NS5A Gene Analysis by Next Generation Sequencing in HCV Nosocomial Transmission Clusters of HCV Genotype 1b Infected Patients

 ,
, 
 ,
,  ,
,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
2.1. Study Population
2.2. HCV Sanger Sequencing
2.3. HCV Next Generation Sequencing
2.4. Consensus Sequences at Different Cutoffs and Viral Haplotypes
2.5. Genetic Diversity Analysis
2.6. Phylogenetic Analysis
2.6.1. Genotype Assignment
2.6.2. Identification of Transmission Clusters
2.6.3. Statistical Analysis
3. Results
3.1. Patient Characteristics
3.2. NS3-NS5A-NS5B Sanger Sequences and NS5A NGS Sequences
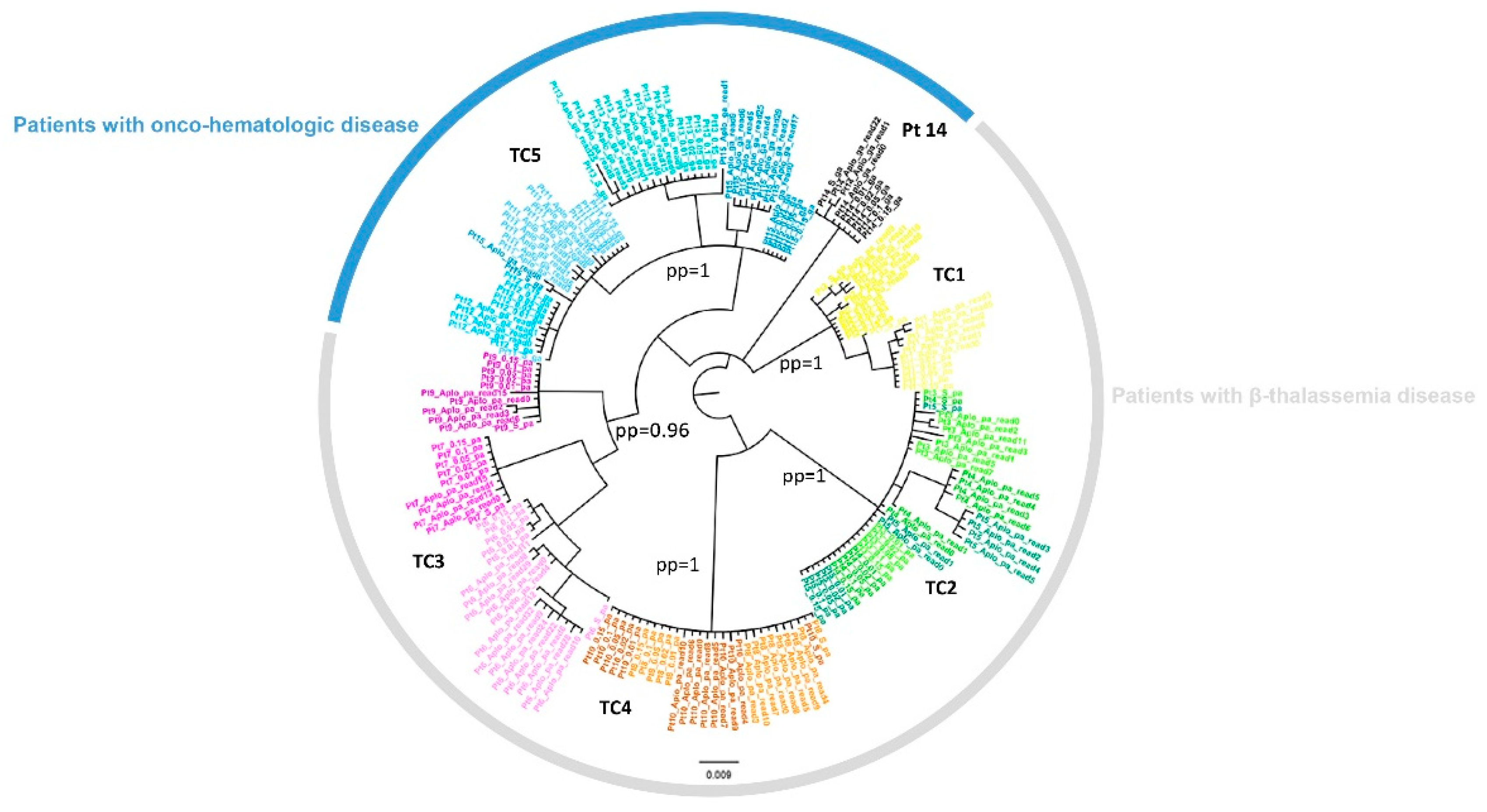
3.3. Cluster Identification
3.4. NS3-NS5A-NS5B RASs in Sanger Sequences and in NS5A NGS Sequences
3.5. Common Patterns of Variants Transmitted within the Same Cluster
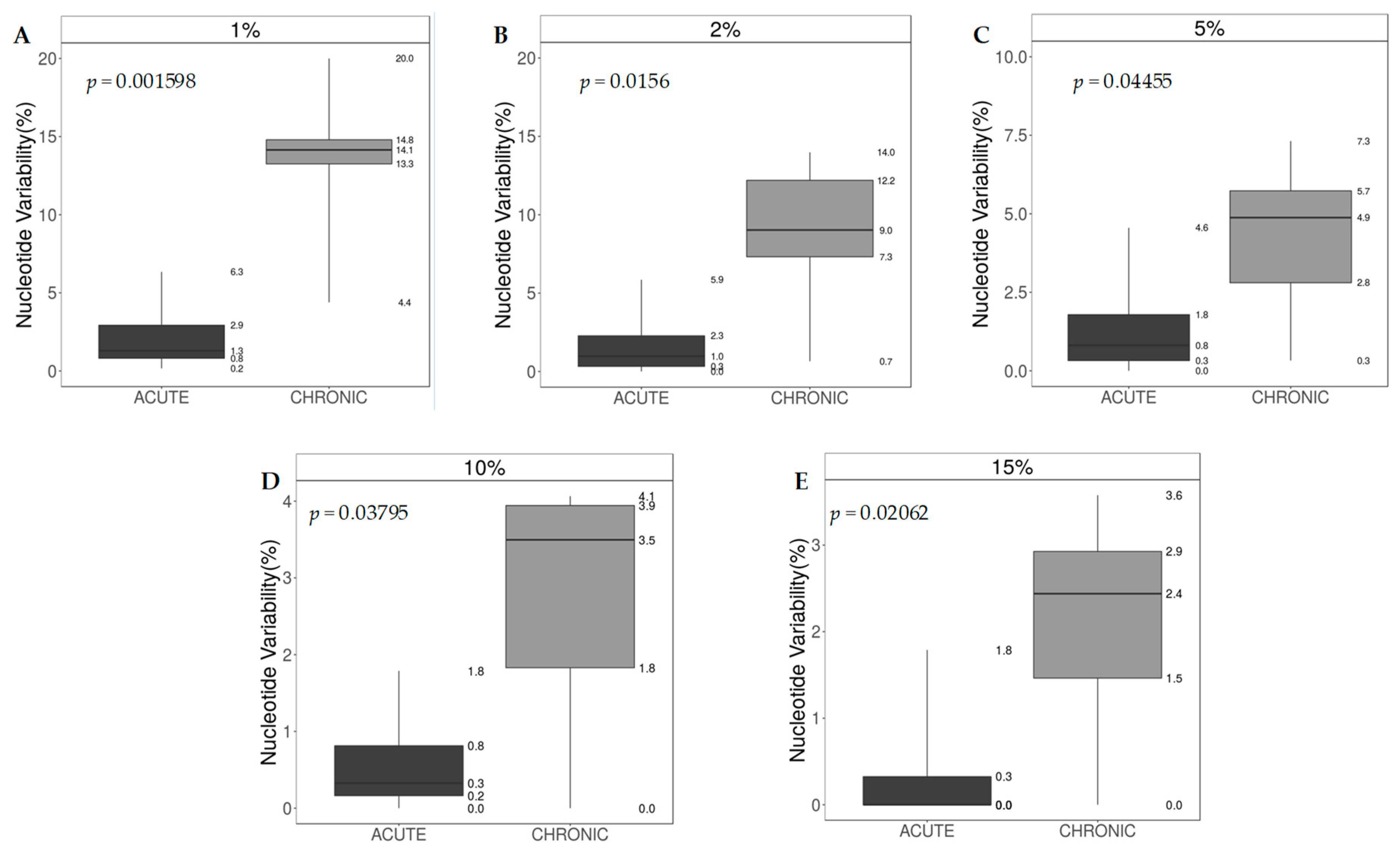
3.6. Nucleotide Sequence Variability and Shannon Entropy
3.7. IL28B Polymorphisms and Intra-Host HCV Variability
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- WHO World Health Organization Hepatitis C Virus Factsheet. Available online: https://www.who.int/en/news-room/fact-sheets/detail/hepatitis-c (accessed on 1 February 2019).
- Centers for Disease Control and Prevention (CDC); National Notifiable Diseases Surveillance System (NNDSS). Hepatitis C, Acute: 2016 Case Definition. Available online: https://wwwn.cdc.gov/nndss/conditions/hepatitis-c-acute/case-definition/2016/ (accessed on 2 April 2019).
- Fox, R.K.; Spach, D.H. Diagnosis of Acute HCV Infection. Available online: https://www.hepatitisc.uw.edu/go/screening-diagnosis/acute-diagnosis/core-concept/all (accessed on 2 April 2019).
- Pawlotsky, J.-M.; Negro, F.; Aghemo, A.; Berenguer, M.; Dalgard, O.; Dusheiko, G.; Marra, F.; Puoti, M.; Wedemeyer, H. EASL Recommendations on Treatment of Hepatitis C. J. Hepatol. 2018, 69, 461–511. [Google Scholar] [CrossRef] [PubMed]
- Thein, H.-H.; Yi, Q.; Dore, G.J.; Krahn, M.D. Estimation of stage-specific fibrosis progression rates in chronic hepatitis C virus infection: A meta-analysis and meta-regression. Hepatology 2008, 48, 418–431. [Google Scholar] [CrossRef] [PubMed]
- Friedman, S.L. Evolving challenges in hepatic fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2010, 7, 425–436. [Google Scholar] [CrossRef] [PubMed]
- Pozzetto, B.; Memmi, M.; Garraud, O.; Roblin, X.; Berthelot, P.; Pozzetto, B.; Memmi, M.; Garraud, O.; Berthelot, P.; Immunité, G. Health care-associated hepatitis C virus infection. World J. Gastroenterol. 2014, 20, 17265–17278. [Google Scholar] [CrossRef] [PubMed]
- Prati, D. Transmission of hepatitis C virus by blood transfusions and other medical procedures: A global review. J. Hepatol. 2006, 45, 607–616. [Google Scholar] [CrossRef] [PubMed]
- Johannessen, I.; Danial, J.; Smith, D.B.; Richards, J.; Imrie, L.; Rankin, A.; Willocks, L.J.; Evans, C.; Leen, C.; Gibson, P.; et al. Molecular and epidemiological evidence of patient-to-patient hepatitis C virus transmission in a Scottish emergency department. J. Hosp. Infect. 2018, 98, 412–418. [Google Scholar] [CrossRef] [PubMed]
- Heikens, E.; Hetem, D.J.; Jousma-rutjes, J.P.W.; Nijhuis, W.; Boland, G.J.; Hommes, N.H.; Thang, O.H.D.; Schuurman, R. Hepatitis C virus transmission in a Dutch haemodialysis unit: Detailed outbreak investigation using NS5A gene sequencing. J. Hosp. Infect. 2019, 101, 333–338. [Google Scholar] [CrossRef]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef]
- Borgia, S.M.; Hedskog, C.; Parhy, B.; Hyland, R.H.; Stamm, L.M.; Brainard, D.M.; Subramanian, M.G.; McHutchison, J.G.; Mo, H.; Svarovskaia, E.; et al. Identification of a Novel Hepatitis C Virus Genotype From Punjab, India: Expanding Classification of Hepatitis C Virus Into 8 Genotypes. J. Infect. Dis. 2018, 218, 1722–1729. [Google Scholar] [CrossRef]
- Hedskog, C.; Parhy, B.; Chang, S.; Zeuzem, S.; Moreno, C.; Shafran, S.D.; Borgia, S.M.; Asselah, T.; Alric, L.; Abergel, A.; et al. Identification of 19 Novel Hepatitis C Virus Subtypes—Further Expanding HCV Classification. Open Forum Infect. Dis. 2019, 6, ofz076. [Google Scholar] [CrossRef]
- Messina, J.P.; Humphreys, I.; Flaxman, A.; Brown, A.; Cooke, G.S.; Pybus, O.G.; Barnes, E. Global distribution and prevalence of hepatitis C virus genotypes. Hepatology 2015, 61, 77–87. [Google Scholar] [CrossRef]
- Bukh, J. The history of hepatitis C virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J. Hepatol. 2016, 65, S2–S21. [Google Scholar] [CrossRef]
- Mancusi, R.L.; Andreoni, M.; d’Angela, D.; Sarrecchia, C.; Spandonaro, F. Epidemiological burden estimates for pathologies with a nonconstant risk: An application to HCV in Italy according to age, Metavir score, and genotype: A systematic review and meta-analysis. Medicine (Baltimore) 2016, 95, e5143. [Google Scholar] [CrossRef]
- Progetto PITER. Available online: http://www.progettopiter.it/ (accessed on 9 May 2019).
- Rossetti, B.; Bai, F.; Tavelli, A.; Galli, M.; Antinori, A.; Castelli, F.; Pellizzer, G.; Cozzi-Lepri, A.; Bonora, S.; d’Arminio Monforte, A.; et al. Evolution of the prevalence of hepatitis C virus infection and hepatitis C virus genotype distribution in human immunodeficiency virus-infected patients in Italy between 1997 and 2015. Clin. Microbiol. Infect. 2018, 24, 422–427. [Google Scholar] [CrossRef]
- Kartashev, V.; Döring, M.; Nieto, L.; Coletta, E.; Kaiser, R.; Sierra, S.; Guerrero, A.; Stoiber, H.; Paar, C.; Vandamme, A.M.; et al. New findings in HCV genotype distribution in selected West European, Russian and Israeli regions. J. Clin. Virol. 2016, 81, 82–89. [Google Scholar] [CrossRef] [PubMed]
- Fondazione Vironet C—Fondazione Italiana per gli Studi di Resistenza ai Farmaci anti HCV. Available online: https://www.vironetc.org/ (accessed on 9 May 2019).
- Domingo, E.; Sheldon, J.; Perales, C. Viral Quasispecies Evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef]
- Spengler, U. Direct antiviral agents ( DAAs )—A new age in the treatment of hepatitis C virus infection. Pharmacol. Ther. 2018, 183, 118–126. [Google Scholar] [CrossRef]
- Bertoli, A.; Sorbo, M.C.; Aragri, M.; Lenci, I.; Teti, E.; Chiara, V.; Maio, D.; Gianserra, L.; Biliotti, E.; Masetti, C.; et al. Prevalence of Single and Multiple Natural NS3, NS5A and Substitutions in Hepatitis C Virus Genotypes 1–4 in Italy. Sci. Rep. 2018, 8, 8988. [Google Scholar] [CrossRef] [PubMed]
- Yerly, S.; Kaiser, L.; Race, E.; Bru, J.-P.; Clavel, F.; Perrin, L. Transmission of antiretroviral-drug-resistant HIV-1 variants. Lancet 1999, 354, 729–733. [Google Scholar] [CrossRef]
- Salpini, R.; Svicher, V.; Cento, V.; Gori, C.; Bertoli, A.; Scopelliti, F.; Micheli, V.; Cappiello, T.; Spanò, A.; Rizzardini, G.; et al. Characterization of drug-resistance mutations in HBV D-genotype chronically infected patients, naïve to antiviral drugs. Antivir. Res. 2011, 92, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Ge, D.; Fellay, J.; Thompson, A.J.; Simon, J.S.; Shianna, K.V.; Urban, T.J.; Heinzen, E.L.; Qiu, P.; Bertelsen, A.H.; Muir, A.J.; et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature 2009, 461, 399–401. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.L.; Thio, C.L.; Martin, M.P.; Qi, Y.; Ge, D.; Colm, O.; Kidd, J.; Kidd, K.; Khakoo, S.I.; Alexander, G.; et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature 2009, 461, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Suppiah, V.; Moldovan, M.; Ahlenstiel, G.; Berg, T.; Weltman, M.; Abate, M.L.; Bassendine, M.; Spengler, U.; Dore, G.J.; Powell, E.; et al. IL28B is associated with response to chronic hepatitis C interferon-α and ribavirin therapy. Nat. Genet. 2009, 41, 1100–1104. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, T.; Pybus, O.G.; Rambaut, A.; Salemi, M.; Cassol, S.; Ciccozzi, M.; Rezza, G.; Gattinara, G.C.; D’Arrigo, R.; Amicosante, M.; et al. Molecular epidemiology: HIV-1 and HCV sequences from Libyan outbreak. Nature 2006, 444, 836–837. [Google Scholar] [CrossRef] [PubMed]
- Aragri, M.; Fabeni, L. Identification of HCV Transmission Clusters in a Group of Thalassemic Patients with Diagnosis of ACUTE HCV Infection [Abstract]. Hepatology 2018, 68, 911A–912A. [Google Scholar]
- Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palù, G. Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 2011, 12, 7861–7884. [Google Scholar] [CrossRef]
- Guinoiseau, T.; Moreau, A.; Hohnadel, G.; Ngo-Giang-Huong, N.; Brulard, C.; Vourc’h, P.; Goudeau, A.; Gaudy-Graffin, C. Deep sequencing is an appropriate tool for the selection of unique Hepatitis C virus (HCV) variants after single genomic amplification. PLoS ONE 2017, 12, e0174852. [Google Scholar] [CrossRef]
- Mansoor, S.; Javed, A.; Ali, A.; Mansoor, A. Heterogeneous genomic locations within NS3, NS4A and NS4B identified for genotyping and subtyping of Hepatitis C virus: A simple genome analysis approach. Infect. Genet. Evol. 2016, 44, 61–68. [Google Scholar] [CrossRef]
- Andre-Garnier, E.; Besse, B.; Rodallec, A.; Ribeyrol, O.; Ferre, V.; Luco, C.; Le Guen, L.; Bourgeois, N.; Gournay, J.; Billaud, E.; et al. An NS5A single optimized method to determine genotype, subtype and resistance profiles of Hepatitis C strains. PLoS ONE 2017, 12, e0179562. [Google Scholar] [CrossRef]
- Brancaccio, G.; Sorbo, M.C.; Frigeri, F.; Rizzo, V.; Cantone, M.; Genderini, F.; Fabeni, L.; Pinto, A.; Perno, C.F.; Ceccherini-Silberstein, F.; et al. Treatment of Acute Hepatitis C With Ledipasvir and Sofosbuvir in Patients With Hematological Malignancies Allows Early Re-start of Chemotherapy. Clin. Gastroenterol. Hepatol. 2018, 16, 977–978. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, V.C.; Cento, V.; Aragri, M.; Paolucci, S.; Pollicino, T.; Coppola, N.; Bruzzone, B.; Ghisetti, V.; Zazzi, M.; Brunetto, M.; et al. Frequent NS5A and multiclass resistance in almost all HCV genotypes at DAA failures: What are the chances for second-line regimens? J. Hepatol. 2018, 68, 597–600. [Google Scholar] [CrossRef] [PubMed]
- Sorbo, M.C.; Cento, V.; Di Maio, V.C.; Howe, A.Y.M.; Garcia, F.; Perno, C.F.; Ceccherini-Silberstein, F. Hepatitis C virus drug resistance associated substitutions and their clinical relevance: Update 2018. Drug Resist. Updat. 2018, 37, 17–39. [Google Scholar] [CrossRef] [PubMed]
- Kalaghatgi, P.; Sikorski, A.M.; Knops, E.; Rupp, D.; Sierra, S.; Heger, E.; Neumann-Fraune, M.; Beggel, B.; Walker, A.; Timm, J.; et al. Geno2pheno [HCV]-A Web-based Interpretation System to Support Hepatitis C Treatment Decisions in the Era of Direct-Acting Antiviral Agents. PLoS ONE 2016, 11, e0155869. [Google Scholar] [CrossRef] [PubMed]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- bamToFreq. Available online: http://github.com/matdoering/bamToFreq (accessed on 15 November 2018).
- Döring, M.; Büch, J.; Friedrich, G.; Pironti, A.; Kalaghatgi, P.; Knops, E.; Heger, E.; Obermeier, M.; Däumer, M.; Thielen, A.; et al. geno2pheno[ngs-freq]: A genotypic interpretation system for identifying viral drug resistance using next-generation sequencing data. Nucleic Acids Res. 2018, 46, W271–W277. [Google Scholar] [CrossRef] [PubMed]
- Töpfer, A.; Zagordi, O.; Prabhakaran, S.; Roth, V.; Halperin, E.; Beerenwinkel, N. Probabilistic inference of viral quasispecies subject to recombination. J. Comput. Biol. 2013, 20, 113–123. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Tavarè, S. Some probabilistic and statistical problems in the analysis of DNA sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- FigTree. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 15 November 2018).
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2018; Available online: http://www.R-project.org/ (accessed on 15 November 2018).
- Kokubo, S.; Horii, T.; Yonekawa, O.; Ozawa, N.; Mukaide, M. A phylogenetic-tree analysis elucidating nosocomial transmission of hepatitis C virus in a haemodialysis unit. J. Viral Hepat. 2002, 9, 450–454. [Google Scholar] [CrossRef] [PubMed]
- Abacioglu, Y.H.; Bacaksiz, F.; Bahar, I.H.; Simmonds, P. Molecular evidence of nosocomial transmission of hepatitis C virus in a haemodialysis unit. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 182–186. [Google Scholar] [CrossRef] [PubMed]
- Duong, C.M.; McLaws, M.-L. An investigation of an outbreak of hepatitis C virus infections in a low-resourced hemodialysis unit in Vietnam. Am. J. Infect. Control 2016, 44, 560–566. [Google Scholar] [CrossRef]
- Garvey, M.I.; Bradley, C.W.; Holden, K.L.; Hewins, P.; Ngui, S.; Tedder, R.; Jumaa, P.; Smit, E. Use of genome sequencing to identify hepatitis C virus transmission in a renal healthcare setting. J. Hosp. Infect. 2017, 96, 157–162. [Google Scholar] [CrossRef]
- Nguyen, D.B.; Gutowski, J.; Ghiselli, M.; Cheng, T.; Bel Hamdounia, S.; Suryaprasad, A.; Xu, F.; Moulton-Meissner, H.; Hayden, T.; Forbi, J.C.; et al. A Large Outbreak of Hepatitis C Virus Infections in a Hemodialysis Clinic. Infect. Control Hosp. Epidemiol. 2016, 37, 125–133. [Google Scholar] [CrossRef]
- Senatore, S.; Galli, C.; Conti, A.; Faccini, M.; Cantoni, S.; Ciconali, G.; Mainardi, G.; Lamberti, A.; Dighera, R.; Radice Trolli, F.; et al. Hepatitis C virus outbreak in a haemodialysis unit: Learning from failures. J. Hosp. Infect. 2016, 3, 249–252. [Google Scholar] [CrossRef]
- Arnold, S.; Melville, S.K.; Morehead, B.; Vaughan, G.; Moorman, A.; Crist, M.B. Notes from the Field: Hepatitis C Transmission from Inappropriate Reuse of Saline Flush Syringes for Multiple Patients in an Acute Care General Hospital—Texas, 2015. MMWR. Morb. Mortal. Wkly. Rep. 2017, 66, 258–260. [Google Scholar] [CrossRef]
- Maria Ricerca, B.; Di Girolamo, A. Infections in Thalassemia a Therapy-Related Complications. Mediterr. J. Hematol. Infect. Dis. 2009, 1, 2009028. [Google Scholar]
- Ahmed Kiani, R.; Anwar, M.; Waheed, U.; Asad, M.J.; Abbasi, S.; Abbas Zaheer, H. Epidemiology of Transfusion Transmitted Infection among Patients with β-Thalassaemia Major in Pakistan. J. Blood Transfus. 2016, 2016, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Franco, S.; Tural, C.; Nevot, M.; Moltó, J.; Rockstroh, J.K.; Clotet, B.; Martinez, M.A. Detection of a sexually transmitted hepatitis C virus protease inhibitor-resistance variant in a human immunodeficiency virus-infected homosexual man. Gastroenterology 2014, 147, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Peiffer, K.-H.; Sommer, L.; Susser, S.; Vermehren, J.; Herrmann, E.; Döring, M.; Dietz, J.; Perner, D.; Berkowski, C.; Zeuzem, S.; et al. Interferon lambda 4 genotypes and resistance-associated variants in patients infected with hepatitis C virus genotypes 1 and 3. Hepatology 2016, 63, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Di Maio, V.C.; Cento, V.; Lenci, I.; Aragri, M.; Rossi, P.; Barbaliscia, S.; Melis, M.; Verucchi, G.; Magni, G.F.; Teti, E.; et al. Multiclass HCV resistance to direct-acting antiviral failure in real-life patients advocates for tailored second-line therapies. Liver Int. 2017, 37, 514–528. [Google Scholar] [CrossRef] [PubMed]
- Ramírez, C.; Gregori, J.; Buti, M.; Tabernero, D.; Camós, S.; Casillas, R.; Quer, J.; Esteban, S.; Homs, M.; Rodriguez-Frías, F. A comparative study of ultra-deep pyrosequencing and cloning to quantitatively analyze the viral quasispecies using hepatitis B virus infection as a model. Antivir. Res. 2013, 98, 273–283. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Patients, N | 28 | |
|---|---|---|
| Patients with β-thalassemia, N | 22 | |
| Patients with onco-hematologic disease, N | 6 | |
| Male, N (%) | 15 (53.5) | |
| Age, Median (IQR) | 43 (34–52) | |
| HCV genotype | 1b (100) | |
| Patients with available rs12979860 IL-28B genotype, N (%) | ||
| CC | 14 (50.0) | |
| CT | 11 (39.2) | |
| TT | 2 (7.1) | |
| Unknown | 1 (3.5) | |
| Acute Infection, N (%) | 17 (61) | |
| Diagnosis of Acute Infection, Year | 2016 | |
| Chronic Infection, N (%) | 11 (39) | |
| Naive * | 27 | |
| HCV-RNA (logIU/mL), Median (IQR) | 4.9 (3.3–6.0) | |
| AST (IU/mL), Median (IQR) ** | 271 (85–547) | |
| ALT (IU/mL), Median (IQR) ** | 485 (255–718) | |
| Cluster HCV | Pt | HCV Infection | HCV-RNA (IU/mL) | IL 28 | RAS | Other Mutations | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| NS3 Sanger | NS5B Sanger | NS5A Sanger | NS5A NGS | % NGS | NS3 Sanger | NS5A Sanger | NS5A NGS 1% | NS5B Sanger | |||||
| TC1 | Pt1 | Acute | 3270 | CT | None | None | Y93H | Y93H | 99.7% | V48I, V51A, A66G, T72I, P86Q, K87A, V132I, F147S, V170I, S174T (5–180aa) | K6R, S17T, K26R, L34V, L37F, K78R, R123QR, V164AE, V174T, Q176M (1–184aa) | K6R, S17T, K26R, L34V, L37F, K78R, V164E, V174T, Q176M (1–205 aa) | T181N, S210A, C213S (153–337aa) |
| Pt2 | Chronic | 2820000 | CC | None | None | Y93H | Y93HY | 97.8% | V48I, V51A, A66G, T72I, P86Q, K87A, V132I, F147LS, V170I, S174T (1–180aa) | K6R, S17ST, K26R, L34V, L37F, G49EG, K78R, V164A, V174T, Q176M (1–211aa) | K6R, V8IV, S17ST, K26KR, L34V, L37FL, G49EG, I63IL, K78R, C98CS, N105NS, R108KR, A114AS, N137NS, V138IMV, A146AT, V164AE, V174T, Q176MT (1–205aa) | S19NS, M57L, K81R, Q90K, R98K, N110NS, V116I, N117KN, K124E, Q127L, T181N, S210A, C213RS, S231NS, T377S, C451H, A513S, R531K (1–540aa) | |
| TC2 | Pt3 | Chronic | 2340000 | CT | None | None | Y93HY | Y93HY | 32.9% | S7A, V48I, Y56F, A66G, P86Q, K87S, S122N, V132I, F147S, A150V (2–180aa) | S3T, K6R, S17T, L34V, K44R, Q54H, T56IT, T64A, H85R, T122V, M133MV, V138LV, R157QR, V164A, V174T (1–213aa) | S3T, K6R, S17T, L34V, K44R, D50DE, I52IDNV, Q54H, T56IT, T64A, H85R, A92AT, T122AV, V124GV, M133MV, V138LIV, R157QR, V164A, V174T (1–205aa) | M57L, V85IV, Q90K, Q127L, N206K, K209A, A252AV, T377S, A513S, T520I, K523MR (1–548aa) |
| Pt4 | Acute | 2090 | CT | None | None | None | None | None | V48I, Y56F, A66G, P86Q, K87S, S122N, V132I, F147S, V170I (15–180aa) | S3T, K6R, S17T, L34V, K44R, Q54H, T64A, H85R, T122V, V138L, R157Q, V164A, V174T (1–187aa) | S3T, K6R, S17T, L34V, K44R, Q54H, T64A, H85R, T122V, V124GV, V138L, R157Q, V164A, V174T (1–205aa) | N206K, K209A, A252V, T377S, I424V, M426T, A513S, T520I, K523R (151–538aa) | |
| Pt5 | Acute | 165 | CC | None | None | None | None | None | V48I, Y56F, A66G, P86Q, K87S, S122N, V132I, F147S, V170I (15–180aa) | S3T, K6R, S17T, L34V, K44R, Q54H, T64A, H85R, T122V, V138L, R157Q, V164A, V174T, C190CG (1–196aa) | S3T, K6R, V15AV, S17T, P32PS, L34V, K44R, Q54H, A61AV, T64A, T83MT, H85R, T122MV, V124GV, G132AG, V138L, R157Q, V164A, V174T, L199I (1–205aa) | N206K, K209A, A252V, R254KR, E258EQ, T377S, A513S, T520I, K523R, S549G, V552A (151–562aa) | |
| TC3 | Pt6 | Chronic | 1800000 | CT | None | None | None | None | None | S7A, C16CW, V48I, S61A, A66G, P86Q, K87AS, F147S (1–180aa) | K6R, S17T, L34V, L37F, T56I, K78R, T79A, V164A, V174T, L183V, S201ST, M202MR, T213AT (1–213 aa) | K6R, S17AT, L34V, L37FL, Y43FY, Q54HQ, T56IT, I63FL, K78R, T79A, T83MT, N105NS, R108KR, V164AT, V174T, L183V, A197AT (1–205aa) | A15S, M57L, Q90K, N117R, R120N, Q127L, T130N, F162Y, G198K, N206NS, C213S, R254K, T377S, V405I, Q464E, V499T, A513S, R531K, S549G (1–565a aa) |
| Pt7 | Chronic | 577000 | CC | None | None | Y93H | Y93H | 99.65% | S7A, V48I, V51A, S61A, A66G, T72I, P86Q, K87A, S122G, F147S, V170I (1–181aa) | K6R, S17T, L34V, L37F, K44R, G49EG, Q54H, K78R, H85N, V138I, V164A, V174T, Q176L, L183V (1–194 aa) | K6R, S17T, L34V, F36FL, L37F, K44R, G49EG, Q54H, T56IT, T64AT, V75AV, K78R, H85NS, V124GV, F127FS, V138I, K139KR, V164A, V174T, Q176L, Q181HQ, L183V (1–205aa) | A15S, M57L, Q90K, V116I, N117R, R120N, Q127L, T130N, V147IV, F162Y, S189PS, G198KR, C213S, R254K, T377S, V405IV, A421V, I424V, T427P, Q464E, V499T, A513S, T520MT, Q544R, S549G, L564V, S565P (1–569 aa) | |
| Pt9 | Chronic | 94600 | CC | None | None | None | None | None | S7A, L14F, V48I, V51A, S61A, A66G, P86Q, K87A, F147S, S174A (1–180aa) | K6R, S17T, L34V, L37F, Q54H, V75A, K78R, T83M, Y161H, V164A, V174T (1–185aa) | K6R, I12IL, S17T, K26KR, L34V, P35LP, L37F, Q54H, V75A, K78R, C80CR, T83M, H85HCRY, A92AS, T99AT, P102LP, R108KR, Y161H, V164A, V174T, A197AT (1–205aa) | A15S, M57L, Q90K, N110S, V116I, N117R, R120N, Q127L, F162Y, K270R, T312S, L314S, V315A, A333AV, S335N, T377S, V405I, K441Q, Q464E, V499T, A513S, K535R, S549G (1–568aa) | |
| TC4 | Pt8 | Chronic | 219000 | CT | None | None | R30Q L31M Y93H | R30Q L31M Y93H | 98.8% 98.8% 99.1% | V48I, A66G, P86Q, K87A, F147S, A150V, I153V (1–180aa) | K6R, S17T, L34V, L37F, Q54H, K78R, R123Q, V124I, M133V, V164A, E171Q, V174T, Q176L, T204TP (1–210aa) | K6R, S17T, L34V, L37F, Q54H, N69NT, K78R, T95MT, R108KR, R123Q, V124I, M133MV, K139KR, V164A, E171Q, V174T, Q176L, A197T, L199V (1–205aa) | A39S, M57L, R65Q, Q90K, K106KR, S113G, Q127L, E131N, I134L, F162Y, S231N, I262V, T377S, A513S, R531K (1–568aa) |
| Pt10 | Acute | 163 | CC | None | None | R30Q L31M Y93H. | R30Q L31M Y93H | 99.4% 99.4% 97.6% | V48I, A66G, P86Q, K87A, F147S, A150V, I153V (16–180aa) | K6R, S17T, L34V, L37F, Q54H, K78R, R123Q, V124I, M133V, V164A, E171Q, V174T, Q176LQ (1–177aa) | K6R, S17T, L23LP, L34V, L37F, Q54H, K78R, R123Q, V124I, M133V, G155EG, V164A, E171Q, V174T, Q176L, A197T, L199V (1–205aa) | F162Y, S231N, I262V, T377S, T403AT, A513S, R531K (153–550aa) | |
| TC5 | Pt11 | Acute | 334000 | CC | None | None | None | None | None | S7A, L14F, S61A, T72TI, D103ND, R118RW, S122T (181aa) | K6R, S17T, L34V, L37F, T83M, V138I, V164A, V174T, A197T (1–213aa) | K6R, S17T, L34V, L37F, V46IV, V75FV, T83M, H85HFLY, V138I, V164A, V174T, A197T (1–205aa) | T19S, L31IV, L36M, L47Q, N117R, R120N, T130N, T132S, F162Y, G198K, E202D, A207T, A210S, A218S, N231S, A300S, V321I E333A, K355Q, Q464E, V499T, R510K, S549G (1–570aa) |
| Pt12 | Acute | 16100 | CC | None | None | Y93HY | Y93H | 99.2% | S7A, L14F, S61A, S122T (181aa) | K6R, S17T, L34V, L37F, T55AT, T83M, P89LP, H128HY, V138I, V164A, V174T, A197T (1–206aa) | K6R, S17AT, L34V, L37F, M53MV, T83M, V138I, V164A, V174T, A197T (1–205aa) | T19S, L36M, L47Q, N117R, R120N, T130N, T132S, G198K, E202D, A207T, A210S, A218S, N231S, A300T, V321I, E333A, K355Q, E437KE, Q464E, V499T, R510K, Q514R, S549G (1–580aa) | |
| Pt13 | Acute | 19690 | CT | None | None | None | None | None | S7A, V48I, S61A, S122T (1–140aa) | K6R, S17T, L34V, L37F, T56IT, T83M, A92T, T135A, V138I, V164A, V174T (1–186aa) | K6R, S17T, L34V, L37F, T83M, A92T, T135A, V138I, V164A, V174T, A197AT, T200AT (1–205aa) | A16TA, T19S, L36M, L47Q, R98K, N117R, R120N, , T130N, E131EG, T132S, F162Y, G198K, E202D, A207T, A210S, A218S, N231S, C242S, A300S, V321I, E333A, K355Q, K441Q, Q464E, V499T, R510K, S549G (1–570aa) | |
| Pt15 | Acute | 1620000 | NA | None | None | None | None | None | S7A, L13LF, L14F, S42SF, S61A, S122T, S93SF, S133SF (181aa) | K6R, S17T, L34V, L37F, T83M, V138I, V174T, A197T (1–213aa) | K6R, S17T, L34V, L37F, Q54HQ, T83M, A92AT, N105NS, V124GV, T135AT, V138I, R157LR, V164AV, V174T, P192PS, A197T, D205DE (1–205aa) | A16T, T19S, L36M, L47Q, R98K, N117R, R120N, T130N, T132S, F162Y, T181N, G189K, E202D, A207T, A210S, A218S, N231S, A300S, V321I, G328EG, E333A, K335Q, K441Q, Q464E, V499T, R510K, S549G, W574L, L588S (1–589aa) | |
| Cluster HCV | Pt | HCV Infection | Cut-off 1% | Cut-off 5% | Cut-off 15% | |||
|---|---|---|---|---|---|---|---|---|
| N Mutations | Common Patterns % | N Mutations | Common Patterns % | N Mutations | Common Patterns % | |||
| TC1 | Pt1 | Chronic | 20 | 50.00 | 13 | 69.23 | 11 | 81.82 |
| Pt2 | Acute | 10 | 10 | 10 | ||||
| TC2 | Pt3 | Chronic | 20 | 70.00 | 16 | 81.25 | 16 | 81.25 |
| Pt4 | Acute | 14 | 13 | 13 | ||||
| Pt5 | Acute | 20 | 14 | 14 | ||||
| TC4 | Pt8 | Chronic | 22 | 81.82 | 20 | 90.00 | 18 | 100 |
| Pt10 | Acute | 20 | 18 | 18 | ||||
| TC3 | Pt6 | Chronic | 17 | 39.11 | 13 | 55.87 | 11 | 56.21 |
| Pt7 | Chronic | 23 | 15 | 15 | ||||
| Pt9 | Chronic | 21 | 14 | 12 | ||||
| TC5 | Pt11 | Acute | 12 | 70.33 | 10 | 80.72 | 10 | 85.91 |
| Pt12 | Acute | 11 | 11 | 11 | ||||
| Pt13 | Acute | 12 | 11 | 11 | ||||
| Pt15 | Acute | 17 | 13 | 10 | ||||
| Patient ID | HCV Infection | Number Cluster | Sn a | Overall HCV Infection | Sn Median (IQR) | p Value b |
|---|---|---|---|---|---|---|
| Patients with β-thalassemia | ||||||
| 1 | Acute | 1 | 0.23 | |||
| 4 | Acute | 2 | 0.35 | |||
| 5 | Acute | 2 | 0.36 | Acute | 0.34 (0.27–0.36) | |
| 10 | Acute | 4 | 0.31 | |||
| 2 | Chronic | 1 | 0.65 | 0.01 c | ||
| 3 | Chronic | 2 | 0.93 | |||
| 6 | Chronic | 3 | 0.66 | |||
| 7 | Chronic | 3 | 0.80 | Chronic | 0.66 (0.60–0.81) | |
| 9 | Chronic | 3 | 0.59 | |||
| 8 | Chronic | 4 | 0.38 | |||
| Patients with onco-hematologic disease | ||||||
| 11 | Acute | 5 | 0.59 | |||
| 12 | Acute | 5 | 0.54 | |||
| 13 | Acute | 5 | 0.41 | Acute | 0.55 (0.54–0.60) | 0.02 d |
| 14 | Acute | Out of cluster | 0.68 | |||
| 15 | Acute | 5 | 0.53 | |||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bellocchi, M.C.; Aragri, M.; Carioti, L.; Fabeni, L.; Pipitone, R.M.; Brancaccio, G.; Sorbo, M.C.; Barbaliscia, S.; Di Maio, V.C.; Bronte, F.; et al. NS5A Gene Analysis by Next Generation Sequencing in HCV Nosocomial Transmission Clusters of HCV Genotype 1b Infected Patients. Cells 2019, 8, 666. https://doi.org/10.3390/cells8070666
Bellocchi MC, Aragri M, Carioti L, Fabeni L, Pipitone RM, Brancaccio G, Sorbo MC, Barbaliscia S, Di Maio VC, Bronte F, et al. NS5A Gene Analysis by Next Generation Sequencing in HCV Nosocomial Transmission Clusters of HCV Genotype 1b Infected Patients. Cells. 2019; 8(7):666. https://doi.org/10.3390/cells8070666
Chicago/Turabian StyleBellocchi, Maria Concetta, Marianna Aragri, Luca Carioti, Lavinia Fabeni, Rosaria Maria Pipitone, Giuseppina Brancaccio, Maria Chiara Sorbo, Silvia Barbaliscia, Velia Chiara Di Maio, Fabrizio Bronte, and et al. 2019. "NS5A Gene Analysis by Next Generation Sequencing in HCV Nosocomial Transmission Clusters of HCV Genotype 1b Infected Patients" Cells 8, no. 7: 666. https://doi.org/10.3390/cells8070666
APA StyleBellocchi, M. C., Aragri, M., Carioti, L., Fabeni, L., Pipitone, R. M., Brancaccio, G., Sorbo, M. C., Barbaliscia, S., Di Maio, V. C., Bronte, F., Grimaudo, S., Mazzucco, W., Frigeri, F., Cantone, M., Pinto, A., Perno, C. F., Craxì, A., Gaeta, G. B., Di Marco, V., & Ceccherini-Silberstein, F. (2019). NS5A Gene Analysis by Next Generation Sequencing in HCV Nosocomial Transmission Clusters of HCV Genotype 1b Infected Patients. Cells, 8(7), 666. https://doi.org/10.3390/cells8070666