Agenesis and Hypomyelination of Corpus Callosum in Mice Lacking Nsun5, an RNA Methyltransferase

Abstract
1. Introduction
2. Materials and Methods
2.1. Generation and Identification of Nsun5-null Mice
2.2. Antibodies
2.3. Histology Analyses
2.4. Western Blot Analysis
2.5. Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.6. Data Analysis/Statistics
3. Results
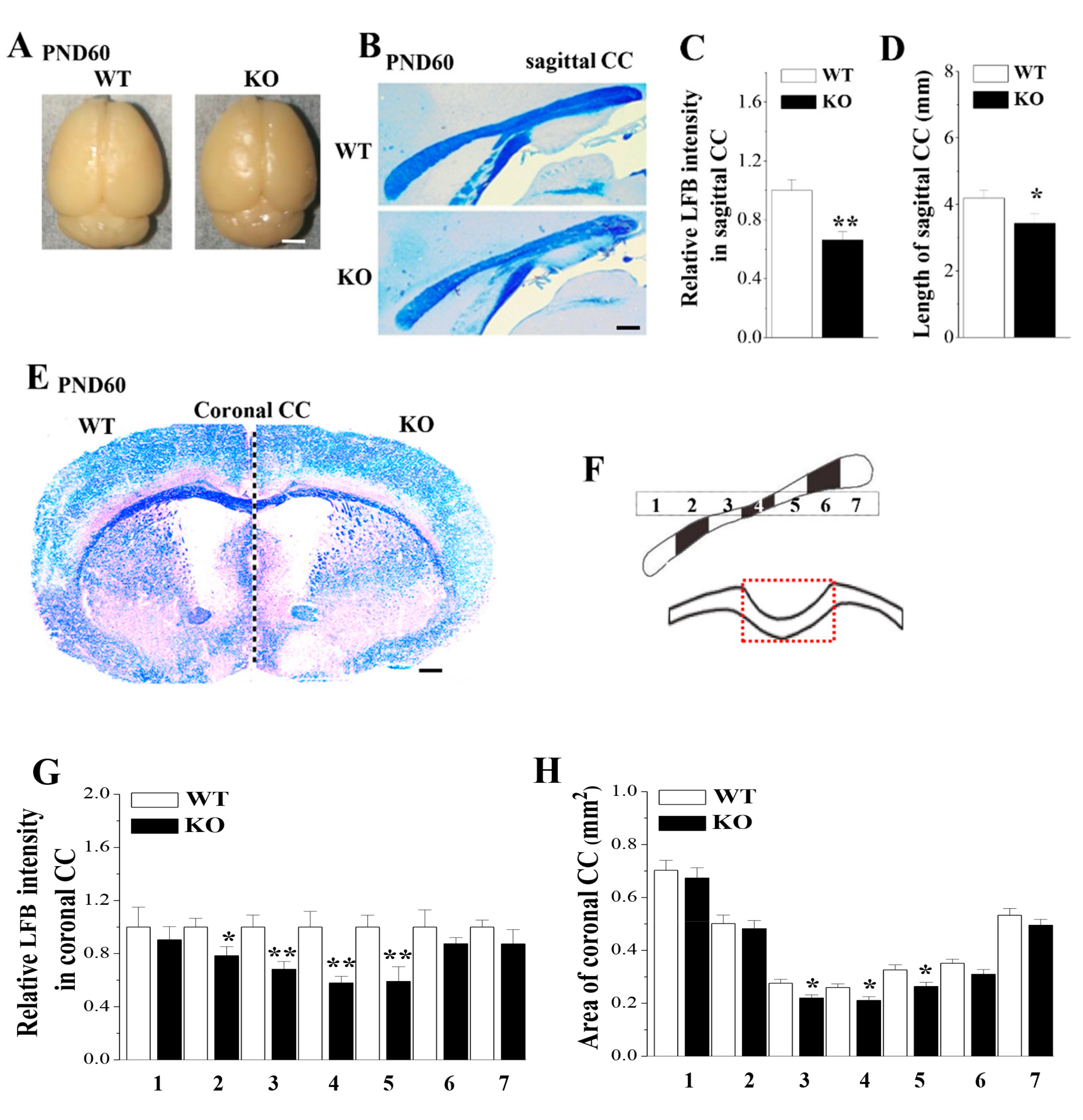
3.1. Loss of Nsun5 causes CC Agenesis
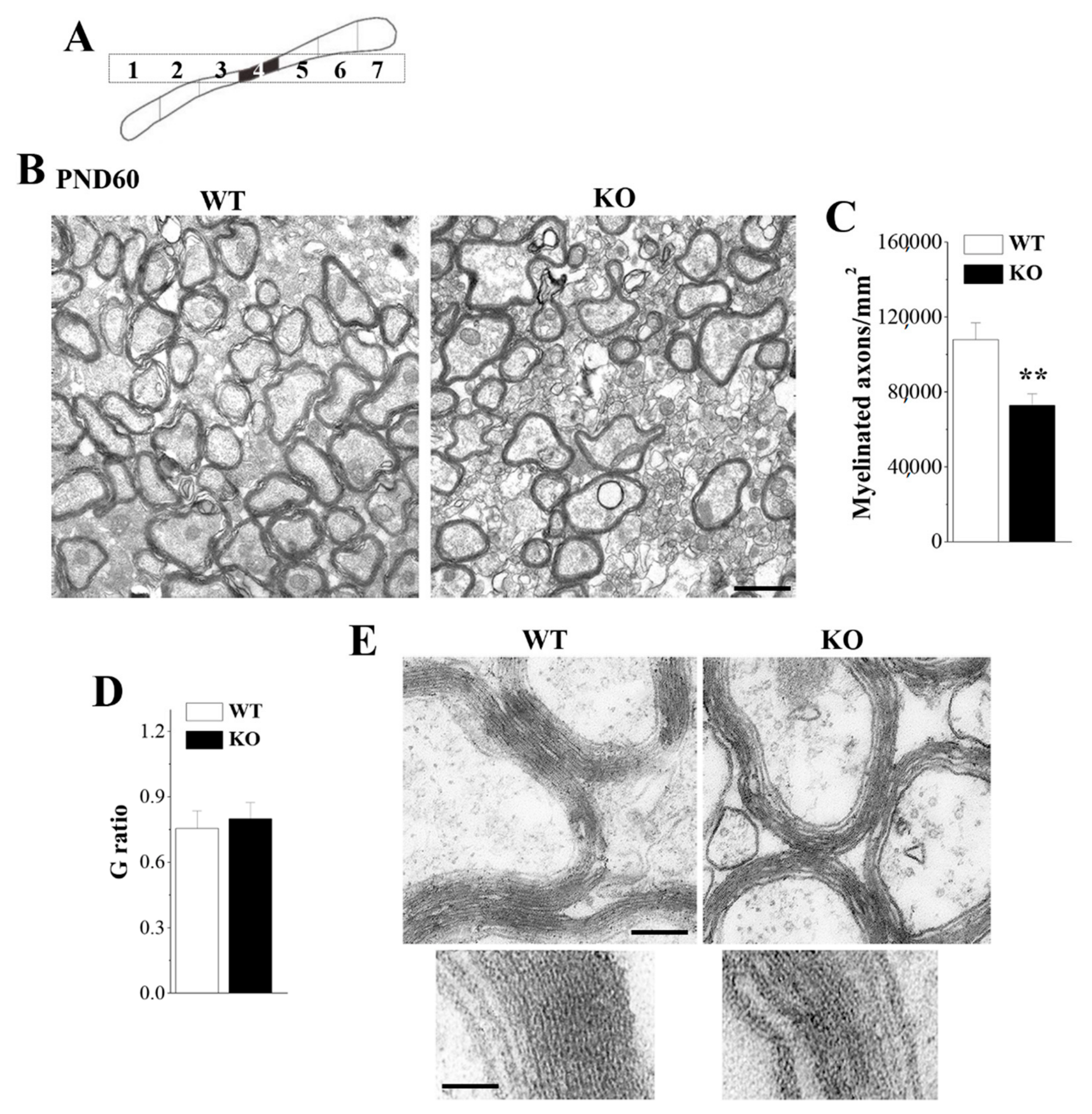
3.2. Loss of Nsun5 Results in Myelination Defects of CC
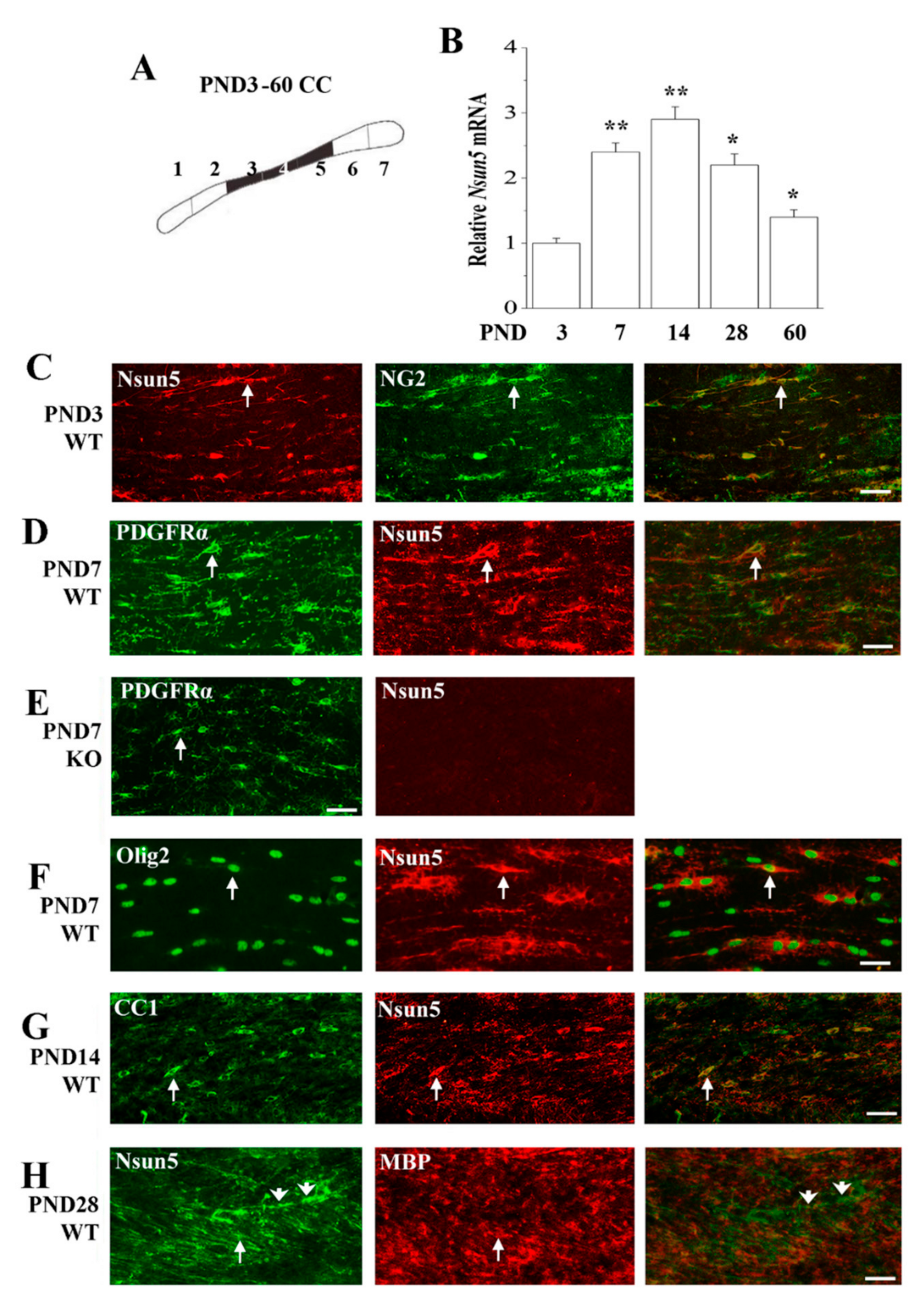
3.3. Nsun5 is Expressed in the OL Lineage of the Developing CC
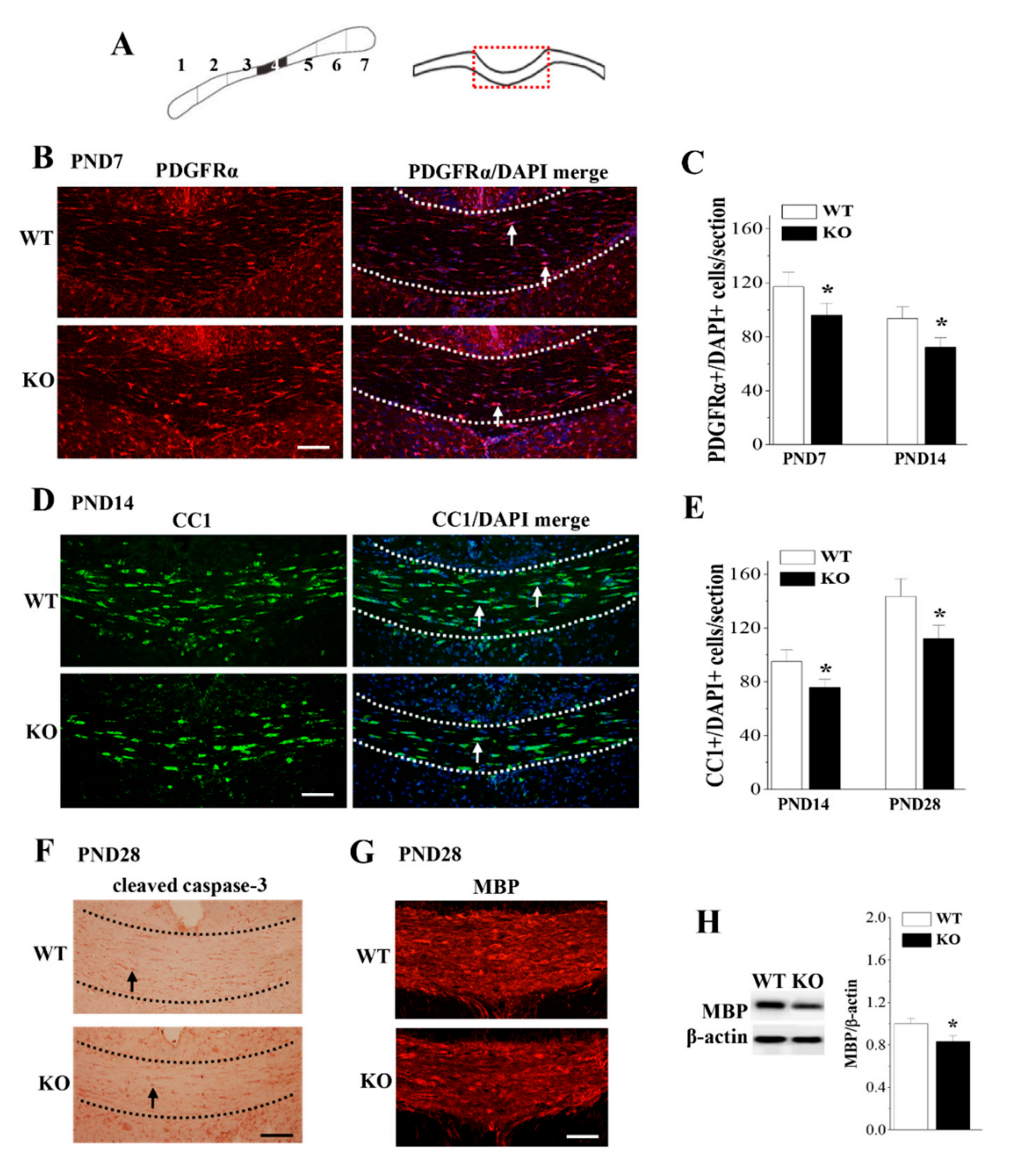
3.4. Loss of Nsun5 Reduces OPCs and OLs Leading to Hypomyelination of CC
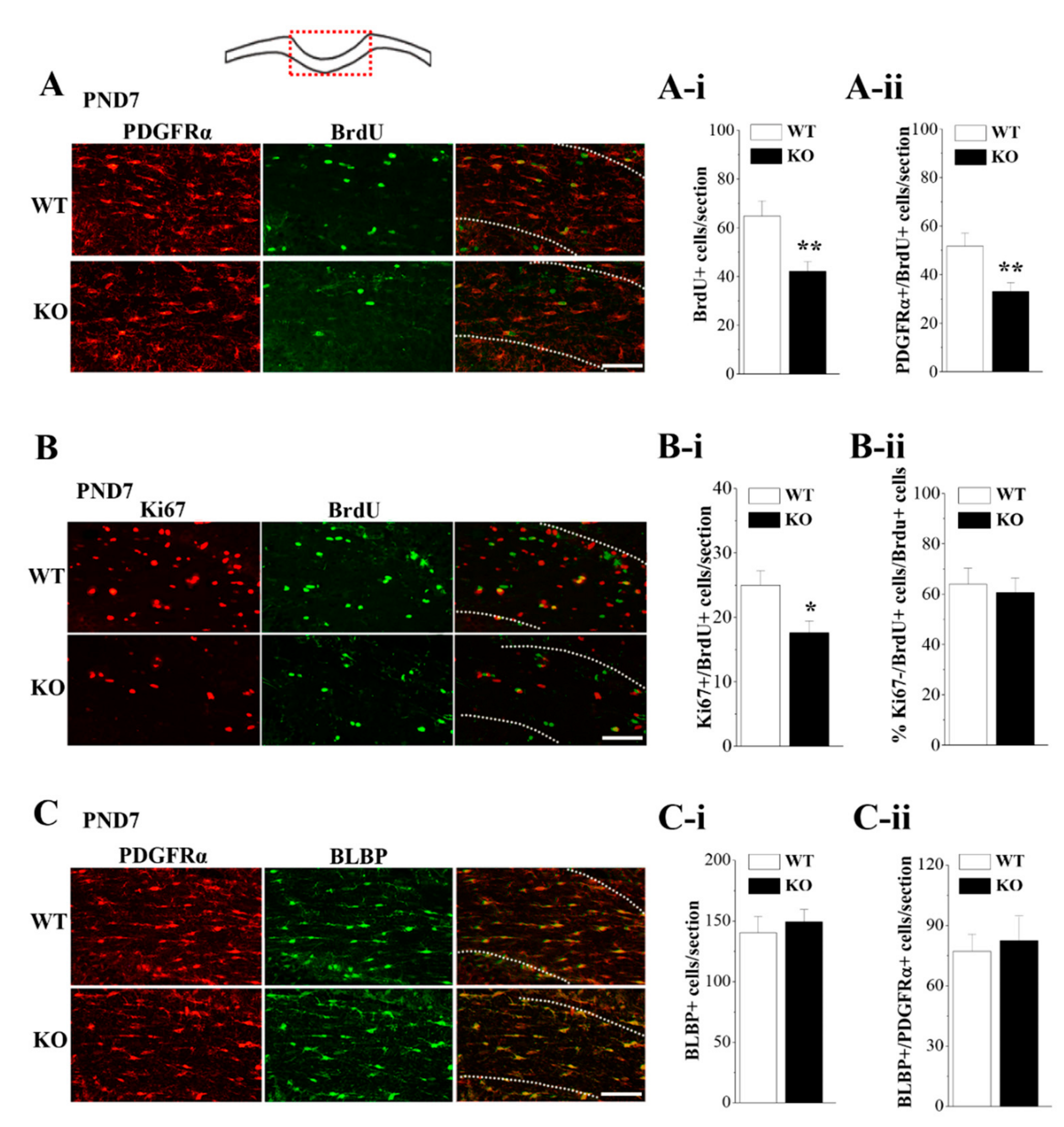
3.5. Loss of Nsun5 Suppresses the Proliferation of OPCs
3.6. Loss of Nsun5 Suppresses CDK1/2 Expression
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Pober, B.R. Williams-Beuren syndrome. N. Engl. J. Med. 2010, 362, 239–252. [Google Scholar] [CrossRef] [PubMed]
- Ewart, A.K.; Morris, C.A.; Atkinson, D.; Jin, W.; Sternes, K.; Spallone, P.; Stock, A.D.; Leppert, M.; Keating, M.T. Hemizygosity at the elastin locus in a developmental disorder, Williams syndrome. Nat. Genet. 1993, 5, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Mila, M.; Carrio, A.; Sanchez, A.; Gomez, D.; Jimenez, D.; Estivill, X.; Ballesta, F. Clinical characterization, molecular and FISH studies in 80 patients with clinical suspicion of Williams-Beuren syndrome. Med. Clin. (Barc) 1999, 113, 46–49. [Google Scholar] [PubMed]
- Van Essen, D.C.; Drury, H.A. Structural and functional analyses of human cerebral cortex using a surface-based atlas. J. Neurosci. 1997, 17, 7079–7102. [Google Scholar] [CrossRef] [PubMed]
- Luders, E.; di Paola, M.; Tomaiuolo, F.; Thompson, P.M.; Toga, A.W.; Vicari, S.; Petrides, M.; Caltagirone, C. Callosal morphology in Williams syndrome: A new evaluation of shape and thickness. Neuroreport 2007, 18, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Torniero, C.; Dalla Bernardina, B.; Novara, F.; Cerini, R.; Bonaglia, C.; Pramparo, T.; Ciccone, R.; Guerrini, R.; Zuffardi, O. Dysmorphic features, simplified gyral pattern and 7q11.23 duplication reciprocal to the Williams-Beuren deletion. Eur. J. Hum. Genet. 2008, 16, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.E.; Eliez, S.; Warsofsky, I.S.; Bellugi, U.; Reiss, A.L. Corpus callosum morphology of Williams syndrome: Relation to genetics and behavior. Dev. Med. Child. Neurol. 2001, 43, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Tomaiuolo, F.; Worsley, K.J.; Lerch, J.; di Paola, M.; Carlesimo, G.A.; Bonanni, R.; Caltagirone, C.; Paus, T. Changes in white matter in long-term survivors of severe non-missile traumatic brain injury: A computational analysis of magnetic resonance images. J. Neurotrauma 2005, 22, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Castiglia, L.; Husain, R.A.; Marquardt, I.; Fink, C.; Liehr, T.; Serino, D.; Elia, M.; Coci, E.G. 7q11.23 microduplication syndrome: Neurophysiological and neuroradiological insights into a rare chromosomal disorder. J. Intellect Disabil. Res. 2018, 62, 359–370. [Google Scholar] [CrossRef]
- Schubert, C. The genomic basis of the Williams-Beuren syndrome. Cell Mol. Life Sci. 2009, 66, 1178–1197. [Google Scholar] [CrossRef]
- Sharma, S.; Yang, J.; Watzinger, P.; Kotter, P.; Entian, K.D. Yeast Nop2 and Rcm1 methylate C2870 and C2278 of the 25S rRNA, respectively. Nucleic Acids Res. 2013, 41, 9062–9076. [Google Scholar] [CrossRef] [PubMed]
- Gigova, A.; Duggimpudi, S.; Pollex, T.; Schaefer, M.; Kos, M. A cluster of methylations in the domain IV of 25S rRNA is required for ribosome stability. RNA 2014, 20, 1632–1644. [Google Scholar] [CrossRef] [PubMed]
- Schosserer, M.; Minois, N.; Angerer, T.B.; Amring, M.; Dellago, H.; Harreither, E.; Calle-Perez, A.; Pircher, A.; Gerstl, M.P.; Pfeifenberger, S.; et al. Methylation of ribosomal RNA by NSUN5 is a conserved mechanism modulating organismal lifespan. Nat. Commun. 2015, 6, 6158. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Chen, P.; Li, W.; Sha, S.; Wang, Y.; Yuan, Z.; Shen, B.; Chen, L. Cognitive deficits in mice lacking Nsun5, a cytosine-5 RNA methyltransferase, with impairment of oligodendrocyte precursor cells. Glia 2018. [Google Scholar] [CrossRef] [PubMed]
- Baumann, N.; Pham-Dinh, D. Biology of oligodendrocyte and myelin in the mammalian central nervous system. Physiol. Rev. 2001, 81, 871–927. [Google Scholar] [CrossRef] [PubMed]
- Levine, J.M.; Reynolds, R.; Fawcett, J.W. The oligodendrocyte precursor cell in health and disease. Trends Neurosci. 2001, 24, 39–47. [Google Scholar] [CrossRef]
- Emery, B. Regulation of oligodendrocyte differentiation and myelination. Science 2010, 330, 779–782. [Google Scholar] [CrossRef]
- Nave, K.A. Myelination and the trophic support of long axons. Nat. Rev. Neurosci. 2010, 11, 275–283. [Google Scholar] [CrossRef]
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402. [Google Scholar] [CrossRef]
- Giera, S.; Deng, Y.; Luo, R.; Ackerman, S.D.; Mogha, A.; Monk, K.R.; Ying, Y.; Jeong, S.J.; Makinodan, M.; Bialas, A.R.; et al. The adhesion G protein-coupled receptor GPR56 is a cell-autonomous regulator of oligodendrocyte development. Nat. Commun. 2015, 6, 6121. [Google Scholar] [CrossRef]
- Zou, Y.; Jiang, W.; Wang, J.; Li, Z.; Zhang, J.; Bu, J.; Zou, J.; Zhou, L.; Yu, S.; Cui, Y.; et al. Oligodendrocyte precursor cell-intrinsic effect of Rheb1 controls differentiation and mediates mTORC1-dependent myelination in brain. J. Neurosci. 2014, 34, 15764–15778. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, V.A.; Tardif, V.; Lyssiotis, C.A.; Green, C.C.; Kerman, B.; Kim, H.J.; Padmanabhan, K.; Swoboda, J.G.; Ahmad, I.; Kondo, T.; et al. A regenerative approach to the treatment of multiple sclerosis. Nature 2013, 502, 327–332. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, E.C.; Pribyl, T.M.; Feng, J.M.; Kampf, K.; Spreur, V.; Campagnoni, C.; Colwell, C.S.; Reyes, S.D.; Martin, M.; Handley, V.; et al. Region-specific myelin pathology in mice lacking the golli products of the myelin basic protein gene. J. Neurosci. 2005, 25, 7004–7013. [Google Scholar] [CrossRef]
- Martinez-Montemayor, M.M.; Otero-Franqui, E.; Martinez, J.; de la Mota-Peynado, A.; Cubano, L.A.; Dharmawardhane, S. Individual and combined soy isoflavones exert differential effects on metastatic cancer progression. Clin. Exp. Metastasis 2010, 27, 465–480. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Lavine, J.A.; Raess, P.W.; Davis, D.B.; Rabaglia, M.E.; Presley, B.K.; Keller, M.P.; Beinfeld, M.C.; Kopin, A.S.; Newgard, C.B.; Attie, A.D. Overexpression of pre-pro-cholecystokinin stimulates beta-cell proliferation in mouse and human islets with retention of islet function. Mol. Endocrinol. 2008, 22, 2716–2728. [Google Scholar] [CrossRef]
- Choi, C.I.; Yoo, K.H.; Hussaini, S.M.; Jeon, B.T.; Welby, J.; Gan, H.; Scarisbrick, I.A.; Zhang, Z.; Baker, D.J.; van Deursen, J.M.; et al. The progeroid gene BubR1 regulates axon myelination and motor function. Aging (Albany NY) 2016, 8, 2667–2688. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, B.; Aguirre, A.; Vandenbosch, R.; Belachew, S.; Berthet, C.; Kaldis, P.; Gallo, V. Cdk2 is critical for proliferation and self-renewal of neural progenitor cells in the adult subventricular zone. J. Cell Biol. 2007, 179, 1231–1245. [Google Scholar] [CrossRef] [PubMed]
- Belachew, S.; Aguirre, A.A.; Wang, H.; Vautier, F.; Yuan, X.; Anderson, S.; Kirby, M.; Gallo, V. Cyclin-dependent kinase-2 controls oligodendrocyte progenitor cell cycle progression and is downregulated in adult oligodendrocyte progenitors. J. Neurosci. 2002, 22, 8553–8562. [Google Scholar] [CrossRef] [PubMed]
- Wolf, R.M.; Wilkes, J.J.; Chao, M.V.; Resh, M.D. Tyrosine phosphorylation of p190 RhoGAP by Fyn regulates oligodendrocyte differentiation. J. Neurobiol. 2001, 49, 62–78. [Google Scholar] [CrossRef]
- Morgan, D.O. Cyclin-dependent kinases: Engines, clocks and microprocessors. Annu. Rev. Cell Dev. Biol. 1997, 13, 261–291. [Google Scholar] [CrossRef]
- Ortega, S.; Prieto, I.; Odajima, J.; Martin, A.; Dubus, P.; Sotillo, R.; Barbero, J.L.; Malumbres, M.; Barbacid, M. Cyclin-dependent kinase 2 is essential for meiosis but not for mitotic cell division in mice. Nat. Genet. 2003, 35, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Aleem, E.; Kiyokawa, H.; Kaldis, P. Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat. Cell Biol. 2005, 7, 831–836. [Google Scholar] [CrossRef] [PubMed]
- Nigg, E.A. Mitotic kinases as regulators of cell division and its checkpoints. Nat. Rev. Mol. Cell Biol. 2001, 2, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Brandeis, M.; Rosewell, I.; Carrington, M.; Crompton, T.; Jacobs, M.A.; Kirk, J.; Gannon, J.; Hunt, T. Cyclin B2-null mice develop normally and are fertile whereas cyclin B1-null mice die in utero. Proc. Natl. Acad Sci. USA 1998, 95, 4344–4349. [Google Scholar] [CrossRef] [PubMed]
- Jablonska, B.; Gierdalski, M.; Chew, L.J.; Hawley, T.; Catron, M.; Lichauco, A.; Cabrera-Luque, J.; Yuen, T.; Rowitch, D.; Gallo, V. Sirt1 regulates glial progenitor proliferation and regeneration in white matter after neonatal brain injury. Nat. Commun. 2016, 7, 13866. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Humenik, J.; Yang, D.; Liebhaber, S.A. PolyC-binding proteins enhance expression of the CDK2 cell cycle regulatory protein via alternative splicing. Nucleic. Acids Res. 2018, 46, 2030–2044. [Google Scholar] [CrossRef] [PubMed]
- Caillava, C.; Vandenbosch, R.; Jablonska, B.; Deboux, C.; Spigoni, G.; Gallo, V.; Malgrange, B.; Baron-Van Evercooren, A. Cdk2 loss accelerates precursor differentiation and remyelination in the adult central nervous system. J. Cell Biol. 2011, 193, 397–407. [Google Scholar] [CrossRef]
- Berthet, C.; Aleem, E.; Coppola, V.; Tessarollo, L.; Kaldis, P. Cdk2 knockout mice are viable. Curr. Biol. 2003, 13, 1775–1785. [Google Scholar] [CrossRef]
- Tuorto, F.; Liebers, R.; Musch, T.; Schaefer, M.; Hofmann, S.; Kellner, S.; Frye, M.; Helm, M.; Stoecklin, G.; Lyko, F. RNA cytosine methylation by Dnmt2 and NSun2 promotes tRNA stability and protein synthesis. Nat. Struct. Mol. Biol. 2012, 19, 900–905. [Google Scholar] [CrossRef]
- Frye, M.; Watt, F.M. The RNA methyltransferase Misu (NSun2) mediates Myc-induced proliferation and is upregulated in tumors. Curr. Biol. 2006, 16, 971–981. [Google Scholar] [CrossRef]
- Xing, J.; Yi, J.; Cai, X.; Tang, H.; Liu, Z.; Zhang, X.; Martindale, J.L.; Yang, X.; Jiang, B.; Gorospe, M.; et al. NSun2 Promotes Cell Growth via Elevating Cyclin-Dependent Kinase 1 Translation. Mol. Cell Biol. 2015, 35, 4043–4052. [Google Scholar] [CrossRef] [PubMed]
- Yuen, T.J.; Silbereis, J.C.; Griveau, A.; Chang, S.M.; Daneman, R.; Fancy, S.P.J.; Zahed, H.; Maltepe, E.; Rowitch, D.H. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell 2014, 158, 383–396. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.R.; Sun, T.; Zhu, Z.; Ma, N.; Garcia, M.; Stiles, C.D.; Rowitch, D.H. Common developmental requirement for Olig function indicates a motor neuron/oligodendrocyte connection. Cell 2002, 109, 75–86. [Google Scholar] [CrossRef]
- Yue, T.; Xian, K.; Hurlock, E.; Xin, M.; Kernie, S.G.; Parada, L.F.; Lu, Q.R. A critical role for dorsal progenitors in cortical myelination. J. Neurosci. 2006, 26, 1275–1280. [Google Scholar] [CrossRef] [PubMed]
- Rajasekharan, S.; Baker, K.A.; Horn, K.E.; Jarjour, A.A.; Antel, J.P.; Kennedy, T.E. Netrin 1 and Dcc regulate oligodendrocyte process branching and membrane extension via Fyn and RhoA. Development 2009, 136, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Draghi, N.A.; Resh, M.D. Signaling from integrins to Fyn to Rho family GTPases regulates morphologic differentiation of oligodendrocytes. J. Neurosci. 2004, 24, 7140–7149. [Google Scholar] [CrossRef] [PubMed]
- Thurnherr, T.; Benninger, Y.; Wu, X.; Chrostek, A.; Krause, S.M.; Nave, K.A.; Franklin, R.J.; Brakebusch, C.; Suter, U.; Relvas, J.B. Cdc42 and Rac1 signaling are both required for and act synergistically in the correct formation of myelin sheaths in the CNS. J. Neurosci. 2006, 26, 10110–10119. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.W.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448. [Google Scholar] [CrossRef] [PubMed]
- Paul, L.K.; van Lancker-Sidtis, D.; Schieffer, B.; Dietrich, R.; Brown, W.S. Communicative deficits in agenesis of the corpus callosum: Nonliteral language and affective prosody. Brain Lang. 2003, 85, 313–324. [Google Scholar] [CrossRef]
- Karmiloff-Smith, A.; Grant, J.; Ewing, S.; Carette, M.J.; Metcalfe, K.; Donnai, D.; Read, A.P.; Tassabehji, M. Using case study comparisons to explore genotype-phenotype correlations in Williams-Beuren syndrome. J. Med. Genet. 2003, 40, 136–140. [Google Scholar] [CrossRef]
- Segura-Puimedon, M.; Sahun, I.; Velot, E.; Dubus, P.; Borralleras, C.; Rodrigues, A.J.; Valero, M.C.; Valverde, O.; Sousa, N.; Herault, Y.; et al. Heterozygous deletion of the Williams-Beuren syndrome critical interval in mice recapitulates most features of the human disorder. Hum. Mol. Genet. 2014, 23, 6481–6494. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| Nsun5 | GAGGGAAGGGTGGATAAGG | GGCACGATGCGGATGTAG |
| CDK2 | GTTGGTGATGGTGCTGTTG | CTGTGGATAACTTAGCGGTCG |
| CDK1 | AAAGCGAGGAAGAAGGAG | GGACAGGAACTCAAAGATGA |
| Cdc42 | GTTGGTGATGGTGCTGTTG | CTGTGGATAACTTAGCGGTCG |
| RhoA | CATTGACAGCCCTGATAGTT | TCGTCATTCCGAAGGTCCTT |
| GAPDH | TGGGTGTGAACCACGAG | ACCACAGTCCATGCCATCAC |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Z.; Chen, P.; Zhang, T.; Shen, B.; Chen, L. Agenesis and Hypomyelination of Corpus Callosum in Mice Lacking Nsun5, an RNA Methyltransferase. Cells 2019, 8, 552. https://doi.org/10.3390/cells8060552
Yuan Z, Chen P, Zhang T, Shen B, Chen L. Agenesis and Hypomyelination of Corpus Callosum in Mice Lacking Nsun5, an RNA Methyltransferase. Cells. 2019; 8(6):552. https://doi.org/10.3390/cells8060552
Chicago/Turabian StyleYuan, Zihao, Peipei Chen, Tingting Zhang, Bin Shen, and Ling Chen. 2019. "Agenesis and Hypomyelination of Corpus Callosum in Mice Lacking Nsun5, an RNA Methyltransferase" Cells 8, no. 6: 552. https://doi.org/10.3390/cells8060552
APA StyleYuan, Z., Chen, P., Zhang, T., Shen, B., & Chen, L. (2019). Agenesis and Hypomyelination of Corpus Callosum in Mice Lacking Nsun5, an RNA Methyltransferase. Cells, 8(6), 552. https://doi.org/10.3390/cells8060552