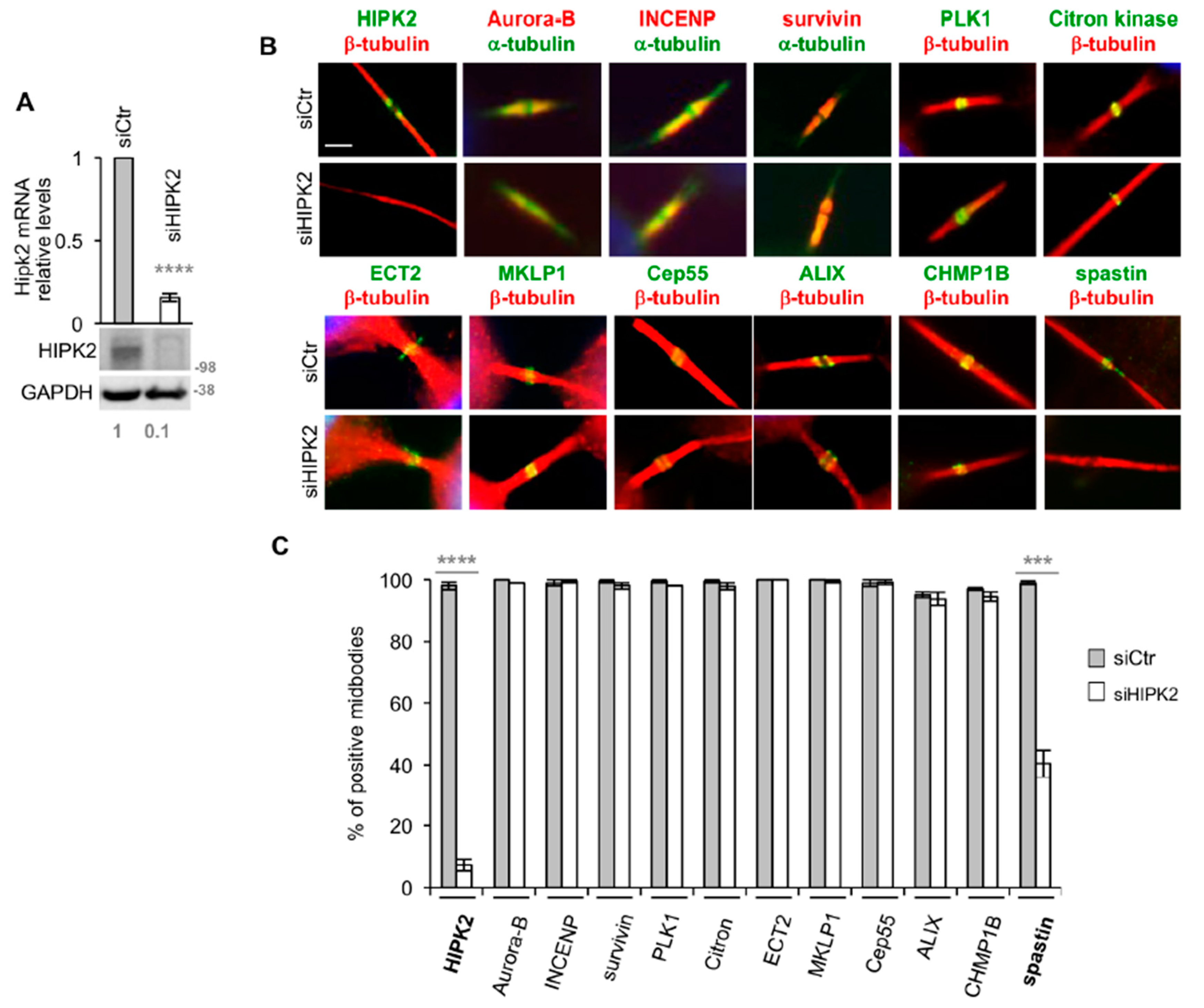{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Culture Conditions and Expression Vectors
2.2. RNA Interference (RNAi) and Real-Time (Reverse Transcription)-PCR (Real-Time (RT)-PCR)
- siHIPK2#1: CCCGAGUCAGUAUCCAGCCCAAUUU;
- siHIPK2#2: CCACCAACCUGACCAUGACCUUUAA;
- siHIPK2#3: CAGGGUUUGCCUGCUGAAUAUUUAU;
- siSpastin#1: CCAGUGAGAUGAGAAAUAUUCGAUU;
- siSpastin#2: CCAGUGAGAUGAGAAAUAUUCGAUU.
2.3. Western Blot (WB) and Co-Immunoprecipitation (co-IP)
2.4. Kinase Assay and Recombinant Proteins
2.5. GST Pull-Down and Binding Assay
2.6. Mass Spectrometry (MS)
2.7. p-S268 Spastin Ab Production and Purification
2.8. Immunofluorescence (IF) and Quantitative Analysis of Signals
2.9. MT Depolymerization Assay
2.10. Statistics
3. Results
3.1. Spastin is Not Detectable at the Midbody in HIPK2-Depleted Cells
3.2. HIPK2-Depleted Cells Show Abscission Defects Similar to Spastin-Depleted Cells
3.3. HIPK2 Phosphorylates Spastin at S268
3.4. Spastin-S268 Phosphorylation Is Present at the Midbody and Is Required for Abscission
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mierzwa, B.; Gerlich, D.W. Cytokinetic abscission: Molecular mechanisms and temporal control. Dev. Cell 2014, 31, 525–538. [Google Scholar] [CrossRef] [PubMed]
- Glotzer, M. Cytokinesis in Metazoa and Fungi. Cold Spring Harb. Perspect. Biol. 2017, 9, a022343. [Google Scholar] [CrossRef] [PubMed]
- Stoten, C.L.; Carlton, J.G. ESCRT-dependent control of membrane remodelling during cell division. Semin. Cell Dev. Biol. 2018, 74, 50–65. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Rismanchi, N.; Renvoisé, B.; Lippincott-Schwartz, J.; Blackstone, C.; Hurley, J.H. Structural basis for midbody targeting of spastin by the ESCRT-III protein CHMP1B. Nat. Struct. Mol. Biol. 2008, 15, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Connell, J.W.; Lindon, C.; Luzio, J.P.; Reid, E. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic 2009, 10, 42–56. [Google Scholar] [CrossRef] [PubMed]
- Claudiani, P.; Riano, E.; Errico, A.; Andolfi, G.; Rugarli, E.I. Spastin subcellular localization is regulated through usage of different translation start sites and active export from the nucleus. Exp. Cell Res. 2005, 309, 358–369. [Google Scholar] [CrossRef] [PubMed]
- D’Avino, P.P.; Capalbo, L. Regulation of midbody formation and function by mitotic kinases. Semin. Cell Dev. Biol. 2016, 53, 57–63. [Google Scholar] [CrossRef]
- Nähse, V.; Christ, L.; Stenmark, H.; Campsteijn, C. The Abscission Checkpoint: Making It to the Final Cut. Trends Cell Biol. 2017, 27, 1–11. [Google Scholar] [CrossRef]
- Mancuso, G.; Rugarli, E.I. A cryptic promoter in the first exon of the SPG4 gene directs the synthesis of the 60-kDa spastin isoform. BMC Biol. 2008, 6, 31. [Google Scholar] [CrossRef]
- Solowska, J.; Garbern, J.; Baas, P.W. Evaluation of loss-of-function as an explanation for SPG4-based hereditary spastic paraplegia. Hum. Mol. Genet. 2010, 19, 2767–2779. [Google Scholar] [CrossRef]
- Guizetti, J.; Schermelleh, L.; Mäntler, J.; Maar, S.; Poser, I.; Leonhardt, H.; Müller-Reichert, T.; Gerlich, D.W. Cortical constriction during abscission involves helices of ESCRT-III-dependent filaments. Science 2011, 331, 1616–1620. [Google Scholar] [CrossRef] [PubMed]
- Goliand, I.; Adar-Levor, S.; Segal, I.; Nachmias, D.; Dadosh, T.; Kozlov, M.M.; Elia, N. Resolving ESCRT-III Spirals at the Intercellular Bridge of Dividing Cells Using 3D STORM. Cell Rep. 2018, 24, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Allison, R.; Lumb, J.H.; Fassier, C.; Connell, J.W.; Ten Martin, D.; Seaman, M.N.; Hazan, J.; Reid, E. An ESCRT-spastin interaction promotes fission of recycling tubules from the endosome. J. Cell Biol. 2013, 202, 527–543. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, C.; Orso, G.; Mancuso, G.; Herholz, M.; Gumeni, S.; Tadepalle, N.; Jüngst, C.; Tzschichholz, A.; Schauss, A.; Höning, S.; et al. Spastin binds to lipid droplets and affects lipid metabolism. PLoS Genet. 2015, 11, e1005149. [Google Scholar] [CrossRef] [PubMed]
- Errico, A.; Claudiani, P.; D’Addio, M.; Rugarli, E.I. Spastin interacts with the centrosomal protein NA14, and is enriched in the spindle pole, the midbody and the distal axon. Hum. Mol. Genet. 2004, 13, 2121–2132. [Google Scholar] [CrossRef]
- Ventimiglia, L.N.; Cuesta-Geijo, M.A.; Martinelli, N.; Caballe, A.; Macheboeuf, P.; Miguet, N.; Parnham, I.M.; Olmos, Y.; Carlton, J.G.; Weissenhorn, W.; et al. CC2D1B Coordinates ESCRT-III Activity during the Mitotic Reformation of the Nuclear Envelope. Dev. Cell 2018, 47, 547–563. [Google Scholar] [CrossRef] [PubMed]
- Saul, V.V.; de la Vega, L.; Milanovic, M.; Krüger, M.; Braun, T.; Fritz-Wolf, K.; Becker, K.; Schmitz, M.L. HIPK2 kinase activity depends on cis-autophosphorylation of its activation loop. J. Mol. Cell Biol. 2013, 5, 27–38. [Google Scholar] [CrossRef]
- Siepi, F.; Gatti, V.; Camerini, S.; Crescenzi, M.; Soddu, S. HIPK2 catalytic activity and subcellular localization are regulated by activation-loop Y354 autophosphorylation. Biochim. Biophys. Acta 2013, 1833, 1443–1453. [Google Scholar] [CrossRef]
- Scaglione, A.; Monteonofrio, L.; Parisi, G.; Cecchetti, C.; Siepi, F.; Rinaldo, C.; Giorgi, A.; Verzili, D.; Zamparelli, C.; Savino, C.; et al. Effects of Y361-auto-phosphorylation on structural plasticity of the HIPK2 kinase domain. Protein Sci. 2018, 27, 725–737. [Google Scholar] [CrossRef]
- D’Orazi, G.; Rinaldo, C.; Soddu, S. Updates on HIPK2: A resourceful oncosuppressor for clearing cancer. J. Exp. Clin. Cancer Res. 2012, 31, 63. [Google Scholar] [CrossRef]
- Blaquiere, J.A.; Verheyen, E.M. Homeodomain-Interacting Protein Kinases: Diverse and Complex Roles in Development and Disease. Curr. Top. Dev. Biol. 2017, 123, 73–103. [Google Scholar] [PubMed]
- Rinaldo, C.; Moncada, A.; Gradi, A.; Ciuffini, L.; D’Eliseo, D.; Siepi, F.; Prodosmo, A.; Giorgi, A.; Pierantoni, G.M.; Trapasso, F.; et al. HIPK2 controls cytokinesis and prevents tetraploidization by phosphorylating histone H2B at the midbody. Mol. Cell 2012, 47, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Valente, D.; Bossi, G.; Moncada, A.; Tornincasa, M.; Indelicato, S.; Piscuoglio, S.; Karamitopoulou, E.D.; Bartolazzi, A.; Pierantoni, G.M.; Fusco, A.; et al. HIPK2 deficiency causes chromosomal instability by cytokinesis failure and increases tumorigenicity. Oncotarget 2015, 6, 10320–10334. [Google Scholar] [CrossRef]
- Monteonofrio, L.; Valente, D.; Ferrara, M.; Camerini, S.; Miscione, R.; Crescenzi, M.; Rinaldo, C.; Soddu, S. HIPK2 and extrachromosomal histone H2B are separately recruited by Aurora-B for cytokinesis. Oncogene 2018, 37, 3562–3574. [Google Scholar] [CrossRef] [PubMed]
- Hu, C.K.; Coughlin, M.; Mitchison, T.J. Midbody assembly and its regulation during cytokinesis. Mol. Biol. Cell 2012, 23, 1024–1034. [Google Scholar] [CrossRef] [PubMed]
- Riano, E.; Martignoni, M.; Mancuso, G.; Cartelli, D.; Crippa, F.; Toldo, I.; Siciliano, G.; Di Bella, D.; Taroni, F.; Bassi, M.T.; et al. Pleiotropic effects of spastin on neurite growth depending on expression levels. J. Neurochem. 2009, 108, 1277–1288. [Google Scholar] [CrossRef]
- Cecchinelli, B.; Porrello, A.; Lazzari, C.; Gradi, A.; Bossi, G.; D’Angelo, M.; Sacchi, A.; Soddu, S. Ser58 of mouse p53 is the homologue of human Ser46 and is phosphorylated by HIPK2 in apoptosis. Cell Death Differ. 2006, 13, 1994–1997. [Google Scholar] [CrossRef]
- Henson, B.J.; Zhu, W.; Hardaway, K.; Wetzel, J.L.; Stefan, M.; Albers, K.M.; Nicholls, R.D. Transcriptional and post-transcriptional regulation of SPAST, the gene most frequently mutated in hereditary spastic paraplegia. PLoS ONE 2012, 7, e36505. [Google Scholar] [CrossRef]
- Canbaz, D.M.; Kırımtay, K.; Karaca, E.; Karabay, A. SPG4 gene promoter regulation via Elk1 transcription factor. J. Neurochem. 2011, 117, 724–734. [Google Scholar] [CrossRef]
- Maddika, S.; Chen, J. Protein kinase DYRK2 is a scaffold that facilitates assembly of an E3 ligase. Nat. Cell Biol. 2009, 11, 409–419. [Google Scholar] [CrossRef]
- Whitehead, E.; Heald, R.; Wilbur, J.D. N-terminal phosphorylation of p60 katanin directly regulates microtubule severing. J. Mol. Biol. 2013, 425, 214–221. [Google Scholar] [CrossRef] [PubMed]
- McNally, F.J.; Roll-Mecak, A. Microtubule-severing enzymes: From cellular functions to molecular mechanism. J. Cell Biol. 2018, 217, 4057–4069. [Google Scholar] [CrossRef]
- Afonso, O.; Figueiredo, A.C.; Maiato, H. Late mitotic functions of Aurora kinases. Chromosoma 2017, 126, 93–103. [Google Scholar] [CrossRef] [PubMed]
- Combes, G.; Alharbi, I.; Braga, L.G.; Elowe, S. Playing polo during mitosis: PLK1 takes the lead. Oncogene 2017, 36, 4819–4827. [Google Scholar] [CrossRef] [PubMed]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pisciottani, A.; Biancolillo, L.; Ferrara, M.; Valente, D.; Sardina, F.; Monteonofrio, L.; Camerini, S.; Crescenzi, M.; Soddu, S.; Rinaldo, C. HIPK2 Phosphorylates the Microtubule-Severing Enzyme Spastin at S268 for Abscission. Cells 2019, 8, 684. https://doi.org/10.3390/cells8070684
Pisciottani A, Biancolillo L, Ferrara M, Valente D, Sardina F, Monteonofrio L, Camerini S, Crescenzi M, Soddu S, Rinaldo C. HIPK2 Phosphorylates the Microtubule-Severing Enzyme Spastin at S268 for Abscission. Cells. 2019; 8(7):684. https://doi.org/10.3390/cells8070684
Chicago/Turabian StylePisciottani, Alessandra, Loredana Biancolillo, Manuela Ferrara, Davide Valente, Francesca Sardina, Laura Monteonofrio, Serena Camerini, Marco Crescenzi, Silvia Soddu, and Cinzia Rinaldo. 2019. "HIPK2 Phosphorylates the Microtubule-Severing Enzyme Spastin at S268 for Abscission" Cells 8, no. 7: 684. https://doi.org/10.3390/cells8070684
APA StylePisciottani, A., Biancolillo, L., Ferrara, M., Valente, D., Sardina, F., Monteonofrio, L., Camerini, S., Crescenzi, M., Soddu, S., & Rinaldo, C. (2019). HIPK2 Phosphorylates the Microtubule-Severing Enzyme Spastin at S268 for Abscission. Cells, 8(7), 684. https://doi.org/10.3390/cells8070684